

SUMMARY
The tripartite AcrAB-TolC multidrug efflux-pump of Escherichia coli is the central conduit for cell-toxic compounds and contributes to antibiotic resistance. While high-resolution structures of all three proteins have been solved, much remains to be learned as to how the individual components come together to form a functional complex. In this study, we investigated the importance of the AcrB β-hairpins belonging to the DN and DC subdomains, which are presumed to dock with TolC, in complex stability and activity of the complete pump. Our data show that the DN subdomain β-hairpin residues play a more critical role in complex stability and activity than the DC subdomain hairpin residues. The failure of the AcrB DN β-hairpin deletion mutant to engage with TolC leads to the drug hypersensitivity phenotype, which is reversed by compensatory alterations in the lipoyl and β-barrel domains of AcrA. Moreover, AcrA and TolC mutants that induce TolC opening also reverse the drug hypersensitivity phenotype of the AcrB β-hairpin mutants, indicating a failure by the AcrB mutant to interact and thus induce TolC opening on its own. Together, these data suggest that both AcrB β-hairpins and AcrA act to stabilize the tripartite complex and induce TolC opening for drug expulsion.
Keywords: Multidrug efflux pump, protein-protein interactions, complex assembly, genetic suppression analysis, site-directed mutagenesis
INTRODUCTION
In Escherichia coli, the major multidrug efflux activity is provided by a tripartite multidrug efflux system TolC-AcrAB composed of the outer membrane channel TolC, the membrane-fusion protein AcrA, and is energized by the inner membrane proton-driven pump AcrB (Misra and Bavro 2009). Strains lacking any one of these proteins display a hypersensitivity phenotype to a wide variety of antibiotics, detergents, and bile salts. The TolC-AcrAB pump is one of the most extensively studied systems and is structurally best understood due to the availability of high-resolution atomic structures of all three proteins (Koronakis et al., 2000; Murakami et al., 2002; Mikolosko et al., 2006).
The trimeric, channel-forming outer-membrane factor (OMF) TolC folds into a unique α/β-barrel, with each protomer contributing one-third of the barrel structure (Koronakis et al., 2000). The outer membrane-embedded structure is a porin-like β-barrel, while the large periplasmic structure assumes a unique α-helical barrel fold which extends 100 Å into the periplasm. The periplasmic aperture of the α-helical barrel of TolC normally remains constitutively closed by the presence of conserved ionic-bridges (Koronakis et al., 2000), but must dilate upon formation of a functional tripartite efflux pump to enable the extrusion of substrate molecules. It is thought that a disruption of these ionic bridges (also called primary gates) occurs upon interaction with the drug-loaded AcrB (Andersen et al., 2002; Bavro et al., 2008).
The inner membrane protein (IMP) component of the efflux system, AcrB, is a drug/proton antiporter belonging to the Resistance Nodulation cell Division (RND) family (Nikaido and Pages, 2012). Similar to TolC, AcrB functions as a trimer: each monomer contains an integral membrane segment consisting of 12 trans-membrane (TM) helices and two large periplasmic loops, extending some 70 Å into the periplasm, inserted between TM 1/2 and TM 7/8 (Murakami et al., 2002 Seeger et al., 2006; Fig. 1). The structure of AcrB also revealed a conformational cycling of subunits, similar to a peristaltic pump mechanism (Murakami et al., 2006; Seeger et al., 2006; Pos, 2009), which is coupled to the proton translocation through the TM domain. The apically-located DN- and DC-hairpin loops of the periplasmic domain of AcrB (Fig. 1) are thought to come in direct contact with the bottom tip of the α-helical barrel of TolC (Murakami et al., 2002). The membrane fusion protein (MFP) AcrA is thought to stabilize interactions and seal the gap between TolC and AcrB, completing the efflux conduit (Zgurskaya et al., 2009; Symmons et al., 2009). The large α-helical hairpin domain of AcrA (Mikolosko et al., 2006) is nestled within an unusually elongated structure, in which the N- and C-termini are held in close proximity to one another. AcrA contains 3 additional domains, namely the membrane proximal (MP), the β-barrel and the lipoyl domains which are discontinuous in their primary sequence. These three domains interact with AcrB (Fernandez-Recio et al., 2004; Symmons et al., 2009).
Figure 1.
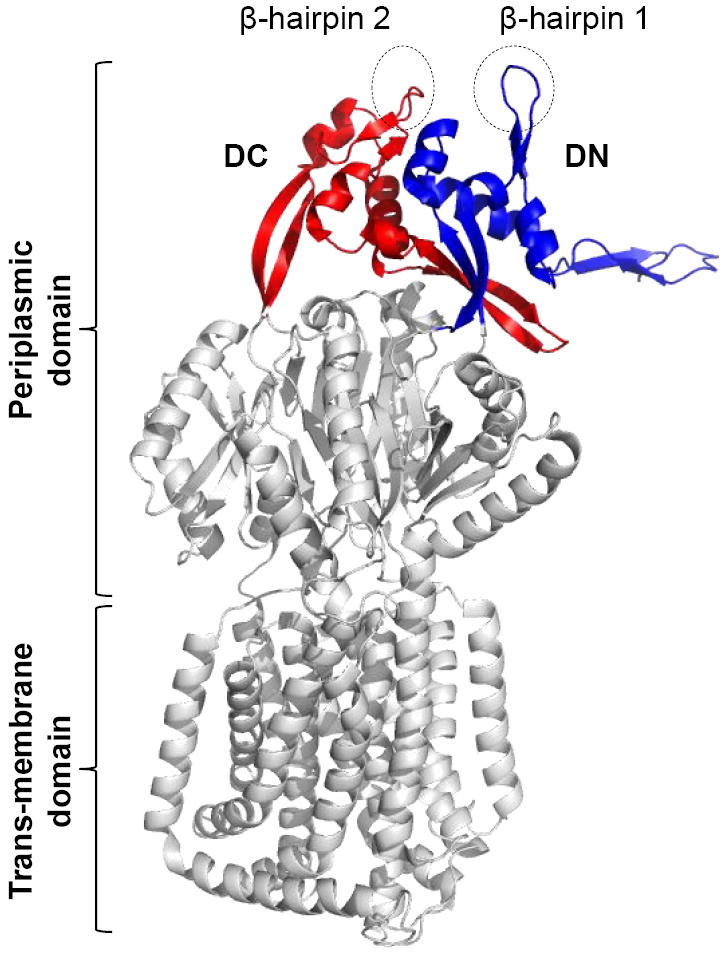
A cartoon showing X-ray structure of AcrB (2GIF). Only AcrB monomer is shown. Positions of two major domains, DN/DC subdomains, and β-hairpins are shown.
The high degree of conformational flexibility observed within the MFPs has led to the suggestion that they play an active role of communicating the conformational changes from the energized IMP to the OMF channel (Ip et al., 2003; Mikolosko et al., 2006; Symmons et al., 2009). This conformational change could potentially allow for the dilation of the OMF aperture via coiled-coil interactions between the deeply interpenetrating α-helices of the MFP hairpin domain and the OMF (Bavro et al., 2008; Symmons et al., 2009; Weeks et al., 2010) bringing the IMP and OMF in direct contact. Recently, an alternative model of pump assembly has been proposed, differing in two major ways: firstly, it invokes a tip-to-tip interaction between the MFP and the OMF and secondly, the IMP is believed not to come in direct contact with the OMF, thus leaving a hollow space above the IMP apex that is enclosed solely by the helical-hairpins of the MFP (Yum et al., 2009; Kim et al., 2010; Xu et al., 2011a; 2011b).
Despite the dramatic molecular transitions associated with the peristaltic cycle of the AcrB, its TolC-docking domain undergoes very little structural change as witnessed by X-ray structures (Murakami et al., 2002; Murakami et al., 2006; Seeger et al., 2006) and numerous molecular dynamics studies (Schultz et al., 2010; Fischer and Kandt, 2013; Yao et al., 2013) suggesting that by itself it may not be sufficient to fully dilate the TolC aperture. This suggests that the role of the interaction between the DN- and DC-hairpins of AcrB with the H3/H4 and H7/H8 turns, respectively, of TolC is to destabilize the primary gates of the TolC, but not to actively open the aperture of the TolC channel (Bavro et al., 2008). This has led us to hypothesize that after initial unlocking of the ion-bridges at the periplasmic end of the channel, the TolC channel reaches full dilation only upon interaction with the α-hairpins of AcrA which perturbs the secondary gate (D371, D374) and drives the full opening of the TolC channel in a two-step reaction (Misra and Bavro 2009; Weeks et al., 2010; Janganan et al., 2013). This suggests an active role of AcrA in transducing conformational signals from AcrB to TolC to fully open the TolC channel.
In this study we focused on the AcrB DN- and DC- β-hairpin loops, which project towards TolC, so as to gain a deeper understanding of the initial steps of AcrB-TolC interactions leading to priming of the TolC channel-opening. We carried out a systematic site-directed mutagenesis and suppression analysis to investigate the contributions of AcrB β-hairpin loop residues in the active pump assembly. Our data suggest that residues of the AcrB β-hairpin DN-loop are critical for docking with TolC and thus functional pump assembly.
RESULTS AND DISCUSSION
AcrB hairpin loop mutants
Two vertical β-hairpins (one from each DN- and DC-subdomains, and here referred to as β-hairpin 1 and 2, respectively), protruding from the tip of the TolC-docking domain of each AcrB protomer (Fig. 1), are thought to come in direct contact with TolC during the functional assembly of the tripartite complex (Murakami et al., 2002). A comprehensive analysis by Tamura et al. (2005) demonstrated proximity of the AcrB β-hairpins and TolC’s periplasmic α-helical turn regions via cysteine-mediated cross-linking of AcrB-TolC complexes in vivo. They specifically detected robust cross-links between TolCC147 and AcrBC255 or AcrBC256, and between TolCC365 and AcrBC795 or AcrBC796. We have previously speculated a role for the AcrB hairpins in opening the primary gates of TolC formed of ion-bridges locking the periplasmic end of the channel (Bavro et al., 2008). In the wild type AcrB protein, conserved aspartate residues occupy positions 256 and 795 at the tip of β-hairpins 1 and 2, and we tested their importance through alanine mutagenesis. Alterations of these two aspartate residues, either individually or concurrently, to alanine produced no drug sensitivity phenotype (data not shown), indicating that the conserved D256 and D795 residues of AcrB hairpins by themselves play no significant role in AcrB function or complex assembly.
We then developed a mutagenesis strategy based on the premise that a combination of side chain- and backbone-mediated interactions between the AcrB hairpin loops and the tip of the TolC channel might facilitate complex assembly. Moreover, since it is possible that multiple and overlapping interactions stabilize the contact between the AcrB hairpins and TolC helical turn regions, we systematically abolished side chain-mediated (by replacing wild type residues with alanine) and/or backbone-mediated (through short deletions) interactions. Minimal inhibitory concentrations (MICs) of erythromycin and novobiocin were significantly lower in a strain expressing the mutant AcrB protein lacking five residues from hairpin 1 (Δ252KVNQD256) compared to a strain expressing wild type AcrB (Table 1). On the other hand, substitution of these five wild type residues with alanine (252AAAAA256) only modestly reduced MICs of the two antibiotics (Table 1). Deletion or alanine substitution of the 793AADGQ797 residues of hairpin 2 had a little effect on MICs (Table 1). Finally, when the five residues of both hairpins 1 and 2 were simultaneously substituted with alanine, the resulting AcrB mutant displayed increased sensitivity to antibiotics (Table 1).
Table 1.
Minimal inhibitory concentration values of various AcrB mutants.
| AcrAB proteinsa | MIC (μg ml-1)b
|
|
|---|---|---|
| Erythromycin | Novobiocin | |
|
| ||
| AcrA+ AcrB+ | >128 | >128 |
| AcrA- AcrB- | 2 | 2 |
| AcrA+ AcrB H1Δ252-256 | 16 | 8-16 |
| AcrA+ AcrB Poly A H1252-256 | 64-128 | 64 |
| AcrA+ AcrB H2Δ793-797 | 128 | 64 |
| AcrA+ AcrB Poly A H2793-797 | 128 | 64 |
| AcrA+ AcrB Poly A H1252-256, Poly A H2793-797 | 32 | 16 |
AcrAB proteins were expressed from pACYC184 in a ΔacrAB background.
MIC, minimal inhibitory concentration.
The hypersensitivity phenotype of the two AcrB mutants, AcrB H1Δ252-256 and AcrB polyalanine-H1 and H2, could be due to impaired interactions of the mutant AcrB proteins with the other two members of the tripartite complex. To test this, we performed complex co-purification assays from total cell lysates to determine whether the pump components still associated successfully in vivo. For this, the His-tagged variants of wild type and mutant AcrB proteins were extracted using a non-ionic detergent, n-dodecyl-β-maltoside (DDM). The solubilized AcrB proteins were then purified by affinity chromatography and the presence of AcrA and TolC in elution fractions containing AcrB was probed by Western blots using AcrA- and TolC-specific antibodies. The results showed that whereas both AcrA and TolC co-purified with wild type AcrB, only AcrA was detected with the two mutant AcrB proteins (Fig. 2). The inability to co-purify TolC with the mutant AcrB protein was not due to destabilization of TolC caused by aberrant TolC-mutant AcrB interactions or formation of an unstable tripartite complex (Fig. S1). In fact, stability of the wild type TolC protein is not reduced in the absence of AcrA and AcrB proteins (Fig. S1).
Figure 2.
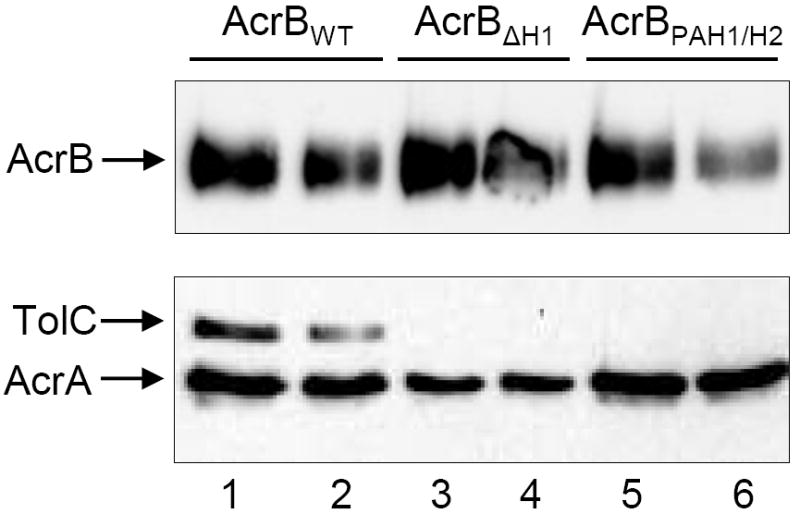
In vivo stability of the AcrAB-TolC complex. The His-tagged wild type and mutant AcrB proteins were extracted from envelopes by DDM and affinity purified as described in the Experimental procedures section. Fractions containing the AcrBHis protein were analyzed by SDS-PAGE and the presence of AcrA, AcrBHis, and TolC were determined by Western blot analysis using antibodies specific to these proteins. Prior to SDS-PAGE analysis, protein samples in a SDS buffer were either boiled (uneven lanes) or kept at room temperature (even lanes).
Collectively, these data pointed to an important role for AcrB hairpins in the functional assembly of the pump complex by stabilizing interactions with TolC. The deletion data suggested a greater importance to the five affected residues of hairpin 1 than those deleted in hairpin 2. The synthetic phenotype of the double polyalanine AcrB mutant reflects overlapping roles of residues from both the hairpins. Moreover, the relative insensitivity of the pump function to substitutions of any particular side-chains strongly supports a model where backbone interactions play an important role in AcrB-TolC binding.
Restoration of wild type residues
Having obtained a drug sensitive AcrB mutant (double polyalanine hairpin substitution) in which the potential for most side chain-mediated, but not backbone-mediated interactions, is abolished, we asked what minimal changes would be required to restore wild type activity. For this we first reintroduced the two aspartate residues that were initially thought to be critical for AcrB-TolC interactions, even though our data proved otherwise. In the double polyalanine mutant background we changed the A256 (hairpin 1) or A795 (hairpin 2) back to D and examined whether this would restore drug resistance. Strikingly, the A256D reintroduction reversed the drug sensitivity phenotype of the double polyalanine mutant, whereas A795D did not (Table 2).
Table 2.
Minimum inhibitory concentration profiles of various AcrB mutants.
| AcrB proteina | MIC (μg ml-1)b
|
|
|---|---|---|
| Erythromycin | Novobiocin | |
|
| ||
| AcrA+ AcrB+ | >128 | >128 |
| AcrA- AcrB- | 2 | 2 |
| AcrA+ AcrB Poly A H1252-256, Poly A H2793-797 | 32 | 16 |
| AcrA+ AcrB Poly A H1252-256(A252K), Poly A H2793-797 | 1 | 4 |
| AcrA+ AcrB Poly A H1252-256(A254N), Poly A H2793-797 | 128 | 128 |
| AcrA+ AcrB Poly A H1252-256(A255Q), Poly A H2793-797 | 32 | 16 |
| AcrA+ AcrB Poly A H1252-256(A256D), Poly A H2793-797 | 128 | 128 |
| AcrA+ AcrB Poly A H1252-256(A256K), Poly A H2793-797 | 16-32 | 16 |
| AcrA+ AcrB Poly A H1252-256, Poly A H2793-797 (A795D) | 16 | 8-16 |
AcrAB proteins were expressed from pACYC184 in a ΔacrAB background.
MIC, minimal inhibitory concentration.
Thus, while standard alanine scanning mutagenesis (one residue at a time) failed to reveal the importance of D256, this was not the case when the contribution from other side chains in the mutated region of AcrB β-hairpin loops was simultaneously abolished. These positive results from hairpin 1 then led us to construct additional variants: in one case we altered A256 to K to test side chain specificity, while in three other cases we altered A252, A254 or A255 to wild type K, N or Q, respectively, to see whether simply returning one of the five polyalanine residues to wild type will restore function. Unlike A256D, the A256K reintroduction did not lower drug sensitivity (Table 2), possibly indicating the importance of a negative charge at this position. The reintroduction of A252 to wild type K in a double polyalanine mutant background failed to reduce antibiotic sensitivity. Interestingly, while A255Q failed to restore function, A252K exacerbated antibiotic sensitivity, indicating a possible charge imbalance. Like A256D, the A254N reintroduction restored wild type activity (Table 2). Finally, restoration of A797 of hairpin 2 to wild type Q did not reduce the drug sensitivity phenotype of the double polyalanine mutant (data not shown). These results indicated that at least some selected side chain-mediated interactions play key roles in the loops’ activity. In summary, by taking the approach of first generating polyalanine changes to produce a phenotype and then restoring function by restoring one side chain at a time, allowed us to identify critical hairpin residues for the complex activity. Our results suggest that the two AcrB hairpins display some functional redundancy and that a combination of both side chain- and backbone-mediated interactions is important for function.
Suppression analysis
The drug hypersensitivity phenotype of AcrB hairpin mutants AcrB H1Δ252-256 and AcrB polyalanine H1/H2 provided a powerful means of conducting genetic suppressor analysis, which should allow us to gain a deeper insight into the various interactions between the TolC-AcrAB proteins leading to the assembly of an active pump. We chose to conduct suppression analysis with AcrB H1Δ252-256 since it contains a deletion of multiple hairpin residues, thus making it unlikely to obtain true revertants.
Cells expressing AcrB H1Δ252-256 grow extremely poorly on plates containing erythromycin/novobiocin (5 μg ml-1 of each) but fail to grow entirely on erythromycin/novobiocin/SDS plates (Fig. S2). Thus we chose the triple inhibitor selection medium for the suppression analysis and plated roughly 5 × 108 cells from 46 independent cultures. Growth after 24 to 36 hours at 37°C yielded inhibitor resistant colonies from roughly half of the independent cultures (21 out 46) at a frequency of roughly 10-8, indicating the prevalence of missense mutations. In the majority of cases (17/21) the suppressor mutations moved with the plasmid expressing acrAB, indicating the location of the mutations to be either in the acrAB promoter, acrA or acrB. Nucleotide sequence determination of the entire acrAB region from the plasmid revealed that in all cases the mutation was located in the acrA gene. In all, 10 different missense mutations from 17 different independent cultures were obtained (Fig. 3).
Figure 3.
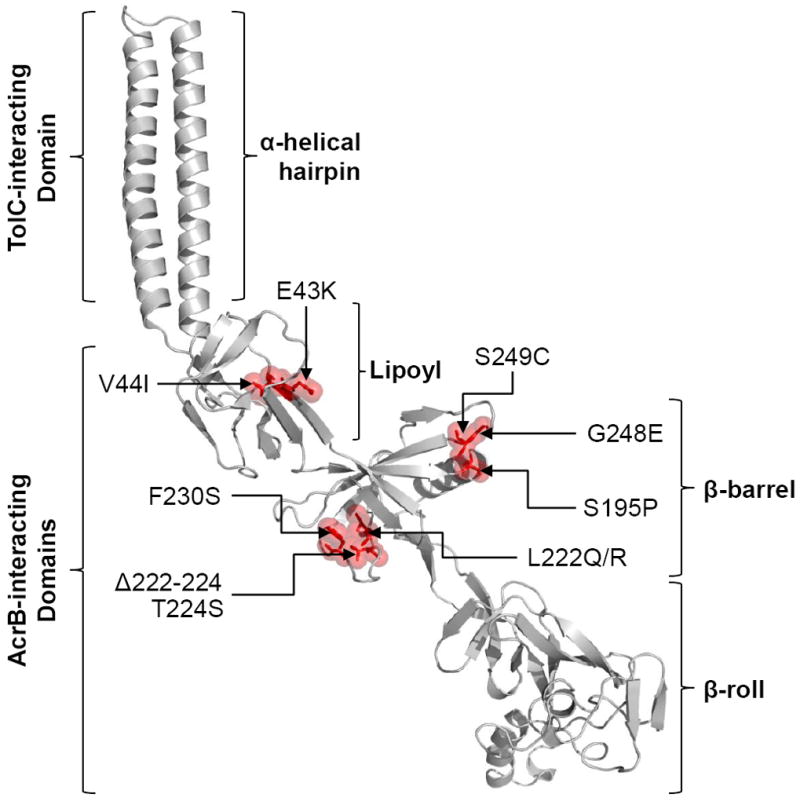
Locations of AcrA suppressor alterations. A cartoon showing the model of full-length AcrA based on the partial X-ray structure of AcrA (2F1M) and the full-length MexA structure (2V4D). Locations of AcrA substitutions, which compensate for the defect of an AcrB β-hairpin mutant, are shown in red with sticks and transparent space-fill representations. AcrA residue numbering corresponds to that of the mature protein. Positions of the four AcrA domains and those that interact with TolC and AcrB are shown.
To facilitate the interpretation of the results, we built a model of the full-length AcrA using MODLLER (Sali and Blundell, 1993) from the available partial structure (Mikolosko et al., 2006) and the full-length MexA structure (Symmons et al., 2009) as structural templates (Fig. 3). Interestingly, 8 of the 10 suppressor alterations were located in the β-barrel domain of AcrA and in only two cases (E43K and V44I) the alterations mapped to the lipoyl domain (Fig. 3). No alterations were found in the α-helical domain or the recently characterized membrane-proximal β-roll domain of AcrA (Symmons et al., 2009). We determined MICs of strains for erythromycin, novobiocin, and SDS in an attempt to phenotypically differentiate various suppressors (Table 3). As expected, all suppressors increased MICs of these inhibitors close to the wild type levels (Table 3). Having obtained AcrA suppressors against the AcrB hairpin 1 mutant, we asked whether they can also suppress the drug sensitivity phenotype of the AcrB polyalanine H1/H2 mutant. For this, we introduced two AcrA suppressor alterations, L222Q and G248E, which efficiently suppress AcrB H1Δ252-256 (Table 3). Both alterations equally effectively reversed the drug sensitivity phenotype of the AcrB polyalanine H1/H2 mutant (data not shown), thus indicating a similar mechanistic defect in the two AcrB hairpin mutants. We also tested whether the two mutant AcrA proteins, bearing L222Q and G248E substitutions, have adapted conformations specifically suited to work with the AcrB hairpin mutants but not with wild type AcrB. The MIC data indicated that both AcrA substitutions work almost as efficiently in the presence of wild type AcrB as they do when the AcrB hairpin mutant is present (Table 3). The apparent lack of allele specificity suggests that the two AcrA mutant proteins, and possibly other AcrA mutants isolated in this study, have not acquired a conformation designed to specially work with the mutant AcrB protein against which they were originally isolated.
Table 3.
Minimal inhibitory concentration profiles of strains expressing various AcrA and AcrB proteins.
| AcrAB proteinsa | AcrA mutant isolation methodb | Minimal inhibitory concentration (μg ml-1)
|
||
|---|---|---|---|---|
| Erythromycin | Novobiocin | SDS | ||
|
| ||||
| AcrA+ AcrB+ | NA | >128 | >128 | >800 |
| AcrA- AcrB- | NA | 2 | 2 | 25 |
| AcrA+ AcrB H1Δ252-256 | SDM | 16 | 8-16 | 100 |
| AcrAE43K AcrB H1Δ252-256 | Spontaneous | >128 | 128 | 400 |
| AcrAV44I AcrB H1Δ252-256 | Spontaneous | >128 | 128 | 200 |
| AcrAS195P AcrB H1Δ252-256 | Spontaneous | >128 | 128 | 400 |
| AcrAL222Q AcrB H1Δ252-256 | Spontaneous | >128 | 128 | 400 |
| AcrAL222R AcrB H1Δ252-256 | Spontaneous | >128 | 128 | 400 |
| AcrAΔ222-224 AcrB H1Δ252-256 | Spontaneous | >128 | 128 | 400 |
| AcrAT224S AcrB H1Δ252-256 | Spontaneous | >128 | 128 | 400 |
| AcrAF230S AcrB H1Δ252-256 | Spontaneous | >128 | 128 | 200-400 |
| AcrAG248E AcrB H1Δ252-256 | Spontaneous | >128 | 128 | 400 |
| AcrAG249C AcrB H1Δ252-256 | Spontaneous | >128 | 128 | 400 |
| AcrAL222Q AcrB+ | SDM | 128 | 128 | ND |
| AcrAG248E AcrB+ | SDM | >128 | 128 | ND |
| AcrAS83G AcrB H1Δ252-256 | SDM | 128 | 64 | 400 |
| AcrAT111P AcrB H1Δ252-256 | SDM | 128 | 64 | 400 |
| AcrAA135T AcrB H1Δ252-256 | SDM | 128 | 64 | 400 |
| AcrAN146T AcrB H1Δ252-256 | SDM | 128 | 64 | 400 |
AcrAB proteins were expressed from pACYC184 in a ΔacrAB background.
NA, not applicable; SDM, site-directed mutagenesis; ND, not determined.
With a few exceptions, the majority of alterations appear to involve drastic changes in the side chain property of the affected residue (e.g. S195P, L222Q or R, F230S, and G248E). Moreover, some of the affected residues (e.g. L222 and F230) are buried in the hydrophobic core of the β-barrel domain and thus a change to a residue with a hydrophilic side chain is expected to produce drastic conformational changes in the domain. Indeed increased in vivo degradation of AcrAE43K, AcrAL222Q, AcrAL222R, and AcrAΔ222-224 points to a dramatic conformational change in the protein’s structure and folding (Fig. 4). We have previously shown that in vivo degradation of AcrAL222Q is dependent on the periplasmic protease DegP and that the stability of the mutant AcrA protein becomes dependent on TolC and AcrB (Gerken and Misra, 2004; Weeks et al., 2010). Despite these seemingly drastic conformational changes, the fact that these suppressor alterations can efficiently reverse the drug sensitivity phenotype of the AcrB hairpin 1 mutant indicates that the mutant AcrA proteins may have adapted conformations that the wild type AcrA normally assumes transiently during the normal course of pump operation, and with all likelihood transmitted to it by AcrB. Hence it seems plausible that these AcrA mutant proteins have assumed a constitutively ‘activated’ state.
Figure 4.

Western blot analysis to determine AcrA levels. Protein extracts from approximately 5 × 107 cells grown overnight at 37°C were analyzed by SDS-PAGE and electro-transferred onto PVDF membranes. Membranes were blotted with primary antibodies against AcrA and LamB. LamB was used as a gel loading control. Protein levels were quantified with Quantity One software (Bio-Rad). AcrA levels were determined relative to LamB and then normalized to the wild type value of 1.
Effects of AcrA suppressor alterations on complex assembly
The data presented so far indicated that certain alterations in the AcrB hairpin regions interfere with the efflux pump activity (Table 1; Fig. S2) by preventing the assembly of a functionally active TolC-AcrAB complex (Fig. 2). AcrA suppressor alterations must then restore functional complex assembly since they significantly reduce the drug hypersensitivity phenotype of the AcrB mutant (Table 3). To test this possibility, we employed a highly sensitive in vivo method that relies on a labile TolC protein, TolCP246R, S350C, whose stability is absolutely dependent on functional interactions with AcrA and AcrB (Gerken and Misra, 2004; Weeks et al., 2010). Without these two proteins, the mutant TolC levels drop to about 2% of that present in an AcrAB+ background (Fig. 5). In an AcrA+ AcrB H1Δ252-256 strain, the mutant TolC level is elevated slightly, but at 6% it remains considerably lower than that present in an AcrAB+ background (Fig. 5).
Figure 5.
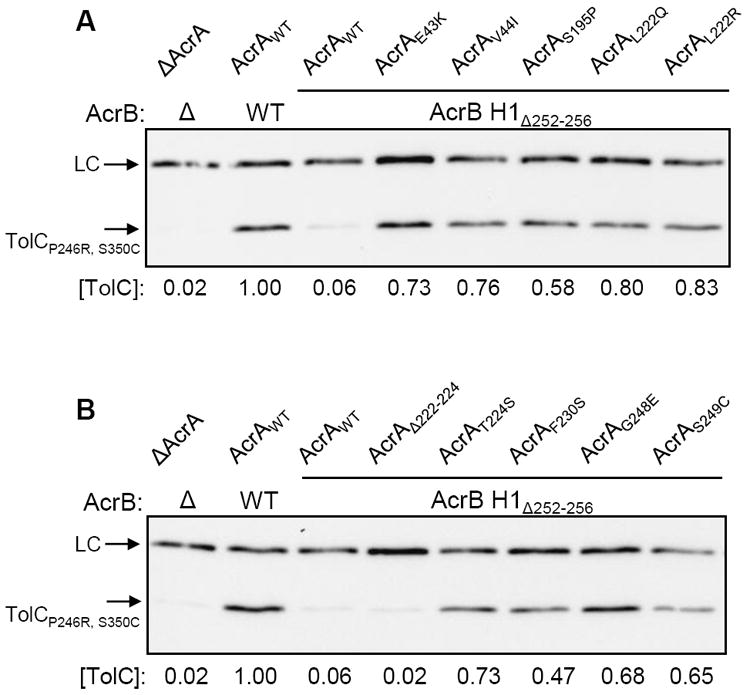
Western blot analyses to determine the in vivo stability of a labile TolC protein carrying P246R and S350C alterations in its mature sequence. Protein extracts from approximately 5 × 107 cells grown overnight at 37°C were analyzed by SDS-PAGE and electro-transferred onto PVDF membranes. Membrane blots were probed with HisProbe-HRP to detect His-tagged TolCP246R, S350C. HisProbe-HRP also recognized an unknown band, which was used as a loading control (LC). Protein levels were quantified with Quantity One software (Bio-Rad). TolCP246R, S350C levels were determined relative to LC and then normalized to the wild type (AcrAB+) value of 1.
All but one of the AcrA suppressor alterations, AcrAΔ222-224, stabilized the mutant TolC protein, elevating its levels between 8 and 14 folds or from 6% in an AcrA+ AcrB H1Δ252-256 background to between 47% and 83% in the presence of the mutant AcrA proteins (Fig. 5). These data are consistent with the notion that AcrA suppressor alterations stabilize the complex formation, leading to reduced drug sensitivity. Interestingly, in one AcrA suppressor background, AcrAΔ222-224, the mutant TolC protein was not stabilized. AcrAΔ222-224 itself is a highly labile protein (Fig. 4), and thus its capacity to stabilize the labile TolC protein is expected to be minimal. Because AcrAΔ222-224 reduces the hypersensitivity phenotype of AcrB H1Δ252-256 (Table 3), it must to some degree restore functional assembly of the complex containing wild type TolC but not the highly labile TolCP246R, S350C protein.
Effects of AcrA α-helical hairpin alterations on the hypersensitivity phenotype of AcrB H1Δ252-256
We have previously reported the isolation of AcrA suppressor alterations using mutant TolC proteins (Gerken and Misra, 2004; Weeks et al., 2010). In addition to alterations in the β-barrel and lipoyl domains of AcrA, four different alterations were discovered in its α-helical hairpin domain. In contrast, in the present study we found that when using a mutant AcrB protein defective in its TolC-docking domain, the AcrA suppressor alterations appeared to be limited solely to the β-barrel and lipoyl domains. A trivial explanation for this observation could be that we were unable to reach saturated mutagenesis, thus did not obtain all possible suppressor mutations in acrA. However, as noted above, we made an exhaustive effort by seeking suppressors from 46 independent cultures of which 21 yielded drug resistant colonies. Nevertheless, we cannot completely rule out this possibility. The second explanation could be that the starting mutant and selective medium favored the isolation of alterations specifically affecting the β-barrel and lipoyl domains of AcrA and selected against alterations mapping at the α-helical hairpin domain of AcrA. To test this possibility, we constructed strains expressing AcrB H1Δ252-256 and AcrA bearing suppressor alterations in its α-helical hairpin domain that were previously obtained using the mutant TolC proteins (Gerken and Misra, 2004; Weeks et al., 2010). The four acrA mutations were introduced via site-directed mutagenesis into a pACYC184-acrAB (AcrB H1Δ252-256) plasmid. Remarkably, all four AcrA α-helical hairpin suppressor alterations significantly increased the MIC values of novobiocin, erythromycin, and SDS (Table 3). However, these values, particularly for erythromycin and novobiocin, were slightly lower than those produced by the AcrA suppressor alterations affecting the β-barrel and lipoyl domains of AcrA (Table 3). Furthermore, AcrB H1Δ252-256 mutants bearing AcrA alterations in its α-helical hairpin domain, but not the one bearing an alteration in the β-barrel domain of AcrA, did not grow well on the medium containing all three inhibitors used in the original selection for suppressors (Fig. S3). Therefore, it appears that slightly lower MIC values of AcrA mutants bearing alterations in the α-helical hairpin domain for erythromycin and novobiocin and their weaker growth on the medium containing all three inhibitors biased against selecting AcrA suppressors with alterations in its α-helical hairpin domain.
Mechanism of suppression
We began with an AcrB β-hairpin mutant defective in its TolC-docking domain and obtained compensatory alterations in the AcrA β-barrel and lipoyl domains. This could suggest that either the two central domains of AcrA directly interact with the AcrB β-hairpin 1 to stabilize the complex or that these AcrA domains indirectly influence TolC recruitment in the complex via influencing the α-helical hairpin domain of AcrA. AcrA is anchored in the inner membrane by N-terminal lipidation, and thus its β-barrel domain, where most of the compensatory mutations are localized (Fig. 3), is unlikely to be able to directly interact with the β-hairpins of AcrB, which are furthermost from the inner membrane. While we cannot fully exclude the possibility of a direct interaction of the β-barrel domain of AcrA with the AcrB-hairpins, our in vivo data are strongly supportive of the latter possibility, namely that β-barrel and lipoyl domains of AcrA indirectly influencing TolC recruitment via modulating the AcrA α-helical hairpin engagement, since the AcrB β-hairpin loop mutants appear to maintain interactions with AcrA but not with TolC (Fig. 2). This notion is further corroborated by our finding that alterations in the membrane-distal AcrA α-helical hairpin region (Weeks et al., 2010), which is shown to primarily interact with TolC helices and not with AcrB (Lobedanz et al., 2007), also reverse the mutant AcrB’s defect (Table 3; Fig. S3). One of these α-helical hairpin alterations, T111P, is located at the tip of the AcrA hairpin, thus furthest from the known AcrB-interacting regions of AcrA. As the AcrA α-helical hairpin region does not directly interact with AcrB, then changes in this region either indirectly influence AcrB-interacting domains of AcrA or bring about their effects by modulating the TolC α-helical domain, the tip of which has been proposed to interact with AcrB β-hairpin loops (Murakami et al., 2002; Fernández-Recio et al., 2004; Tamura et al., 2005; Bavro et al., 2008). Therefore, it is not inconceivable that suppressor alterations in AcrA influence TolC to stabilize TolC-AcrB interactions, likely by influencing the engagement of the α-helical hairpin domain of AcrA with TolC, and permit TolC aperture to transition from closed to open state for drug expulsion. Indeed the primacy of the α-helical hairpin domain (Janganan et al., 2013) and the ability of the membrane-fusion protein (Weeks et al., 2010) to induce opening of the outer membrane channel have been demonstrated.
The AcrB β-hairpins have been previously proposed to engage the primary gate of the TolC channel, namely the ion bridges involving Y362 and R367 residues (Bavro et al., 2008) Accordingly, if a defect in complex assembly in the AcrB β-hairpin loop mutant ultimately prevents the TolC channel aperture to transition from a closed to open state, then TolC mutants with a constitutively open channel aperture might partially compensate for the mutant AcrB’s defect and reverse the drug hypersensitivity phenotype. To test this, we used two different TolC mutants carrying an R367E or R390E alteration that causes partial opening of the TolC channel (Augustus et al., 2004; Bavro et al., 2008). Remarkably, and in support of such interpretation, both TolC alterations reversed the hypersensitivity phenotype of AcrB H1Δ252-256 (Table 4; Fig. S4). This indicates that complex assembly must be restored to some degree in the presence of R367E and R390E alterations. However, the TolCR367E and TolCR390E mutants, which remained partially sensitive to SDS (Table 4), failed to support growth of the AcrB H1Δ252-256 mutant on medium containing SDS, novobiocin and erythromycin (Fig. S4). It is possible that the presence of SDS may adversely affect the assembly of the active tripartite pump containing the mutant forms of both AcrB and TolC. This would explain why such types of TolC mutants were not obtained in our original suppression analysis on the medium containing all three inhibitors.
Table 4.
Effects of TolC alterations on minimal inhibitory concentration profiles of the AcrB H1Δ252-256 mutant.
| AcrAB proteinsa | TolC proteina | Minimal Inhibitory Concentration (μg ml-1)b
|
||
|---|---|---|---|---|
| Erythromycin | Novobiocin | SDS | ||
|
| ||||
| AcrA+ AcrB+ | TolC+ | >64 | 32 | 400 |
| AcrA+ AcrB+ | TolCR367E | 64 | 32 | 400 |
| AcrA+ AcrB+ | TolCR390E | 64 | 32 | 400 |
| AcrA+ AcrB H1Δ252-256 | TolC+ | 8 | 8 | 50 |
| AcrA+ AcrB H1Δ252-256 | TolCR367E | 64 | 32 | 200 |
| AcrA+ AcrB H1Δ252-256 | TolCR390E | 64 | 32 | 200 |
AcrAB and TolC were expressed from pACYC184 and pTrc99A plasmids, respectively, in a ΔacrAB ΔtolC background.
Reduced MIC values of the strain expressing wild type AcrAB and TolC may be due to the expression of TolC from a plasmid replicon as compared to its expression from the chromosomal locus in strains used in Table 3.
CONCLUSIONS
Collectively, the data presented here show that alterations in the AcrB β-hairpins prevent normal assembly with TolC (Fig. 2), thus precluding the TolC aperture to transition from a closed to a drug-efflux-competent open state. This is consistent with the proposed role of AcrB β-hairpins in controlling TolC aperture opening. Compensatory alterations mapping in the β-barrel and lipoyl domains of AcrA stabilize the complex and indirectly facilitate TolC opening by influencing TolC-AcrB engagement, probably at the level of TolC-recruitment via the α-helical hairpin domain of AcrA. In contrast, alterations in the AcrA α-helical hairpin region, which is known to directly interact with TolC (Lobedanz et al., 2007) and influence TolC aperture opening (Week et al., 2010; Janganan et al., 2013) and in the lower part of the TolC α-helix (Augustus et al., 2004; Bavro et al., 2008), reverse the hypersensitivity phenotype towards novobiocin and erythromycin by directly influencing TolC opening. These alterations must also stabilize complex assembly.
Experimental Procedures
Strains, culture conditions and chemicals
Bacterial strains and plasmids used in the study are listed in Table S1. Bacterial strains used here were derived from MC4100 (Casadaban, 1976) bearing a Δara714 allele (Werner and Misra, 2005). Luria broth (LB) and LB agar (LBA) media were prepared as described by Silhavy et al. (1984). When necessary, ampicillin (50 μg ml-1), chloramphenicol (12.5 μg ml-1), novobiocin (5 μg ml-1), erythromycin (5 μg ml-1), or sodium dodecyl sulfate (SDS; 0.1% w/v) were added to bacterial cultures and/or medium. All chemicals were of analytical grade.
DNA manipulations
Plasmids were purified using kits from Qiagen. The construction of a pACYC184 plasmid (Chang and Cohen, 1978) clone expressing the acrAB locus from their native promoter has been previously described (Augustus et al., 2004). AcrBΔ252KVNQD256, AcrB252AAAAA256, AcrBΔ793AADGQ797 and AcrB793AAAAA797 mutants were engineered by site-directed mutagenesis using the Quick Change Lightning Site-Directed Mutagenesis kit (Agilent Technologies). In order to construct the AcrB252AAAAA256/793AAAAA797 double polyalanine mutant, pACYC184-acrAB (AcrB252AAAAA256) was used as template. AcrB Loop 1 restoration mutants were constructed using site-directed mutagenesis of the pACYC-acrAB (AcrB252AAAAA256/793AAAAA797) plasmid. AcrA α-helical mutants were similarly introduced in the pACYC-acrAB (AcrBΔ252KVNQD256) template by site-directed mutagenesis, as were AcrA β-barrel mutants in the pACYC-acrAB (AcrB252AAAAA256/793AAAAA797) template. DNA nucleotide sequencing was carried out for the entire acrAB region for all mutants engineered through site-directed mutagenesis to ensure no additional mutations were present. Primers used for site-directed mutagenesis are listed in Table S2.
The pTrc99a-tolC (NcoI) plasmid clone expressing the His-tagged TolCP246R, S350C protein has previously been described (Gerken and Misra, 2004). This plasmid was used to amplify the mutant tolC gene using the primers (Table S2) that contain a unique BspHI (Forward) and HindIII (Reverse) restriction site. The reverse primer also contains six consecutive histidine codons. BspHI and HindIII double digested PCR-amplified DNA was ligated into pTrc99a digested with NcoI and HindIII. Chromosomal deletions of acrAB and tolC were created using the Datsenko and Wanner (2000) method and have been described previously (Augustus et al., 2004).
Determination of AcrAB-TolC complex stability
Overnight cultures of RAM1197 (ΔacrAB) containing pACYC184-acrAB (AcrBHis and mutant derivatives) were subcultured into fresh LB medium supplemented with chloramphenicol (12.5 μg ml-1) to OD600 ~1.2-1.5. Cells were pelleted and washed with 10 mM Tris-HCl (pH 7.5), 0.5 mM PMSF. After washing, cells were resuspended in a modified plasmolysis buffer as described by Morona and Reeves (1981) with the following modifications. Cells were resuspended in 3.2 ml of 20% (w/v) sucrose in 30 mM Tris-HCl (pH 7.5). The following chemicals were added to the cell suspension: 0.4 ml of 0.1 M PMSF in DMSO, 0.4 ml of 10 mg ml-1 lysozyme in 0.1 M EDTA (pH 8), and 20 μl of 10 mg ml-1 DNase I. The mixture was incubated on ice for 30 minutes, diluted in 16 ml of 3 mM EDTA (pH 8), and then cells were lysed by French press (three passages) at 700 PSI. Following a low speed centrifugation to remove unlysed cells, cell lysates were centrifuged for 1 hour at 105,000 × g at 4°C to separate soluble from insoluble fractions. The insoluble fraction was resuspended in Buffer A (20 mM Tris-HCl [pH 8], 500 mM NaCl, 0.5 mM PMSF) supplemented with 5 mM imidazole and 1% (w/v) DDM. Solubilization was performed overnight at 4°C. The insoluble fraction was separated by centrifugation for 1 hour at 105,000 × g at 4°C. AcrBHis purification was carried out essentially as described by Tikhonova and Zgurskaya (2004) with slight modifications. Briefly, solubilized proteins were applied to a Ni2+ charged HisBind (GE Healthcare) column equilibrated with Buffer A supplemented with 5 mM imidazole and 0.03% (w/v) DDM. Ten column volumes of Buffer A with 0.03% (w/v) DDM, 2 mM PMSF were applied to wash the column and then non-specifically bound proteins were removed by applying gradually increasing imidazole concentrations (5 mM, 20 mM, 50 mM, and 100 mM). AcrAB-TolC complexes were eluted in Buffer A containing 500 mM imidazole and 0.03% (w/v) DDM. Peak fractions were dialyzed twice in Buffer A containing 0.03% DDM and 0.1 mM PMSF to remove imidazole. After purification, the complex composition was analyzed by Western blots.
Western blots
Proteins were analyzed by mini sodium dodecyl sulfate (SDS)-polyacrylamide (11%) gel electrophoresis (PAGE) and transferred onto Immobilon-P PVDF membranes (Millipore). Membranes were blocked overnight in 5% (w/v) non-dairy cream. After blocking, membranes were incubated with primary antibodies for 1.5 hours. Primary antibodies used were raised against AcrB-MBP, AcrAHis, TolC-MBP, or LamB. After incubation with primary antibodies, membranes were washed twice for 15 min and finally incubated for 1 hr in secondary antibodies (goat anti-rabbit horseradish peroxidase-conjugated IgG). When visualizing TolCP246R, S350C-6His, membranes were blocked overnight in 5% (w/v) non-dairy cream followed by 1.5 hr incubation with HisProbe (Pierce). Detection of HisProbe and HRP-conjugated secondary antibodies was performed using immunostar HRP substrate (Pierce). Protein bands were visualized with Bio-Rad Molecular Imager ChemiDoc XRS System. Protein bands were quantified using Quantity One Software (Bio-Rad).
Antibiotic sensitivity assays
Minimal inhibitory concentrations against erythromycin, novobiocin, and SDS were determined by a two-fold serial dilution method using 96-well microtiter plates. Approximately 105 cells were used in each well containing 200 μl of LB or LB supplemented with different amounts of the inhibitor. Plates were incubated for 18 hours at 37°C on a gently rocking platform. Optical densities were measured at 600 nm using VersaMax ELISA Microplate Reader from Molecular Devices. MICs were determined from two independent cultures and each culture was tested in duplicate.
Additionally, antibiotic sensitivity was assessed by monitoring bacterial growth on solid media. For this, cultures were streaked onto LBA plates containing just chloramphenicol (12.5 μg ml-1) or in addition containing novobiocin and erythromycin (5 μg ml-1 each), or novobiocin and erythromycin (5 μg ml-1 each) with SDS (0.1% w/v). Comparative growth was carried out by streaking out a single colony onto each of the media. Control strains either expressing or not expressing pump proteins were grown in addition to all strains expressing mutant pump proteins. Comparative growth assays were done in triplicate.
AcrA modeling and visualization of AcrB-Δ252KVNQD256 compensatory mutations
Modeling of full-length AcrA (corresponding to the E. coli K-12 MG1655 – UniProt ID P0AE06) was undertaken using MODELLER (Sali and Blundell, 1993) and based on the available experimental X-ray partial structure of AcrA (2F1M.pdb; Mikolosko et al., 2006) and the full-length structure of MexA (2V4D.pdb; Symmons et al., 2009). Visualization of the full-length AcrA model (in Fig. 3) was performed using PyMol (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC).
Supplementary Material
Acknowledgments
VNB is supported by a Birmingham University Fellowship. Support for this research in part came from a grant from the National Institutes of the Health to RM.
References
- Andersen C, Koronakis E, Bokma E, Eswaran J, Humphreys D, Hughes C, et al. Transition to the open state of the TolC periplasmic tunnel entrance. Proc Natl Acad Sci USA. 2002;99:11103–11108. doi: 10.1073/pnas.162039399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustus AM, Celaya T, Husain F, Humbard M, Misra R. Antibiotic-sensitive TolC mutants and their suppressors. J Bacteriol. 2004;186:1851–1860. doi: 10.1128/JB.186.6.1851-1860.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavro VN, Pietras Z, Furnham N, Pérez-Cano L, Fernández-Recio J, Pei XY, et al. Assembly and channel opening in a bacterial drug efflux machine. Mol Cell. 2008;30:114–121. doi: 10.1016/j.molcel.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadaban MJ. Transposition and fusion of the lac genes to select promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol. 1976;141:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- Chang ACY, Cohen SN. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Recio J, Walas F, Federici L, Venkatesh Pratap J, Bavro VN, Miguel RN, et al. A model of a transmembrane drug-efflux pump from Gram-negative bacteria. FEBS Lett. 2004;578:5–9. doi: 10.1016/j.febslet.2004.10.097. [DOI] [PubMed] [Google Scholar]
- Fischer N, Kandt C. Porter domain opening and closing motions in the multi-drug efflux transporter AcrB. Biochim Biophys Acta. 2013;1828:632–641. doi: 10.1016/j.bbamem.2012.10.016. [DOI] [PubMed] [Google Scholar]
- Gerken H, Misra R. Genetic evidence for functional interactions between TolC and AcrA proteins of a major antibiotic efflux pump of Escherichia coli. Mol Microbiol. 2004;54:620–631. doi: 10.1111/j.1365-2958.2004.04301.x. [DOI] [PubMed] [Google Scholar]
- Husain F, Humbard M, Misra R. Interaction between the TolC and AcrA proteins of a multidrug efflux system of Escherichia coli. J Bacteriol. 2004;186:8533–8536. doi: 10.1128/JB.186.24.8533-8536.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip H, Stratton K, Zgurskaya H, Liu J. pH-induced conformational changes of AcrA, the membrane fusion protein of Escherichia coli multidrug efflux system. J Biol Chem. 2003;278:50474–50482. doi: 10.1074/jbc.M305152200. [DOI] [PubMed] [Google Scholar]
- Janganan TK, Bavro VS, Zhang L, Borges-Walmsley MI, Walmsley AR. Tripartite efflux pumps: energy is required for dissociation, but not assembly or opening of the outer membrane channel of the pump. Mol Microbiol. 2013;88:590–602. doi: 10.1111/mmi.12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, Xu Y, Lee M, Piao S, Sim SH, Ha NC, Lee K. Functional Relationships between the AcrA Hairpin Tip Region and the TolC Aperture Tip Region for the Formation of the Bacterial Tripartite Efflux Pump AcrAB-TolC. J Bacteriol. 2010;192:4498–4503. doi: 10.1128/JB.00334-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- Lobedanz S, Bokma E, Symmons MF, Koronakis E, Hughes C, Koronakis V. A periplasmic coiled-coil interface underlying TolC recruitment and the assembly of bacterial drug efflux pumps. Proc Natl Acad Sci USA. 2007;104:4612–4617. doi: 10.1073/pnas.0610160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolosko JK, Bobyk K, Zgurskaya HI, Ghosh P. Conformational flexibility in the multidrug efflux system protein AcrA. Structure. 2006;14:577–587. doi: 10.1016/j.str.2005.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra R, Bavro VN. Assembly and transport mechanism of tripartite drug efflux systems. Biochim Biophys Acta. 2009;1794:817–825. doi: 10.1016/j.bbapap.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morona R, Reeves P. Molecular Cloning of the tolC locus of Escherichia coli K-12 with the use of Transposon Tn10. Mol Gen Genet. 1981;184:430–433. doi: 10.1007/BF00352517. [DOI] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- Nikaido H, Pagès JM. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol Rev. 2012;36:340–363. doi: 10.1111/j.1574-6976.2011.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pos KM. Drug transport mechanism of the AcrB efflux pump. Biochim Biophys Acta. 2009;1794:782–793. doi: 10.1016/j.bbapap.2008.12.015. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Schulz R, Vargiu AV, Collu F, Kleinekathöfer U, Ruggerone P. Functional rotation of the transporter AcrB: insights into drug extrusion from simulations. PLoS Comput Biol. 2010;6:e1000806. doi: 10.1371/journal.pcbi.1000806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Klaas M, et al. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- Silhavy TJ, Berman M, Enquist L. Experiments with gene fusions. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1984. [Google Scholar]
- Symmons MF, Bokma E, Koronakis E, Hughes C, Koronakis V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc Natl Acad Sci USA. 2009;106:7173–7178. doi: 10.1073/pnas.0900693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura N, Murakami S, Oyama Y, Ishiguro M, Yamaguchi A. Direct interaction of multidrug efflux transporter AcrB and outer membrane channel TolC detected via site-directed disulfide cross-linking. Biochem. 2005;44:11115–11121. doi: 10.1021/bi050452u. [DOI] [PubMed] [Google Scholar]
- Tikhonova EB, Zgurskaya HI. AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J Biol Chem. 2004;279:32116–32124. doi: 10.1074/jbc.M402230200. [DOI] [PubMed] [Google Scholar]
- Weeks JW, Celaya-Kolb T, Pecora S, Misra R. AcrA suppressor alterations reverse the drug hypersensitivity phenotype of a TolC mutant by inducing TolC aperture opening. Mol Microbiol. 2010;75:1468–1483. doi: 10.1111/j.1365-2958.2010.07068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner J, Misra R. YaeT (Omp85) affects the assembly of lipid-dependent and lipid-independent outer membrane proteins of Escherichia coli. Mol Microbiol. 2005;57:1450–1459. doi: 10.1111/j.1365-2958.2005.04775.x. [DOI] [PubMed] [Google Scholar]
- Xu Y, Lee M, Moeller A, Song S, Yoon BY, Kim HM, et al. Funnel-like hexameric assembly of the periplasmic adapter protein in the tripartite multidrug efflux pump in gram-negative bacteria. J Biol Chem. 2011a;286:17910–17920. doi: 10.1074/jbc.M111.238535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Song S, Moeller A, Kim N, Piao S, Sim SH, et al. Functional implications of an intermeshing cogwheel-like interaction between TolC and MacA in the action of macrolide-specific efflux pump MacAB-TolC. J Biol Chem. 2011b;286:13541–13549. doi: 10.1074/jbc.M110.202598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao XQ, Kimura N, Murakami S, Takada S. Drug uptake pathways of multidrug transporter AcrB studied by molecular simulations and site-directed mutagenesis experiments. J Am Chem Soc. 2013;135:7474–7485. doi: 10.1021/ja310548h. [DOI] [PubMed] [Google Scholar]
- Yum S, Xu Y, Piao S, Sim SH, Kim HM, Jo WS, et al. Crystal structure of the periplasmic component of a tripartite macrolide-specific efflux pump. J Mol Biol. 2009;387:1286–1297. doi: 10.1016/j.jmb.2009.02.048. [DOI] [PubMed] [Google Scholar]
- Zgurskaya HI, Yamada Y, Tikhonova EB, Ge Q, Krisnamoorthy G. Structural and functional diversity of bacterial membrane fusion proteins. Biochim Biophysi Acta. 2009;1794:794–807. doi: 10.1016/j.bbapap.2008.10.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.