

Abstract
Cluster 2b streptokinase (SK2b), secreted by invasive skin-trophic strains of Streptococcus pyogenes (GAS), is a human plasminogen (hPg) activator that optimally functions when human plasma hPg is bound, via its kringle-2 domain, to cognizant bacterial cells through the a1a2 domain of the major cellular hPg receptor, Plasminogen-binding group A streptococcal M-like protein (PAM). Another class of streptokinases (SK1), secreted primarily by GAS strains that possess affinity for pharyngeal infections, does not require PAM-bound hPg for optimal activity. We find herein that replacement of the central β-domain of SK2b with the same module from SK1 reduces the dependency of SK2b on PAM, and the converse is true when the β-domain of SK1 is replaced with this same region of SK2b. These data suggest that simple evolutionary shuttling of protein domains in GAS can be employed by GAS to rapidly generate strains that differ in tissue tropism and invasive capability and allow the bacteria to survive different challenges by the host.
Keywords: Streptokinase, Streptococcus pyogenes, bacterial virulence, human plasminogen, protein domains
1. Introduction
The conversion of the inactive single-chain 791-amino acid residue protein, human plasma plasminogen (hPg), to the disulfide-linked two-chain serine protease, plasmin (hPm), occurs upon mandatory cleavage of a single peptide bond, Arg561-Val562, in the hPg zymogen [1]. Thus, physiological mammalian hPg activators are themselves serine proteases with very limited specificity for this peptide bond. On the other hand, hPg is also efficiently converted to hPm by a secreted 414-amino acid residue streptococcal protein, streptokinase (SK), that possesses no inherent proteolytic activity. This process occurs via complexes of SK and hPg that specifically generate highly specific enzymatic activity initiated through conformational alterations in these SK/hPg complexes, ultimately leading to a robust hPg activator [2].
Streptococcus pyogenes (GAS) is a strict human pathogen, and >250 serotypes have been identified through polymorphic and immunologic differences in surface-exposed M- and M-like protein [3]. These strains are pathologically distinguished based on their dermal or pharyngeal specificity, as well as their virulence and ability to disseminate in the human host. Most of the ~700 M GAS infections reported annually are of a superficial nature and are readily treated by antibiotics. However, several strains of GAS have been isolated from patients with highly invasive infections, such as necrotizing fasciitis and streptococcal toxic shock syndrome. In the strains that have been characterized, e.g., AP53 and NS88.2, those that result in highly invasive skin infections contain the hPg/hPm direct binding M-like protein PAM [4], and also secrete a specific type of SK needed to activate PAM-bound hPg, cluster 2b streptokinase (SK2b) [5,6]. Several pharyngeal-specific strains of GAS, e.g., SF370, contain a distinct M-protein, M1, encoded by the emm1 gene, in place of PAM that does not directly interact with hPg, but with fibrinogen (Fg), which then interacts with hPg. This strain secretes a variant SK (SK2a) that optimally activates hPg bound to cells via Fg [5,7]. Lastly, many strains of GAS possess a M-protein that does not interact with hPg or Fg, e.g., NS931, and also produce a variant of SK (SK1) that optimally activates hPg in solution [8]. While highly homologous to each other, critical differences between PAM and M1 reside in a small amino-terminal ~30 amino acid region, which in PAM contains the a1a2 locus that is fully responsible for the tight interaction between hPg/hPm and PAM. This form of M-protein, viz., PAM, appears to be coinherited in strains that produce SK2b [6,8].
In the current study, we have exchanged each of the three known domains of SK2b into SK1 in order to identify the region(s) of this protein that confers PAM-dependent SK2b. The results of this investigation are reported herein.
2. Materials and Methods
2.1. Proteins and Cell Lines
The recombinant (r) SK chimeric mutants, with α, β, or γ domain exchanges between SK1 from GAS strain NS931 and SK2b from GAS strain NS88.2, were expressed and purified from cDNA clones as described [9].
rPAMAP53 (residues 42–392, lacking the C-terminal membrane insertion domain) was expressed and purified as described [8].
Human full-length recombinant Glu1-plasminogen (hPg) was expressed in Drosophila Schneider S2 cells and purified as described [10].
GAS strain AP53 was obtained from M. J. Walker (Queensland, Australia).
2.2. Activation of Glu1-hPg in Solution
Activation rates of rhPg were continuously monitored in 96-well Corning NBS non-binding microwell plates in Cl− buffer to maintain the closed (tight) conformation of hPg. Catalytic levels of rSK (50:1, m:m, hPg/rSK) were employed to accelerate the activation of hPg. For these assays, 200 μl of a solution containing 0.2 μM hPg/± 0.5 μM rPAM/0.25 mM S2251 (final concentrations) in 10 mM Hepes/150 mM NaCl, pH 7.4, was added to the wells, followed by addition of 2 μl of 0.5 μM rSK (5 nM, final concentration). The amidolytic activity of the generated plasmin (hPm) was monitored by the absorbance (A) at 405 nm from release by plasminolysis of p-nitroanilide (pNA) from the chromogenic substrate, S2251 (H-D-Val-Leu-Lys-pNA; Chromogenix, Milan, Italy) [8].
2.3. Activation of hPg on GAS Cells
GAS AP53 cells were grown in THY medium to A600nm ~0.6 and collected by centrifugation. The cells were washed with 10 mM Hepes/150 mM NaCl, pH 7.4, and then resuspensed in this buffer to A600nm ~1.0. For hPg activation assays, 20 μl (~1 x 107 cfu) of cells was added to wells of a 96-well Corning NBS non-binding microwell plate, followed by 180 μl of 0.22 μM hPg/0.28 mM S2251 (to reach final concentrations of 0.2 μM and 0.25 mM, respectively) in the same buffer. Finally, 5 nM rSK (final concentration) was added and the A405nm was continually measured as above [8].
3. Results and Discussion
3.1. hPg activation ability of SK likely evolved along with the M-protein serotype
It is clear that host hPg/hPm is a necessary invasive GAS pathogenicity factor [11–13]. Thus, one important virulence mechanism employed by GAS involves the secretion of SK which then specifically activates the host fibrinolytic system, producing the extracellular serine protease, hPm, thereby allowing the microorganism to acquire extracellular protease activity. hPm possesses activities are important to bacterial virulence, such as dissolution of fibrin that encapsulates the bacteria and digestion of extracellular matrix components and basement membrane, e.g., laminin, fibronectin, either directly or indirectly via activation of matrix metalloproteinases [14,15].
Some highly invasive strains of GAS, such as skin tropic AP53, possess hPg/hPm cell surface receptors. One such receptor of paramount importance is PAM [16]. Binding of hPg to a small N-terminal region (a1a2) within PAM, enhances its activation rate. Binding of hPm to this same protein protects this enzyme from inhibition by its natural plasma inhibitor, α2-antiplasmin (AP) [17], thus, in a coherent manner utilizing the host to acquire proteolytic activity to combat one feature of its own innate immune response. Other less invasive GAS strains that do not possess PAM, e.g., pharyngeal-tropic NS931, also do not bind hPg/hPm strongly [18], a feature that appears to be general. Another group of GAS strains, e.g., SF370, possess a M-protein, that interacts strongly with Fg, which in-turn interacts with hPg, and also show enhanced virulence.
These different receptor bound and free forms of host hPg encountered by GAS would benefit the virulence of the microorganism by GAS providing a hPg activator that optimized activation under the circumstances presented. Indeed this seems to be the case. SK, not being a protease, is not inactivated by host protease inhibitors, and is an optimal activator for hPg. This protein is secreted by all GAS strains thus-far encountered and indeed several general variants of SK are produced. Non-invasive strains of GAS, e.g., NS931, produce SK cluster 1 (SK1) [5], which optimally activates hPg in solution. The hPm thus produced would be systemic in this case and would be rapidly inactivated by AP. These conditions are not optimal for promoting dissemination. On the other hand, skin-tropic invasive strains of GAS, e.g., NS88.2, AP53, secrete a different SK, subcluster SK2b [5], which has very low activity toward hPg in solution and much higher activity with hPg bound to PAM [8]. Lastly, SK subcluster 2a (SK2a) is produced in strains in which the M-protein (M1) interacts with hPg through assembly of Fg [19,20]. One known GAS strain of this type SF370 is not highly virulent, but a clonal variant of SF370, M1T1, is virulent and depends on the presence of hPg.
3.2. The β2b domain is a major determinant for PAM-dependency while α2b, β2b play synergistic roles
Three domains exist in the 414-amino-acid SK, viz., an amino-terminal α-domain (residues 1–146), a central β-domain (residues 147–290), and a carboxyl terminal γ-domain (residues 291–414). In order to attempt to understand the molecular evolution of SK in GAS, we exchanged each of the domains in SK1 and SK2b and assessed whether PAM dependence could be incorporated into SK1, or eliminated from SK2b, by limited exchanges between SK1 and SK2b. In this study, we employed 0.5 μM rPAMAP53, a concentration that saturates its effect on hPg activation by all SKs employed (not shown), along with rSK1NS931 and rSK2bNS88.2, prototypical SK1 and SK2b subtypes [5,9].
The large differences in activation rate of hPg in the absence of rPAMAP53 between rSK1NS931 (α1β1γ1) and rSK2bNS88.2 (α2bβ2bγ2b) are shown in Fig. 1 A–C, where, in the absence of rPAMAP53, SK2bNS88.2 is 35-fold lower than rSK1NS931 in the hPg activation activity, in agreement with our earlier data [8]. Upon addition of rPAMAP53, rSK1NS931 is stimulated by 2–3-fold, whereas rSK2bNS88.2 is stimulated by 19-fold, again in agreement with our earlier data [8]. This confirms that SK2b is much more highly stimulated by rPAMAP53, as compared to SK1. The data of Figure 1 A–C also demonstrate that substitution of the entire α1 (in rSKM2) or γ1 domains (in rSKM6) in place of the corresponding α2b and γ2b domains in rSK2bNS88.2, thus creating α1β2bγ2b and α2bβ2bγ1 rSK domain chimeras, respectively, did not elevate the hPg activator activity in the absence of rPAMAP53 significantly, compared to rSK2bNS88.2, yet still raised the stimulation by rPAMAP53 by 46-fold for rSKM2 and 43-fold for rSKM6. Conversely, replacing the complete β1 domain in rSKNS931 with the β2b module from rSK2bNS88.2, generating chimera α1β2bγ1 (rSKM3), resulted in diminished hPg activator activity of this variant in the absence of rPAMAP53 to a value nearly equal to that of rSK2bNS88.2, and also led to stimulation by rPAMAP53 (43-fold) to a value similar to those of rSKM2 and rSKM6. Thus, variants that contain β2b in rSK possess rSK2b-type activity, whereas the α1 and γ1 domains do not generate this property. Undoubtedly, however, the α- and γ-domains play a synergistic role with the β-domain in dictating the properties of SK, since each of the chimeric variants of Fig. 1 are more effectively stimulated by rPAMAP53 than the native rSK2bNS88.2.
Figure 1.
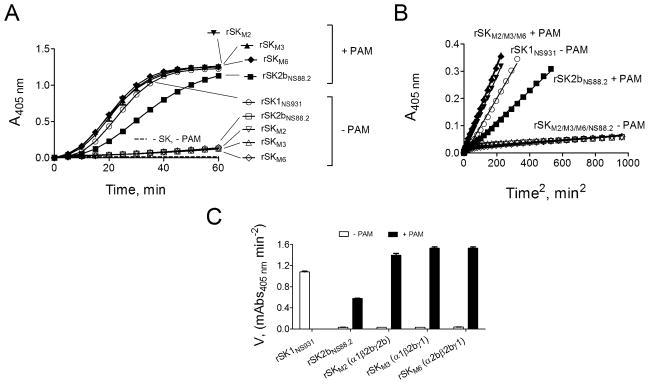
Activation of rhPg by purified rSK and rSK variants in the absence and presence of rPAMAP53. (A). Activation of 200 nM of hPg by 5 nM of purified rSKs, as labeled in Panel A, without PAM (-PAM) and with 0.5 μM rPAMAP53 (+ PAM). The unfilled symbols are assays absent PAM, and filled circles are assays in the presence of 0.5 μM rPAMAP53. The control (dashed line) was performed under the same conditions, except without the addition of rSK and rPAMAP53. The generation of amidolytic activity was monitored continuously by the absorbance at 405 nm (A405nm) versus time at 37 °C using 0.25 mM (final concentration) S2251 (H-D-Val-Leu-Lys-pNA). (B). A405 nm versus time2 transformed from Panel A with linear regression of the linearized region. The symbols are as in Panel A. (C). The initial velocities calculated from the linear regions of plots in Panel B and shown as m(illi)A405 nm versus time2.
The above results have been generally reproduced using the same rSKs, with a PAM-producing cell line, GAS-AP53, from which the rPAMAP53 has been cloned, as compared to rPAMAP53 (Fig. 2). While the extents of stimulation by cell-expressed PAM are slightly different from that of rPAMAP53 protein, the general principles remain the same. In addition, use of the cell line lacking PAM, AP53/γpam, only shows a 1.5 fold stimulation hPg activation by rSK2b, suggesting a minor importance in this regard of cellular receptors other than PAM, whereas stimulations of hPg activation activity by WT-AP53 cells with rSKM2, rSKM3, and rSKM6 are 22-fold, 21-fold, and 26-fold, respectively.
Figure 2.
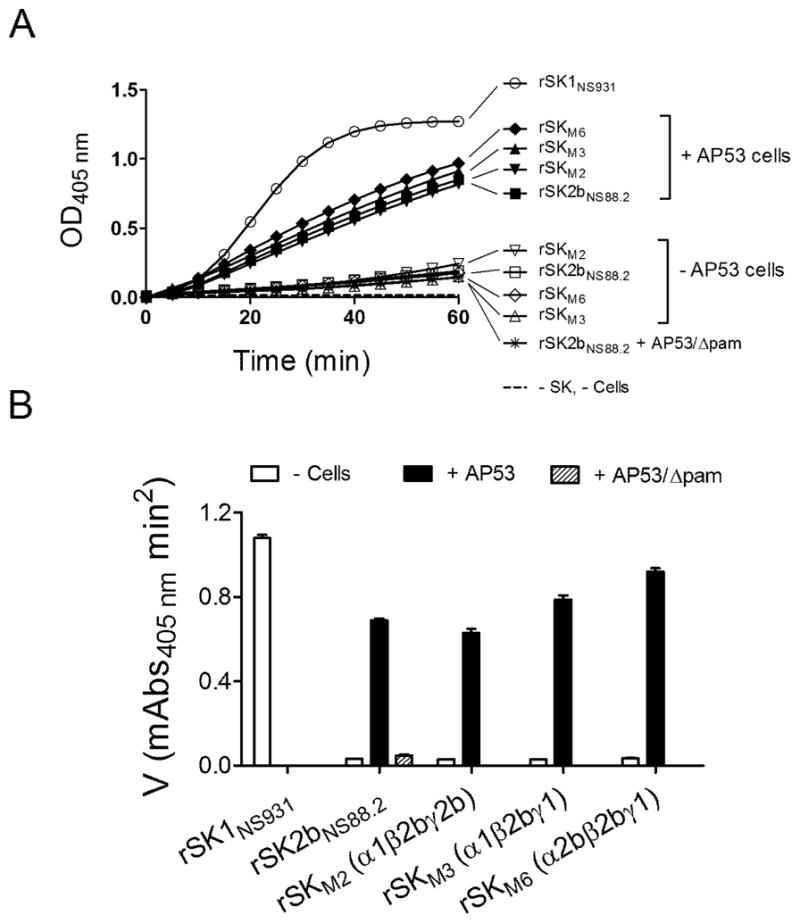
Activation of rhPg by purified rSK and rSK mutants in absence and in presence of GAS WT-AP53 cells or AP53/Δpam cells. (A). Activation of 200 nM of hPg by 5 nM (final concentrations) of purified rSKs, as labeled in Panel A. The unfilled symbols are assays absent cells, and the filled circles are assays in the presence of 20 μl of GAS WT-AP53 cells (A600 nm, 1.0). The control (dashed line) was performed under the same conditions, except without the addition of rSK and cells. The generation of amidolytic activity was monitored continuously by the absorbance at 405 nm (A405nm) versus time at 37 °C using 0.25 mM (final concentration) of S2251 as the hPm substrate. (B). The data from A were replotted as initial velocities of activation (as in Figure 1B).
In conclusion, our results show that primary structural differences in the β-domains dictate hPg activation differences between SK1 and SK2b, while sequence variations in the SK α- and γ-domains result in more minor and synergistic effects on the β-domain. This report on the primary structure-functional relationships between naturally-occurring SK1 and SK2b sheds light on the different manners in which GAS adapts to host factors to enhance its virulence.
Domains were switched between cluster 1 (SK1) and cluster 2b (SK2b) streptokinases
The dependency on PAM protein of the resulting streptokinase variants was assessed
The β domain of SK2b is the determinate domain for the dependency of SK2b on PAM
The α and γ domains play synergistic roles in dictating the properties of SK
Acknowledgments
This work was supported, in whole or in part, by National Institutes of Health Grant HL013423.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Castellino FJ, Ploplis VA. Plasminogen Structure, activation, and regulation. Kluwer Academic/Plenum Publishers; 2003. Human Plasminogen: Structure, activation, and function; pp. 3–17. [Google Scholar]
- 2.Castellino FJ, Ploplis VA. Plasminogen and streptokinase. In: Bachmann F, editor. Handbook of Pharmacology. Springer-Verlag; 2001. pp. 25–56. [Google Scholar]
- 3.Smeesters PR, McMillan DJ, Sriprakash KS. The streptococcal M protein: a highly versatile molecule. Trends Microbiol. 2010;18:275–82. doi: 10.1016/j.tim.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Berge A, Sjobring U. PAM, a novel plasminogen-binding protein from Streptococcus pyogenes. J Biol Chem. 1993;268:25417–24. [PubMed] [Google Scholar]
- 5.McArthur JD, McKay FC, Ramachandran V, et al. Allelic variants of streptokinase from Streptococcus pyogenes display functional differences in plasminogen activation. FASEB J. 2008;22:3146–53. doi: 10.1096/fj.08-109348. [DOI] [PubMed] [Google Scholar]
- 6.Kalia A, Bessen DE. Natural selection and evolution of streptococcal virulence genes involved in tissue-specific adaptations. J Bacteriol. 2004;186:110–21. doi: 10.1128/JB.186.1.110-121.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker MJ, McArthur JD, McKay F, Ranson M. Is plasminogen deployed as a Streptococcus pyogenes virulence factor? Trends Microbiol. 2005;13:308–13. doi: 10.1016/j.tim.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Liang Z, Hsueh HT, Ploplis VA, Castellino FJ. Characterization of streptokinases from Group A Streptococci reveals a strong functional relationship that supports the coinheritance of plasminogen-binding M protein and cluster 2b streptokinase. J Biol Chem. 2012;287:42093–103. doi: 10.1074/jbc.M112.417808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Liang Z, Glinton K, Ploplis VA, Castellino FJ. Functional differences between Streptococcus pyogenes cluster 1 and cluster 2b streptokinases are determined by their beta-domains. FEBS Lett. 2013;587:1304–9. doi: 10.1016/j.febslet.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nilsen SL, Castellino FJ. Expression of human plasminogen in Drosophila Schneider S2 cells. Protein Expr Purif. 1999;16:136–43. doi: 10.1006/prep.1999.1045. [DOI] [PubMed] [Google Scholar]
- 11.Boyle MD, Lottenberg R. Plasminogen activation by invasive human pathogens. Thromb Haemost. 1997;77:1–10. [PubMed] [Google Scholar]
- 12.Sun H, Ringdahl U, Homeister JW, et al. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science. 2004;305:1283–6. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]
- 13.Sanderson-Smith ML, Dinkla K, Cole JN, et al. M protein-mediated plasminogen binding is essential for the virulence of an invasive Streptococcus pyogenes isolate. FASEB J. 2008;22:2715–22. doi: 10.1096/fj.07-105643. [DOI] [PubMed] [Google Scholar]
- 14.Wong AP, Cortez SL, Baricos WH. Role of plasmin and gelatinase in extracellular matrix degradation by cultured rat mesangial cells. Am J Physiol. 1992;263:F1112–8. doi: 10.1152/ajprenal.1992.263.6.F1112. [DOI] [PubMed] [Google Scholar]
- 15.Zwijsen A, van Grunsven LA, Bosman EA, et al. Transforming growth factor beta signalling in vitro and in vivo: activin ligand-receptor interaction, Smad5 in vasculogenesis, and repression of target genes by the deltaEF1/ZEB-related SIP1 in the vertebrate embryo. Mol Cell Endocrinol. 2001;180:13–24. doi: 10.1016/s0303-7207(01)00505-6. [DOI] [PubMed] [Google Scholar]
- 16.Ringdahl U, Svensson M, Wistedt AC, et al. Molecular co-operation between protein PAM and streptokinase for plasmin acquisition by Streptococcus pyogenes. J Biol Chem. 1998;273:6424–30. doi: 10.1074/jbc.273.11.6424. [DOI] [PubMed] [Google Scholar]
- 17.Lottenberg R, DesJardin LE, Wang H, Boyle MD. Streptokinase-producing streptococci grown in human plasma acquire unregulated cell-associated plasmin activity. J Infect Dis. 1992;166:436–40. doi: 10.1093/infdis/166.2.436. [DOI] [PubMed] [Google Scholar]
- 18.Liang Z, Zhang Y, Agrahari G, et al. A natural inactivating mutation in the CovS component of the CovRS regulatory operon in a pattern D Streptococcal pyogenes strain influences virulence-associated genes. J Biol Chem. 2013;288:6561–73. doi: 10.1074/jbc.M112.442657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ringdahl U, Svensson HG, Kotarsky H, et al. A role for the fibrinogen-binding regions of streptococcal M proteins in phagocytosis resistance. Mol Microbiol. 2000;37:1318–26. doi: 10.1046/j.1365-2958.2000.02062.x. [DOI] [PubMed] [Google Scholar]
- 20.Macheboeuf P, Buffalo C, Fu CY, et al. Streptococcal M1 protein constructs a pathological host fibrinogen network. Nature. 2011;472:64–8. doi: 10.1038/nature09967. [DOI] [PMC free article] [PubMed] [Google Scholar]