

Abstract
Approximately 30% of current drinkers in the US drink excessively, and are referred to as problem/hazardous drinkers. These individuals, who may not meet criteria for alcohol abuse or dependence, comprise binge, heavy drinkers, or both. Given their high prevalence, interventions that reduce the risk of binge and heavy drinking have important public health implications. Impulsivity has been repeatedly associated with excessive drinking in the clinical literature. As impulsivity is correlated with, and may play a critical role in the initiation and maintenance of excessive drinking, this behavior may be an important target for therapeutic intervention. Hence, a better understanding of pharmacological treatments capable of attenuating excessive drinking models and impulsivity may markedly improve clinical outcomes. The high alcohol-preferring (HAP) mice represent an optimal rodent model to study the relationship between impulsivity and excessive alcohol drinking, as recent evidence indicates they consume high levels of alcohol throughout their active cycle and are innately impulsive. Using this model, the present study demonstrates that the triple monoamine uptake inhibitors (TUIs) amitifadine and DOV 102, 677 effectively attenuate binge drinking, heavy drinking assessed via a 24-hour free-choice assay, and impulsivity measured by the delay discounting procedure. In contrast, 3-PBC, a GABA-A α1 preferring ligand, with mixed agonist-antagonist properties attenuates excessive drinking without affecting impulsivity. These findings suggest in the HAP mice, monoamine pathways may predominate as a common mechanism underlying impulsivity and excessive drinking, while the GABAergic system may be more salient in regulating excessive drinking. We further propose that TUIs such as amitifadine and DOV 102, 677 may be used to treat the co-occurrence of impulsivity and excessive drinking.
Keywords: Alcohol use disorders, impulsivity, triple uptake inhibitor, delay discounting, HAP mice
INTRODUCTION
Excessive alcohol drinking is the third leading lifestyle-related cause of death in the US, and has been suggested to kill approximately 75,000 people annually. It results in 2.3 million years of potential life lost, about 30 years of life lost per death (CDC-Prevention, 2001; Chikritzhs et al., 2001; Town et al., 2006). Excessive drinkers are considered to be binge drinkers, heavy drinkers, or both (Town et al., 2006). Binge drinkers include men who consume 5 or more drinks and women who consume 4 or more drinks on one or more occasions in the past month. For the typical adult, this pattern often results in a blood alcohol concentration (BAC) of 0.08 gram percent or above in a 2 hr. period (NIAAA, 2004). Heavy drinking includes those who have consumed 60 drinks in the past month for men, and 30 for women (Town et al., 2006). While excessive drinkers are at significantly increased risks for serious medical conditions [e.g., hypertension, cardiomyopathy, obesity, and liver diseases], (CDC-Prevention, 2001; Chikritzhs et al., 2001), most excessive drinkers do not meet criteria for alcohol abuse, or dependence (Dawson et al., 2005). Moreover, emerging evidence suggests that, in contrast to alcohol dependence, excessive drinking contributes to most alcohol-related problems in the US (Institute of Medicine, 1990; Town et al., 2006; Dawson et al., 2005; Courtney and Polich, 2009). However, few medications have been investigated for treatment of such populations despite the high prevalence of excessive drinking.
Interestingly, the trait of impulsivity is correlated with addiction to virtually all drugs of abuse (Kirby et al., 1999; Hoffman et al., 2006), but particularly with dimensions of excessive alcohol drinking (Rubio et al., 2008; Dick et al., 2010; Vanderveen et al., 2012). Impulsivity is involved in vulnerability to alcohol-use initiation, onset of binge drinking behaviors, early-stage alcohol problems, and end-stage diagnoses of alcohol dependence and abuse (reviewed in Lejuez et al., 2010). Therefore, this behavior may be an important target of therapeutic intervention. However, current clinical treatments are ineffective in treating both impulsivity and excessive alcohol drinking (Oberlin et al., 2010). Thus, a better understanding of the pharmacological treatments capable of regulating excessive alcohol drinking as well as impulsivity may improve clinical outcomes.
Research in our laboratories has focused on impulsive choice, also referred to as “cognitive impulsivity” (Rachlin and Green, 1972; Winstanley et al., 2004). This definition is experimentally assessed using a task called delay discounting (DD). The DD task is widely used in human and animal studies, and is similar between species, lending good face validity to assessments of impulsivity in experimental animal models (Bickel et al., 1999; Richards et al., 1997; Petry, 2001). In the present study, we focused on investigating the potential pharmacological overlap between cognitive impulsivity and two modes of excessive drinking: binge drinking and heavy drinking assessed via 24 hr. free-choice. Both types of excessive drinking have been shown to result in alcohol dependence/use disorders in some individuals (King et al, 2011).
Preclinical research employing alcohol-preferring rodent lines has consistently demonstrated that the association between impulsivity and alcohol use is genetically mediated (e.g., Wilhelm and Mitchell, 2008; Oberlin and Grahame, 2009). Employing high-alcohol drinking (HAD) and low-alcohol drinking (LAD) rats, Wilhelm and Mitchell (2008), using the DD assay, demonstrated that HAD rats were more impulsive than LAD rats. Using replicate selected lines of outbred high-alcohol preferring (HAP) mice, Oberlin and Grahame (2009) showed that both HAP2 and HAP1 lines of mice were more impulsive than the LAP2 and HS/Ibg lines, respectively. Together, these results in naïve, alcohol-preferring rodents suggest impulsivity is a heritable difference that precedes the initiation of alcohol use. However, despite the genotypic correlation of impulsivity and alcohol use, no research has been able to identify a single therapeutic modality capable of attenuating both phenotypes (Mitchell et al., 2007; Oberlin et al., 2010). Hence, it is plausible that differential, though genetically linked, neurobiological mechanisms contribute to the development of impulsivity and alcohol drinking.
In an effort to identify both neurochemical targets and a single pharmacotherapy capable of attenuating both excessive drinking and impulsivity, our laboratories have employed a series of compounds referred to as both “broad spectrum” antidepressants (ADs) and triple uptake inhibitors (TUIs) (Skolnick and Basile, 2007). Unlike currently available ADs, these agents inhibit the uptake of dopamine (DA), norepinephrine (NE), and serotonin (5-HT), with varying potencies at their respective monoaminergic transporters. The exact neural mechanisms regulating cognitive impulsivity are not known; however, the monoamines DA, NE, and 5-HT are widely implicated in modulating impulsivity based on the clinical effects of drugs that increase the activity of the relevant pathways, and by evidence that dopaminergic, noradrenergic, and/or serotonergic neurotransmission is deficient in patients and animal models of impulse control disorders (Bevilacqua, et al., 2010; Economidou et al., 2012). Further evidence suggests that hypofunctional mesolimbic monoaminergic pathways contribute to the clinical manifestations of alcoholism (Johnson, 2008; Simon O’Brien et al., 2011). The γ-amino butyric acid-A (GABAA) receptors are also an established target for excessive drinking (Harris et al., 2008). Recent research indicates a significant role for both the GABA α1 and α2 subunits in regulating binge drinking (Liu et al., 2011; Yang et al., 2011). Significant support for the role of these subunits in regulating excessive drinking in the clinical literature is also well established (see Liu et al., 2011). However, few studies have investigated a pharmacologic association between excessive drinking and impulsivity using GABAergic and monoaminergic ligands. Because of the critical role of impulsivity in alcohol use disorders, this behavior may be an important target of therapeutic intervention, and hence improve clinical outcomes.
In the present research, based on our previous findings that the two TUIs, amitifadine (formerly DOV 21, 947) and DOV 102, 677, and 3-PBC, a GABA-A α1 preferring ligand with mixed agonist-antagonist properties (Harvey et al., 2002), reduced excessive drinking in P rats (June and Eiler, 2007; Yang et al., 2012; Warnock et al., 2012), we hypothesized that the three ligands would reduce two modes of excessive drinking [i.e. binge drinking and heavy drinking] and impulsivity in HAP mice. The HAP mouse model was an optimal model to use for these studies, because these mice are both excessive drinkers and “innately” impulsive (Matson and Grahame 2011; Oberlin and Grahame, 2009).
MATERIALS AND METHODS
Experiment 1: Effects of Amitifadine, DOV 102, 677, and 3-PBC on Binge Alcohol and Sucrose Drinking
Subjects
Two cohorts of male and female HAP2/HAP3 mice (N = 105) were used to model binge alcohol drinking in humans using the reverse light cycle as previously reported in rats (Liu et al., 2011; Warnock et al., 2012). The first cohort comprised 27 male and 20 female HAP2 mice of the 34th generation, and 14 male and 14 female HAP2 mice of the 37th generation. The second cohort comprised 15 male HAP2 mice of the 35th generation, and 15 male HAP3 mice of the 14th generation. The second cohort, while drug-naïve, had previously participated in a study evaluating affect-related behaviors (see Can et al., 2012). The amount of time between the two studies was approximately four weeks. Animals were approximately 4 – 5 months of age at the beginning of the experiments. All subjects for all experiments were individually housed. The treatment of subjects for experiment 1 was approved by the institutional review board of the University of Maryland School of Medicine.
Compounds
Amitifadine and DOV 102, 677 were obtained from DOV Pharmaceutical, Somerset, N.J. 3-PBC was obtained from Dr. James Cook of the University of Wisconsin-Milwaukee, Milwaukee, WI. Drug formulations were prepared immediately before each test session in a volume of 10 ml/kg using deionized (DI) water. They were administered by oral gavage 25 min prior to binge and locomotor activity experiments due to the half-life/estimated half-life in the previously published studies (see June and Eiler, 2007; Tizzano et al., 2008). Animals were habituated to the gavage procedures by administering DI water alone over a number of experimental sessions.
Equipment
Binge drinking procedures were tested in standard mouse operant chambers (Coulbourn Instruments, Inc., Lehigh Valley, PA) as previously described (June et al., 2007). The dipper cup size was 0.1 mL, and contained 10% (v/v) alcohol or 1% (w/v) sucrose reinforcers. The Coulbourn Graphic State “3” operant software was used (June et al., 2007).
Drinking in the Dark Multiple Scheduled Access (DIDMSA) Paradigm
The DIDMSA protocol was used to initiate binge drinking with HAP mice. Identical and complete training procedures have recently been employed in alcohol-preferring (P) rats (Liu et al., 2011; Warnock et al., 2012). To initiate the DIDMSA protocol in mice, the subjects were given a 30 min operant session using an FR-4 schedule. After the initial 30 min session had elapsed, mice were placed in the home cage with food and water ad libitum for 1 h. Mice then received two additional 30 min alcohol access periods, spaced 1 h apart over the 21 consecutive day time course. In total, animals received three daily 30-min access periods, each spaced 1 h apart. Other cohorts of mice were trained in an identical manner for 1% (w/v) sucrose. The sucrose concentration was selected so response rates would be relatively similar, eliminating the potential confound of a difference in reinforcer efficacy (June and Gilpin, 2010).
BAC Measurement
To ensure the HAP mice were consuming pharmacologically relevant amounts of ethanol to effectively model human binge drinking (e.g., Naimi et al., 2003), BACs were taken as previously reported (June et al., 2007) on day 21 from a subset of mice randomized into the drug treatment groups. The BAC levels at 90 min were consistent with the NIAAA definition of binge alcohol consumption in humans (NIAAA, 2004).
Procedural Summary
On Day 22, mice in the drug treatment groups were randomly administered their respective treatments to evaluate effects on binge alcohol drinking. 28 mice comprising 24 males and 4 females of the 34th generation were selected to receive amitifadine. Mice were randomly divided into four (n=7) dosage groups [vehicle, 25, 50, and 75 mg/kg]. After completion of the amitifadine treatment for binge alcohol drinking and a 7-day washout period, 24 of the 28 mice that participated in the alcohol study were then randomly divided into four (n=6) dosage groups [vehicle, 25, 50, and 75 mg/kg], and retrained on the binge drinking procedure using sucrose as a reinforcer.
Thirty-five mice comprising 3 males and 16 females of the 34th generation HAP2 line, and 14 males and 2 females of the 37th generation HAP2 line were tested using DOV 102, 677. Mice were randomly divided into five (n=7) dosage groups [vehicle, 12.5, 25, 50 and 75 mg/kg]. After completion of the DOV 102, 677 treatment for binge alcohol drinking and a 7-day washout period, the 35 mice that participated in the alcohol study were then randomly divided into five (n=7) dosage groups [vehicle, 12.5, 25, 50 and 75 mg/kg] and retrained using sucrose.
Forty-two mice comprising 15 females of the 35th generation HAP2 line, 12 females of the 37th generation HAP2 line, and 15 males of the 14th generation HAP3 line were tested using 3-PBC. Mice were then randomly divided into seven (n=6) dosage groups [vehicle, 30, 60, 80, 100, 200, and 300 mg/kg]. After completion of the 3-PBC treatment for binge alcohol drinking and a 10-day washout period, 35 of the mice that participated in the alcohol study were randomly divided into six (n=7) sucrose dosage groups [vehicle, 60, 80, 100, 200, and 300 mg/kg] and retrained using sucrose.
Statistical Analysis
Given the number of male and female mice in the DOV 102, 677 and 3-PBC treatment groups, responding was initially analyzed using a mixed ANOVA for sex × dose (2 × 4) collapsed over generation. However, because no sex or interaction effects were seen, data were re-analyzed using a univariate ANOVA for only dose. Thus, data obtained using amitifadine, DOV 102, 677 and 3-PBC were analyzed by separate univariate ANOVAs for binge alcohol or sucrose drinking followed by Dunnett’s post hoc tests. BAC and responding were analyzed by Pearson correlation and repeated measures ANOVAs. The dissimilar composition of male/female mice selected to receive amitifadine in Experiment 1 was due to the availability of mice at the time the experiment was being conducted, many of whom were obtained from a moderately large study evaluating affect-related behaviors (see Can et al., 2011) at one of the researchers’ institutions.
Effects of Amitifadine on Locomotor Activity
Amitifadine effects on locomotor activity were evaluated using mice randomly selected from the three binge sucrose experiments [See Supplemental Data Section].
Experiment 2: Effects of Amitifadine and DOV 102, 677 on Heavy [Free-Choice] Drinking
Subjects
Thirty-six male and female HAP1 mice from the 44th generation were tested using amitifadine and a separate cohort of 36 male and female HAP1 mice from the 49th generation were tested using DOV 102, 677. Lights were on from 20:00 to 08:00 hours, and drinking was measured during the dark part of the cycle using red illumination. Mice had ad lib access to food and water. Procedures were approved by the IACUC of IUPUI, and were conducted in strict adherence with the National Institutes of Health Guide for the Care and Use of Laboratory Animals at all institutions.
Compounds
Amitifadine was dissolved in isotonic saline to concentrations of 0.8 to 2.5 mg/ml. 3-PBC was first dissolved in 0.1 ml DMSO and then brought to concentrations of 0.4 to 1.2 mg/ml in 1% DMSO/saline vehicle. DOV 102, 677 was dissolved in saline to concentrations of 1.5, 3.0, and 4.0 mg/ml. Injection volumes of all agents were 10 ml/kg administered intraperitoneally.
Procedural Summary
Prior to administration of compounds, mice were given free-choice access to 10% alcohol for 3 weeks. Intakes were read and bottles side-switched every other day. During drug testing, alcohol drinking was measured in the home cage beginning at the start of the dark part of the cycle. Drugs were given 30 min prior to measuring drinking. Intakes were measured directly on the cage to + 0.05 ml every 2 hours from 0800–2000 using graduated sipper tubes. A single dose of amitifadine (0 to 25 mg/kg) was given on Tuesday and Thursday, while each dose of DOV 102, 677 (0 to 40 mg/kg) was given to all subjects in a Latin Square design on Mondays and Thursdays.
Statistical Analysis
Daily bihourly and overall drinking data were analyzed by mixed ANOVAs for dose × time × sex (4 × 4 × 2) to examine the effects of amitifadine on g/kg alcohol and ml/kg water drinking. Dose × sex (4 × 2) repeated measures ANOVAs were used to analyze DOV 102, 677 drinking on g/kg alcohol and ml/kg water drinking due to the Latin Square design. A repeated-measures ANOVA of the bihourly data examined the changes in these variables with time for each compound. Dunnett’s post hoc tests were performed to assess individual dose-response effects in comparison to saline control in amitifadine testing, and planned pairwise comparisons were used in DOV 102, 677 testing. All analyses for Experiments 3 and 4 were performed using SPSS version 18 (IBM, Chicago, IL).
Experiment 3: Effects of Amitifadine, DOV 102, 677, and 3-PBC on Delay Discounting
Subjects
Forty-eight male and female HAP2 mice from the 39th generation were tested using all three compounds. Amitifadine study required an additional 49 male and female HAP2 mice from the 37th generation following the observation of a trend toward an effect after the first cohort. Half of these mice had previously received alcohol access and were counterbalanced considering this factor. All mice were counterbalanced across sex and litter for run order and drug treatment and otherwise handled identically to those in Experiment 2.
Compounds
Amitifadine was dissolved in isotonic saline to concentrations of 0.4 to 1.2 mg/ml, while DOV 102, 677 was used in concentrations of 3.0 and 4.0 mg/ml in saline. 3-PBC was first dissolved in 0.1 ml DMSO and then brought to concentrations of 1.5 to 6.0 mg/ml in 1% DMSO/saline vehicle. Injection volumes were 10 ml/kg administered intraperitoneally.
Procedural Summary
Operant boxes consisted of a nosepoke light, two levers, a house light, and a descending sipper tube for saccharin reinforcement (0.032% w/v) (for details, see Oberlin and Grahame, 2009). Boxes were controlled using MedPC IV software. Mice underwent 5 stages of behavioral shaping, with the final, fifth stage serving as 0-second delay testing (Oberlin and Grahame, 2009), serving as a reinforcer magnitude discrimination task prior to introduction of any delay to the large reward. Immediate reward amount started at 1 s of saccharin access, and was adjusted upwards and downwards by 0.1 s based on the mouse’s choices. Forced trials of the non-selected reward followed two consecutive identical choices. Average adjusted amounts of the reward over the last 20 trials of the session served as the measure of adjusted amount. If mice failed to complete 20 trials during Stages 1–4 of shaping, or achieve adjusted amounts of 1.6 seconds or higher on 3 consecutive days during Stage 5, they were excluded from testing. Additionally, a 6th stage with a 10-second delay was performed to acclimate mice to the 10-s delay to the large reward used during drug testing. To ensure stable responding, mice that failed to achieve adjusted amounts of 0.6 s or lower on 3 consecutive days were excluded from testing. Use of a long delay to the large reinforcer ensured an impulsive baseline of behavior on which to detect any drug–induced attenuation of impulsivity (Oberlin et al., 2010).
All mice received 2-hour water access in their home cage at the end of daily testing. Mice received only one dose of each compound, but all mice received all compounds, except for replication 2 of amitifadine that only received this compound. Animals were injected immediately prior to placement in operant boxes and the commencement of DD testing. 3-PBC was first administered to mice in doses of 0 to 120 mg/kg, but the 120 mg/kg group was reduced to a 15 mg/kg dose following the observation of markedly decreased responding on the first day of testing. 3-PBC was tested for four days, as the 15 mg/kg group was run on one additional day to have equal data points for all groups. Amitifadine was administered in 0 to 12 mg/kg doses for four days during each replicate. DOV 102, 677 was tested for four days at 0 to 40 mg/kg doses.
Statistical Analysis
Adjusted amounts were analyzed using factorial ANOVA of sex × dose (2 × 4) using SPSS. As no sex or interaction effects were seen, data were re-examined by collapsing across sex using a univariate ANOVA for evaluation of dose. Dunnett’s post hoc tests were performed to assess individual dose-response effects. The second replicate of amitifadine testing was analyzed separately to assess any alcohol pre-exposure effects, then in tandem with the first to assess effects of replication.
RESULTS
Experiment 1: Operant Binge Drinking and BACs
Amitifadine dose-dependently reduced binge alcohol responding rates, as shown by a significant dose effect [F(3,18) = 15.284, p < 0.001] (Fig. 1A). In contrast, sucrose responding was not significantly altered by any of the doses (Fig. 1B). DOV 102, 677 also dose-dependently reduced responding rates, as shown by a significant dose effect [F(4,24) = 7.604, p < 0.001] (Fig. 2A). As with amitifadine, responding maintained by sucrose was not affected with DOV 102, 677 (Fig. 2B). Similar to the TUIs, 3-PBC also dose-dependently reduced binge alcohol responding [F(6,36) = 27.457, p < 0.001] (Fig. 3A). However, responding maintained by sucrose was not affected p > 0.05 (Fig. 3B).
Fig. 1.

Amitifadine dose-response on binge alcohol (10% v/v) responding with the vehicle and 25 to 75 mg/kg doses [N = 7 per dose group] [A], and sucrose (1% w/v) responding with the vehicle and 25 to 75 mg/kg doses [N = 6 per dose group] [B] in HAP mice. Data were analyzed by between group ANOVAs, and Dunnett’s post-hoc test. Drinking is measured as lever presses over 90 min [three 30-min sessions], and the data are presented as mean ± SEM. *, p≤0.05, compared with vehicle control.
Fig. 2.

DOV 102, 677 dose-response on binge alcohol (10% v/v) responding with the vehicle and 12.5 to 75 mg/kg doses [N = 7 per dose group] [A], and sucrose (1% w/v) responding with the vehicle and 12.5 to 75 mg/kg doses [N = 7 per dose group] [B] in HAP mice. Data were analyzed by between group ANOVAs, and Dunnett’s post-hoc test. Drinking is measured as lever presses over 90 min [three 30-min sessions], and the data are presented as mean ± SEM. *, p≤0.05, compared with vehicle control.
Fig. 3.
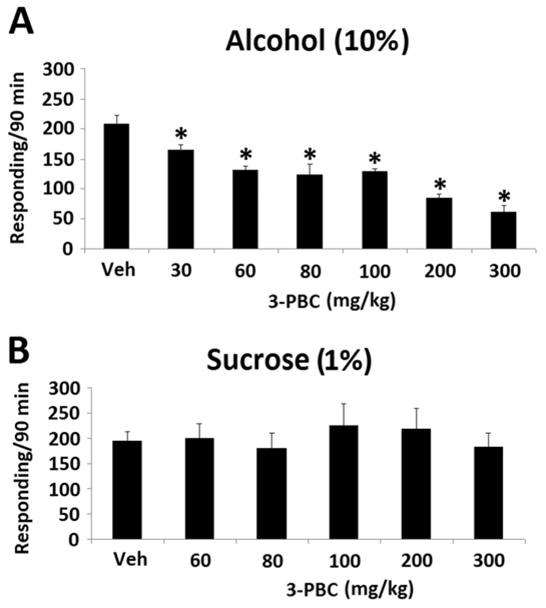
3-PBC dose-response on binge alcohol (10% v/v) responding with the vehicle, and 30 to 300 mg/kg doses [N = 6 per dose group] [A], and sucrose (1% w/v) responding with the vehicle, and 60 to 300 mg/kg doses [N = 7 per dose group] [B] in HAP mice. Data were analyzed by between group ANOVAs, and Dunnett’s post-hoc test. Drinking is measured as lever presses over 90 min [three 30-min sessions], and the data are presented as mean ± SEM. *, p≤0.05, compared with vehicle control.
Compared with BACs (N=6) taken after the initial 30 min session, there was a profound increase in BAC after the final 90 min session [F(1,5) = 200.13, p < 0.001] (Fig. 4A). Responding maintained by alcohol was also markedly increased from the initial 30 min to the final 90 min session [F(1,5) = 63.12, p < 0.001] (Fig. 4B). To further evaluate the relationship between BAC and responding, Pearson product correlations were performed between BACs and level of responding for the first 30 min and total 90 min sessions. Significant positive correlations were observed between the BACs and level of responding for the first 30 min session [r = 0.849, p < 0.03], and the total 90 min session [r = 0.774, p < 0.05]
Fig. 4.
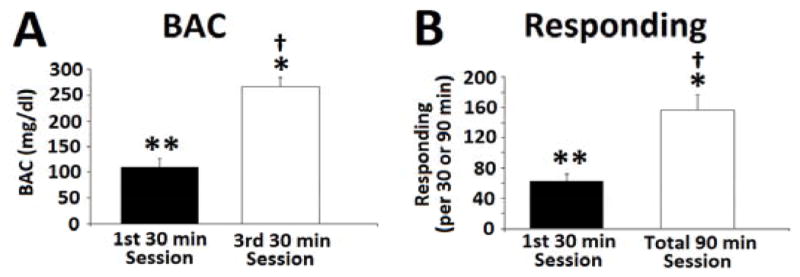
[A] BAC of HAP2 mice after the first and third 30 min operant binge alcohol drinking sessions [N = 6]. [B] Number of lever presses of mice during the first 30 min session [N = 6] and the total responding after the entire 90 min session. Data are presented as mean ± SEM. Data were analyzed using repeated measures ANOVA with Dunnett’s post-hoc test. Pearson correlations were also performed. *, p < 0.01, compared to first 30 min session. **, positive correlation between BAC and responding for 1st 30 min session p < 0.03. †, positive correlation between BAC and responding after entire 90 min session, p < 0.05.
Supplemental Figure 1 illustrates the effects of oral amitifadine on horizontal activity (Fig. S1A) and stereotypy (Fig. S1B) 30 min prior to exposure in the open field. Amitifadine was without effect on either type of activity.
Experiment 2: Heavy [Free-Choice] Drinking
Over the course of 12 hours of drinking, amitifadine dose-dependently reduced alcohol consumption, as shown by a main effect of dose [F(3, 28) = 25.95, p < 0.001], with no effect of sex and/or interaction (Fig. 5A). Post hoc tests showed that the 8, 14, and 25 mg/kg doses all decreased alcohol intake relative to saline (p < 0.01). Effects of amitifadine were most pronounced early in the dark cycle, and began to wane over time; a repeated-measures ANOVA showed an hour × dose interaction on g/kg/hr [F(15,160) = 5.44, p < 0.001], justifying follow-up one-way ANOVAs at each time point, which revealed dose-dependent effects as indicated by the symbols in Fig. 5B. Interestingly, as alcohol intake decreased, mice appeared to compensate by increasing water intake, as demonstrated by a main effect of dose on ml/kg water intake [F(3, 28) = 11.31, p < 0.001], and Dunnett’s post hoc tests showing that the 14 and 25 mg/kg doses increased water intake relative to saline (p < 0.001; Fig. 5C).
Fig. 5.
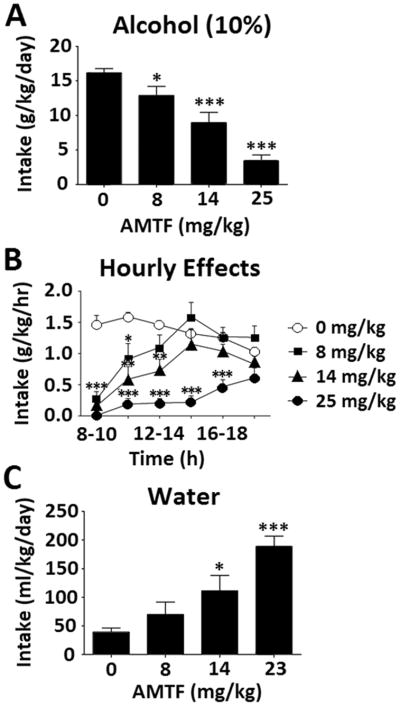
Effects of amitifadine on ethanol intake [N=12] [A], hourly alcohol (10% v/v) intake [N=12] [B], and water intake [N=12] [C]. Data expressed as mean ± SEM; significance expressed as compared to saline, *p < .05, **p < .01, ***p < .001.
Six mice were lost during data collection using DOV 102, 677 to unknown illness (final n = 18). Similar to amitifadine, DOV 102, 677 decreased alcohol intake as demonstrated by a dose main effect in a repeated-measured ANOVA on g/kg/day [F(3,51) = 64.208, p < 0.001] (Fig. 6A). Pairwise comparisons showed that the 15, 30, and 40 mg/kg doses all differed from saline (p < 0.001). A sex effect was observed [F(1,17) = 6.600 p = 0.020], consistent with prior data demonstrating that female mice drink more alcohol, and a dose × sex interaction was also seen [F(3,51) = 4.456, p = 0.007]; however, this effect was driven by the higher baseline consumption of females, as a repeated-measures ANOVA run without the saline dose revealed no interaction effect. Thus, data were collapsed across sex for additional analyses. Effects were again most pronounced early in the dark cycle; a repeated-measures ANOVA showed an hour × dose interaction [F(15,270) = 7.754, p < 0.001], justifying pairwise comparisons of saline to all doses at all times of day which revealed dose-dependent effects as indicated by the symbols in Fig. 6B. Similar to amitifadine, DOV 102, 677 caused increased water intake that appeared to compensate for reduced alcohol consumption. A repeated-measures ANOVA on ml/kg/day showed a main effect of dose [F(3,51) = 2.990, p = .039], and pairwise comparisons showed that the 30 and 40 mg/kg doses were significantly different than saline (p < .05; Fig. 6C).
Fig. 6.
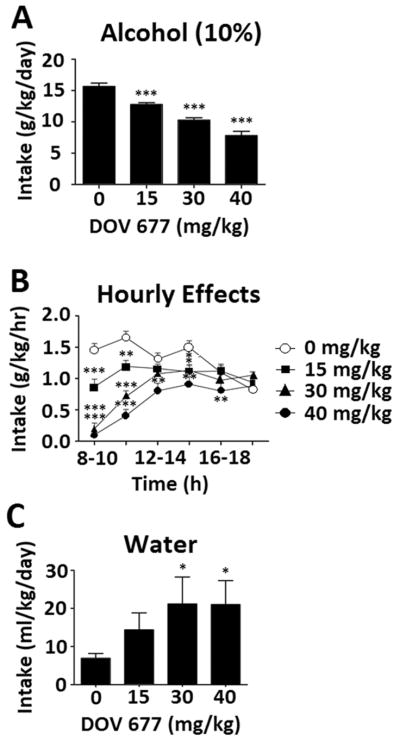
Effects of DOV 102, 677 on ethanol intake [N=12] [A], hourly alcohol (10 % v/v) intake [N=12] [B], and water intake [N=12] [C]. Data expressed as mean ± SEM; significance expressed as compared to saline, *p < .05, **p < .01, ***p < .001.
Experiment 3: Delay Discounting
Three subjects were excluded from the experiment after stage 4 of shaping due to an inability to complete a sufficient number of trials to generate adjusted amount data. Additionally, animals that did not complete 20 trials on at least two days of drug testing were excluded, resulting in the ns listed in the figure captions. 3-PBC did not affect impulsivity, as shown by no effect of dose on adjusted amount, (p > 0.5) (Fig. 7C). Amitifadine did reduce impulsivity, as shown by a main effect of dose on adjusted amount [F(3, 71) = 3.92, p = 0.012]. Dunnett’s post hoc testing showed that the 8 mg/kg dose increased the adjusted amount relative to saline, demonstrative of decreased impulsivity (p < = 0.005) (Fig. 7A). Surprisingly, the 12 mg/kg dose did not differ from saline, suggesting a non-dose-dependent effect for this compound. In contrast, DOV 102,677 dose-dependently decreased impulsivity as indicated by increases in the adjusted amount (Fig. 7B). This was supported by main effect of dose, [F(2, 23) = 6.33, p = .006], and post hoc tests showing higher adjusted amounts in both the 30 and 40 mg/kg doses compared with the vehicle condition (p < 0.005) (Fig. 7B). No sex effects or interaction effects were seen.
Fig. 7.
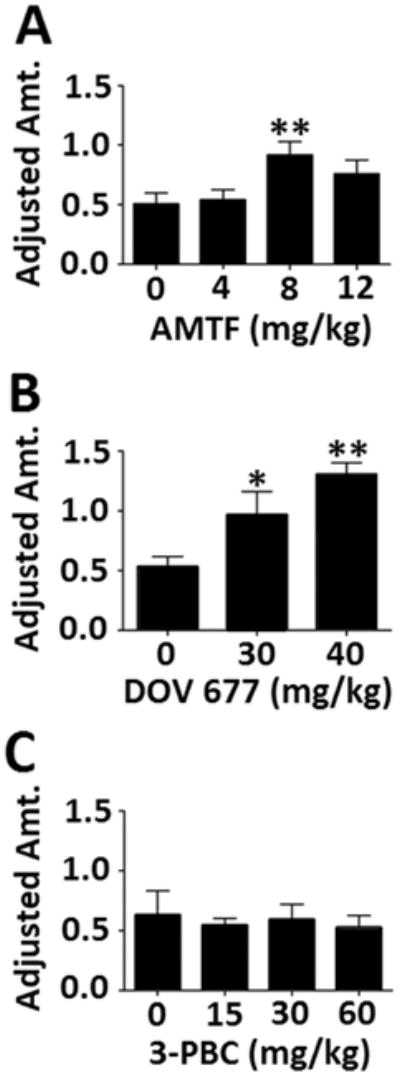
Effects of the three pharmacoactive compounds on delay discounting. Adjusted amounts are listed in seconds; a higher adjusted amount is representative of decreased impulsivity. [N’s for each group: A. 7, 19, 23, 12; B. 9, 10, 7; C. 9, 10, 10, 9]. Data expressed as mean ± SEM; significance expressed as compared to saline, *p < .05, **p < .01, ***p < .001
DISCUSSION
Using an established model of binge alcohol drinking (Liu et aI., 2011; Warnock et aI., 2012), the present study demonstrated that the TUls, amitifadine and DOV 102, 677, and the GABA-A α1 preferring ligand, 3-PBC, which exerts agonist effects on some, but antagonist effects at other GABA receptor subunits (Harvey et al., 2002; June and Eiler, 2007), effectively reduce binge alcohol, but not binge sucrose drinking in HAP mice. These results indicate that increased activity in the dopaminergic, noradrenergic, and serotonergic pathways may cause marked attenuation of binge drinking. In addition, given the modulatory actions of 3-PBC (Harvey et al., 2002; June and Eiler, 2007), along with the recent siRNAs findings in P and HAD rats (Liu et al., 2011; Yang et al., 2011) the findings with 3-PBC provide critical support for the hypothesis that the GABA-A α1 subunit is important in the regulation of binge drinking.
Similar to the findings with binge drinking, both TUls produced a selective dose-dependent reduction on free-choice drinking, which was used to emulate heavy drinking. These findings were accompanied by a concomitant dose-related elevation of water intake. The concomitant elevation in water intake provides support for the behavioral specificity of these compounds on alcohol consumption, consistent with specificity on binge drinking. While we did not assess BACs during free-choice drinking, intake amounts and patterns in saline-treated animals were similar to our prior reports where BACs levels exceeded 100 mg/dl by 2 h after the onset of drinking (Matson and Grahame, 2011). In the present study, because mice had access to alcohol continuously, we were able to accurately measure the duration of action of these drugs. All DOV 102, 677 doses significantly suppressed alcohol intake for up to 6 hrs., while all amitifadine doses reduced intake for up to 8 hrs. Amitifadine was the most efficacious antagonist of the two compounds, reducing drinking by 70 to 100% of control levels early in the 12 hr. period. To our knowledge, no approved antidepressant has produced such potent, prolonged, and selective suppression on excessive alcohol drinking. Given the microdialysis time course for the three monoamines following administration of the two agents (approx 4 hrs) (Popik et al., 2006; Golembiowska et aI., 2012), the prolonged duration of suppression on alcohol drinking for amitifadine and DOV 102, 677 may suggest the utility of further evaluation of both TUls as putative alcohol antagonists in humans. Also, the compensatory dose-related elevations in water intake with the TUIs in the heavy drinking model suggest the absence of toxic effects.
Consistent with their effects on binge and free-choice drinking, the two TUls significantly attenuated cognitive impulsivity. However, while the effective dose response profiles for impulsivity, free-choice, and binge drinking were relatively similar for DOV 102, 677, a different profile emerged for amitifadine. Specifically, the lowest effective amitifadine dose in reducing impulsivity and free-choice drinking was 8 mg/kg, while the lowest effective dose in suppressing binge drinking was 25 mg/kg. This difference in dose response for amitifadine may in part be explained by the usage of i.p. dosing in the impulsivity and free-choice drinking studies compared with using oral dosing in the binge studies. It is well-documented that i.p. and oral routes differ significantly with respect to the absolute magnitude and the time course of increases in extracellular catecholamines and behavioral effects. Intraperitoneal injection has been reported to be twice as potent, as oral in increases in extracellular catecholamines such as dopamine, and neurobehavioral effects (Gerasimov et al., 2000). In addition, it is also well established that the quantitatively different response profiles between oral and i.p. are a function of bioavailability (Chan et al., 1981). Specifically, the lower bioavailability for the oral route is presumably due to the slower absorption from the gastrointestinal tract, and a greater degree of metabolism. Nevertheless, while more research is required to clarify the differences in dose response profiles of amitifadine across behavioral assays and genotypes, the seminal oral dose-response function which produced antidepressant-like effects (5–20mg/kg) (Skolnick, 2012) were comparable to i.p. doses which produced effectiveness in the impulsivity and free-choice drinking studies in mice. Hence, these findings suggest that in some behavioral assays, comparable potency is observed with amitifadine, independent of route of drug administration and rodent used. Interestingly, the dose-response function seen in our data for DOV 102, 677 on impulsivity, free-choice and binge drinking was similar across routes of administration.
Amitifadine is an unbalanced TUI, showing preferential inhibition to the serotonin transporter (i.e., SERT, NET, and DAT; 1:2:8) (Skolnick and Basile, 2007). Amitifadine (10 mg/kg) markedly increases extracellular levels of DA, NE, and 5-HT in the prefrontal cortex to 208%, 274%, and 412%, above baseline 100 min after administration, respectively. Unlike amitifadine, DOV 102, 677 is a balanced inhibitor of DA, NE, and 5-HT transporters with potency close to a 1:1:1 ratio (Popik et al., 2006). DOV 102, 677 (20 mg/kg i.p.) increased extracellular levels of 5-HT and DA in the prefrontal cortex to 280% and 320% respectively, above baseline 100 min after administration (Popik et al., 2006). NE levels increased linearly to a maximum of 348% at 240 min post-dosing. Thus, relative to amitifadine, DOV 102, 677 is a preferential DAT inhibitor, and our results are consistent with dopaminergic agonists that decrease impulsivity (Sagvolden, 2000; Oberlin et al., 2010). Thus, taken together, while route of administration may be a salient factor to explain the differential dose response between the two TUls, amitifadine’s capacity to modulate both DA and 5-HT and DOV 102, 677’s capacity to primarily modulate DA may also contribute to the differential dose response function. Moreover, it is well established that 5-HT regulates the activity of many neurotransmitters, particularly DA-neurons of the mesoaccumbens circuitry (Fink and Gothert, 2007; Dalley and Roiser, 2012). Hence, given the greater activity of 5-HT, along with the i.p. route, lower amitifadine doses would be needed to attenuate impulsivity and heavy drinking in the present study.
A general consensus in the preclinical and clinical studies is that a reduction in brain 5-HT appears to be important for increasing impulsivity (Bevilacqua et al., 2010; Dalley and Roiser, 2012). In the present study, the TUls may decrease impulsive behavior via increasing release of DA/NE via serotonergic activity in addition to their direct effects on the neurotransmitter transporters (see Skolnick and Basile, 2007). Furthermore, increased 5-HT tone has been associated with a reduced potential for reinforcement among drugs that increase monoaminergic neurotransmission, lessening abuse potential (Wee et al., 2006). The failure of amitifadine to exert activating effects in the present study is also consistent with a greater engagement of the 5-HT transporters relative to DA, suggesting that amitifadine may be a good candidate for impulsivity reduction without motor or rewarding side effects (Howell et al., 2007). The lack of motor activating effects is also consistent with a recent preclinical report from others (Golembiowska et al., 2012), as well as our laboratory (Warnock et al., 2012).
The inverted U-shaped dose-response curve observed with amitifadine on impulsivity, however, is worth noting, and similar effects on cognitive-related behaviors have been reported in the literature (Baldi and Bucherelli, 2005) with agents used to treat impulsivity (Tannock et al., 1995). Unfortunately, it is not easy to elucidate the mechanisms on which this effect is based (see Baldi and Bucherelli, 2005). However, in relation to the present study, it is possible the neuropharmacological effects related to the descending limb of the dose-response curve may be related to stereotypy, nonspecific excitation, or serotonergic related effects unrelated to impulsivity. Any, or several of the neuropharmacological effects noted, may have interfered with the attenuation of impulsivity.
In contrast to the TUIs, the GABA-A α1 preferring ligand, 3-PBC, was ineffective in altering cognitive impulsivity. The rationale for this is not clear at present; however, it may reflect the fact that the neural mechanism(s) regulating impulsivity in the HAP mice may be mediated via only monoamines, while excessive drinking in the HAP mice may be regulated via GABA, monoamines, as well as other neurotransmitters (see McBride and Li, 1998). Nevertheless, the finding with 3-PBC in the present study contrasts the report by Murphy et al., (2012) where the GABA-A receptor antagonist bicuculline blocked the increase in impulsivity produced by the agonist muscimol. However, it should be noted that this study used a five-choice serial reaction time task, which measures motor impulsivity, unlike the cognitive impulsivity-based DD assay of the present study. Different measures of impulsivity may have distinct underlying neuropharmacological profiles (Dalley and Roiser, 2012).
In conclusion, the data of the present study provides compelling evidence that a pharmacologic association exits between two modes of excessive drinking [i.e., binge and heavy drinking] and cognitive impulsivity using two TUls, while a dissociation exists using a GABAergic ligand. These findings suggest while the neuronal mechanism(s) that regulate excessive drinking may directly involve DA, NE, 5-HT, and GABAergic activity, the neuronal mechanism(s) regulating cognitive impulsivity appear to involve only DA, NE, and 5-HT activity. Furthermore, the data from the present and our previous study (Yang et al., 2012) provide support for the hypothesis that elevations in 5-HT and DA neurotransmission may be critical in the prolonged suppression of alcohol drinking, as the highest TUI doses were effective in the free-choice model for 6 to 8 hrs. From a clinical perspective, these findings are important because a “single” TUI dose may be capable of sustained reduction of heavy drinking in the absence of untoward effects, increasing efficacy in human alcoholics (Yang et al., 2012). Finally, given the safety profile of amitifadine and DOV 102, 677 (Skolnick, 2012; Tran et al., 2012), and their proposed low abuse liability, we hypothesize that both agents would be effective in treating the co-occurrence of excessive drinking and impulsivity in humans.
Supplementary Material
Acknowledgments
Experiments 1 and 2 were financed by grant AA10406 to HLJ from the National Institute of Alcohol Abuse and Alcoholism (NIAAA). HAP mice were provided by AA015512 to Lawrence Lumeng. Experiments 3 and 4 were supported by grant AA017611 to David Crabb.
Footnotes
DISCLOSURE
The authors declare that over the past 5 years DOV Pharmaceutical, Inc., the original manufacturer of amitifadine (DOV 21, 947) and DOV 102, 677, has provided funding for past projects performed in the laboratory of HLJ; however, it provided no funds for any of the studies included in this manuscript. HLJ is currently a consultant for Euthymics Bioscience, the current licensee of amitifadine and DOV 102, 677.
References
- Baldi E, Bucherelli C. The inverted “u-shaped” dose-effect relationships in learning and memory: modulation of arousal and consolidation. Nonlinearity Biol Toxicol Med. 2005;3(1):9–21. doi: 10.2201/nonlin.003.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua L, Doly S, Kaprio J, Yuan Q, Tikkanen R, Paunio T, Zhou Z, Wedenoja J, Maroteaux L, Diaz S, Belmer A, Hodgkinson CA, Dell’osso L, Suvisaari J, Coccaro E, Rose RJ, Peltonen L, Virkkunen M, Goldman D. A population-specific HTR2B stop codon predisposes to severe impulsivity. Nature. 2010;468(7327):1061–6. doi: 10.1038/nature09629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickel WK, Odum AL, Madden GJ. Impulsivity and cigarette smoking: delay discounting in current, never, and ex-smokers. Psychopharmacology (Berl) 1999;146:447–454. doi: 10.1007/pl00005490. [DOI] [PubMed] [Google Scholar]
- Can A, Grahame NJ, Gould TD. Affect-related behaviors in mice selectively bred for high and low voluntary alcohol consumption. Behav Genet. 2012;42(2):313–22. doi: 10.1007/s10519-011-9505-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Davison SC, Dehghan A, Hyman N. The effect of neostigmine on pyridostigmine bioavailability in myasthenic patients after oral administration. Methods Find Exp Clin Pharmacol. 1981;3(5):291–6. [PubMed] [Google Scholar]
- Chikritzhs TN, Jonas HA, Stockwell TR, Heal PF, Dietze PM. Mortality and life-years lost due to alcohol: a comparison of acute and chronic causes. Medical J Australia. 2001;174:281–284. doi: 10.5694/j.1326-5377.2001.tb143269.x. [DOI] [PubMed] [Google Scholar]
- Center for Disease Control and Prevention. Alcohol-related mortality and years lost. US, [1990–2000] MMWR Morb Mortal Wkly Rep. 2001;50:1064–1065. [Google Scholar]
- Courtney KE, Polich J. Binge drinking in young adults: Data, definitions, and determinants. Psychol Bull. 2009;135(1):142–56. doi: 10.1037/a0014414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Roiser JP. Dopamine, serotonin and impulsivity. Neuroscience. 2012;215:42–58. doi: 10.1016/j.neuroscience.2012.03.065. [DOI] [PubMed] [Google Scholar]
- Dawson DA, Grant BF, Li T-K. Quantifyoing the risks associated with exceeding recommended drinking limits. Alcohol Clin Exp Res. 2005;34:1334–1345. doi: 10.1097/01.alc.0000164544.45746.a7. [DOI] [PubMed] [Google Scholar]
- Dick DM, Smith G, Olausson P, Mitchell SH, Leeman RF, O’Malley SS, Sher K. Understanding the construct of impulsivity and its relationship to alcohol use disorders. Addict Biol. 2010;15(2):217–26. doi: 10.1111/j.1369-1600.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economidou D, Theobald DE, Robbins TW, Everitt BJ, Dalley JW. Norepinephrine and dopamine modulate impulsivity on the five-choice serial reaction time task through opponent actions in the shell and core sub-regions of the nucleus accumbens. Neuropsychopharmacology. 2012;37(9):2057–66. doi: 10.1038/npp.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink KB, Göthert M. 5-HT receptor regulation of neurotransmitter release. Pharmacol Rev. 2007;59(4):360–417. doi: 10.1124/pr.107.07103. [DOI] [PubMed] [Google Scholar]
- Gerasimov MR, Franceschi M, Volkow ND, Gifford A, Gatley SJ, Marsteller D, Molina PE, Dewey SL. Comparison between intraperitoneal and oral methylphenidate administration: A microdialysis and locomotor activity study. J Pharmacol Exp Ther. 2000;295(1):51–7. [PubMed] [Google Scholar]
- Golembiowska K, Kowalska M, Bymaster FP. Effects of the triple reuptake inhibitor amitifadine on extracellular levels of monoamines in rat brain regions and on locomotor activity. Synapse 2012. 2012;66(5):435–44. doi: 10.1002/syn.21531. [DOI] [PubMed] [Google Scholar]
- Harris RA, Trudell JR, Mihic SJ. Ethanol’s molecular targets. Sci Signal. 2008;1(28):re7. doi: 10.1126/scisignal.128re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, Carroll MR, Foster KL, McKay PF, Collette Grey C, McCane S, Rancia C, Mason D, Seyoum R, Jones CM, Ma C, Cook JM, June HL. Journal of Neuroscience. 2002;22:3765–3775. doi: 10.1523/JNEUROSCI.22-09-03765.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman WF, Moore M, Templin R, McFarland B, Hitzemann RJ, Mitchell SH. Neuropsychological function and delay discounting in methamphetamine-dependent individuals. Psychopharmacology (Berl) 2006;188(2):162–70. doi: 10.1007/s00213-006-0494-0. [DOI] [PubMed] [Google Scholar]
- Howell LL, Carroll FI, Votaw JR, Goodman MM, Kimmel HL. Effects of combined dopamine and serotonin transporter inhibitors on cocaine self-administration in rhesus monkeys. J Pharmacol Exp Ther. 2007;320(2):757–65. doi: 10.1124/jpet.106.108324. [DOI] [PubMed] [Google Scholar]
- Institute of Medicine. Broadening the base of treatment for alcohol problems. Washington, DC: National Academy Press; 1990. [PubMed] [Google Scholar]
- Johnson BA. Update on neuropharmacological treatments for alcoholism: scientific basis and clinical findings. Biochem Pharmacol. 2008;75(1):34–56. doi: 10.1016/j.bcp.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June HL, Eiler WLA. Handbook of contemporary neuropharmacology. In: Sibley DR, Hanin I, Kuhar M, Skolnick P, editors. Dopaminergic and GABAergic Regulation of Alcohol-Motivated Behaviors: Novel Neuroanatomical Substrates. Hoboken, NJ: Wiley & Sons; 2007. pp. 1–72. [Google Scholar]
- June HL, Sr, Foster KL, Eiler WJ, 2nd, Goergen J, Cook JB, Johnson N, Mensah-Zoe B, Simmons JO, June HL, Jr, Yin W, Cook JM, Homanics GE. Dopamine and benzodiazepine-dependent mechanisms regulate the EtOH-enhanced locomotor stimulation in the GABAA alpha1 subunit null mutant mice. Neuropsychopharmacology. 2007;32(1):137–52. doi: 10.1038/sj.npp.1301097. [DOI] [PubMed] [Google Scholar]
- June HL, Gilpin NW. Operant self-administration models for testing the neuropharmacological basis of ethanol consumption in rats. Curr Protoc Neurosci. 2010;Chapter 9(Unit 9.12):1–26. doi: 10.1002/0471142301.ns0912s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AC, de Wit H, McNamara PJ, Cao D. Rewarding, stimulant, and sedative alcohol responses and relationship to future binge drinking. Arch Gen Psychiatry. 2011;68(4):389–399. doi: 10.1001/archgenpsychiatry.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby KN, Petry NM, Bickel WK. Heroin addicts have higher discount rates for delayed rewards than non-drug-using controls. J Exp Psychol Gen. 1999;128(1):78–87. doi: 10.1037//0096-3445.128.1.78. [DOI] [PubMed] [Google Scholar]
- Lejuez CW, Magidson JF, Mitchell SH, Sinha R, Stevens MC, de Wit H. Behavioral and biological indicators of impulsivity in the development of alcohol use, problems, and disorders. Alcohol Clin Exp Res. 2010;34:1334–1345. doi: 10.1111/j.1530-0277.2010.01217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yang AR, Kelly T, Puche A, Esoga C, June HL, Jr, Elnabawi A, Merchenthaler I, Sieghart W, June HL, Sr, Aurelian L. Binge alcohol drinking is associated with GABAA alpha2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala. Proc Natl Acad Sci U S A. 2011;108(11):4465–70. doi: 10.1073/pnas.1019020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson LM, Grahame NJ. Pharmacologically relevant intake during chronic, free-choice drinking rhythms in selectively bred high alcohol-preferring mice. Addict Biol. 2011 doi: 10.1111/j.1369-1600.2011.00412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride WJ, Li TK. Animal models of alcoholism: neurobiology of high alcohol-drinking behavior in rodents. Crit Rev Neurobiol. 1998;12(4):339–69. doi: 10.1615/critrevneurobiol.v12.i4.40. [DOI] [PubMed] [Google Scholar]
- Mitchell JM, Tavares VC, Fields HL, D’Esposito M, Boettiger CA. Endogenous opioid blockade and impulsive responding in alcoholics and healthy controls. Neuropsychopharmacology. 2007;32:439–449. doi: 10.1038/sj.npp.1301226. [DOI] [PubMed] [Google Scholar]
- Murphy ER, Fernando AB, Urcelay GP, Robinson ES, Mar AC, Theobald DE, Dalley JW, Robbins TW. Impulsive behaviour induced by both NMDA receptor antagonism and GABAA receptor activation in rat ventromedial prefrontal cortex. Psychopharmacology (Berl) 2012;219(2):401–10. doi: 10.1007/s00213-011-2572-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naimi TS, Brewer RD, Mokdad A, Denny C, Serdula MK, Marks JS. Binge drinking among US adults. JAMA. 2003;289(1):70–5. doi: 10.1001/jama.289.1.70. [DOI] [PubMed] [Google Scholar]
- NIAAA. National Institute on Alcohol Abuse and Alcoholism Council approves definition of binge drinking. NIAAA Newsletter. 2004;3 (cited 2012 Sept 12). Available from: http://pubs.niaaa.nih.gov/publications/Newsletter/winter2004/Newsletter_Number3.pdf. [Google Scholar]
- Oberlin BG, Grahame NJ. High alcohol preferring mice are more impulsive than low alcohol preferring mice as measured in the delay discounting task. Alcohol Clin Exp Res. 2009;33:1–10. doi: 10.1111/j.1530-0277.2009.00955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlin BG, Bristow RE, Heighton ME, Grahame NJ. Pharmacologic dissociation between impulsivity and alcohol drinking in high alcohol preferring mice. Alcohol Clin Exp Res. 2010;34(8):1363–75. doi: 10.1111/j.1530-0277.2010.01220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petry NM. Delay discounting of money and alcohol in actively using alcoholics, currently abstinent alcoholics, and controls. Psychopharmacology. 2001;154:243–250. doi: 10.1007/s002130000638. [DOI] [PubMed] [Google Scholar]
- Popik P, Krawczyk M, Golembiowska K, Nowak G, Janowsky A, Skolnick P, Lippa A, Basile AS. Pharmacological profile of the “triple” monoamine neurotransmitter uptake inhibitor, DOV 102,677. Cell Mol Neurobiol. 2006;26(4–6):857–73. doi: 10.1007/s10571-006-9012-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachlin H, Green L. Commitment, choice and self-control. J Exp Anal Behav. 1972;17:15–22. doi: 10.1901/jeab.1972.17-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JB, Mitchell SH, de Wit H, Seiden LS. Determination of discount functions in rats with an adjusting-amount procedure. J Exp Anal Behav. 1997;67:353–366. doi: 10.1901/jeab.1997.67-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio G, Jiménez M, Rodríguez-Jiménez R, Martínez I, Avila C, Ferre F, Jiménez-Arriero MA, Ponce G, Palomo T. The role of behavioral impulsivity in the development of alcohol dependence: a 4-year follow-up study. Alcohol Clin Exp Res. 2008;32:1681–7. doi: 10.1111/j.1530-0277.2008.00746.x. [DOI] [PubMed] [Google Scholar]
- Sagvolden T. Behavioral validation of the spontaneously hypertensive rat (SHR) as an animal model of attention-deficit/hyperactivity disorder (AD/HD) Neurosci Biobehav Rev. 2000;24(1):31–39. doi: 10.1016/s0149-7634(99)00058-5. [DOI] [PubMed] [Google Scholar]
- Simon O’Brien E, Legastelois R, Houchi H, Vilpoux C, Alaux-Cantin S, Pierrefiche O, André E, Naassila M. Fluoxetine, desipramine, and the dual antidepressant milnacipran reduce alcohol self-administration and/or relapse in dependent rats. Neuropsychopharmacology. 2011;36(7):1518–30. doi: 10.1038/npp.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skolnick P, Basile AS. Triple reuptake inhibitors (“broad spectrum” antidepressants) CNS Neurol Disord Drug Targets. 2007;6:141–9. doi: 10.2174/187152707780363285. [DOI] [PubMed] [Google Scholar]
- Skolnick P. Triple-Uptake Inhibitors (Broad Spectrum Antidepressants) In: Peters JU, editor. Polypharmacology in Drug Discovery. Wiley & Sons Inc; New York: 2012. pp. 361–380. [Google Scholar]
- Tannock R, Schachar R, Logan G. Methylphenidate and cognitive flexibility: dissociated dose effects in hyperactive children. J Abnorm Child Psychol. 1995;23(2):235–66. doi: 10.1007/BF01447091. [DOI] [PubMed] [Google Scholar]
- Tizzano JP, Stribling DS, Perez-Tilve D, Strack A, Frassetto A, Chen RZ, Fong TM, Shearman L, Krieter PA, Tschöp MH, Skolnick P, Basile AS. The triple uptake inhibitor (1R,5S)-(+)-1-(3,4-dichlorophenyl)-3-azabicyclo[3.1.0] hexane hydrochloride (DOV 21947) reduces body weight and plasma triglycerides in rodent models of diet-induced obesity. J Pharmacol Exp Ther. 2008;324(3):1111–26. doi: 10.1124/jpet.107.133132. [DOI] [PubMed] [Google Scholar]
- Town M, Naimi TS, Mokdad AH, Brewer RD. Health care access among U.S. adults who drink alcohol excessively: missed opportunities for prevention. Prev Chronic Dis. 2006;3(2):A53. [PMC free article] [PubMed] [Google Scholar]
- Tran P, Skolnick P, Czobor P, Huang NY, Bradshaw M, McKinney A, et al. Efficacy and tolerability of the novel triple reuptake inhibitor amitifadine in the treatment of patients with major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Psychi Res. 2012;46(1):64–71. doi: 10.1016/j.jpsychires.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Vanderveen JW, Cohen LM, Watson NL. Utilizing a multimodal assessment strategy to examine variations of impulsivity among young adults engaged in co-occurring smoking and binge drinking behaviors. Drug Alcohol Depend. 2012 doi: 10.1016/j.drugalcdep.2012.06.026. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Warnock KT, Yang AR, Yi HS, June HL, Jr, Kelly T, Basile AS, Skolnick P, June HL. Amitifadine, a triple monoamine uptake inhibitor reduces binge drinking and negative affect in an animal model of co-occurring alcoholism and depression symptomatology. Pharmacol Biochem Behav. 2012;103(1):111–118. doi: 10.1016/j.pbb.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S, Carroll FI, Woolverton WL. A reduced rate of in vivo dopamine transporter binding is associated with lower relative reinforcing efficacy of stimulants. Neuropsychopharmacology. 2006;31(2):351–62. doi: 10.1038/sj.npp.1300795. [DOI] [PubMed] [Google Scholar]
- Wilhelm CJ, Mitchell SH. Rats bred for high alcohol drinking are more sensitive to delayed and probabilistic outcomes. Genes Brain Behav. 2008;7:705–713. doi: 10.1111/j.1601-183X.2008.00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstanley CA, Dalley JW, Theobald DE, Robbins TW. Fractionating impulsivity: contrasting effects of central 5-HT depletion on different measures of impulsive behavior. Neuropsychopharmacology. 2004;29:1331–1343. doi: 10.1038/sj.npp.1300434. [DOI] [PubMed] [Google Scholar]
- Yang AR, Liu J, Yi HS, Warnock KT, Wang M, June HL, Jr, Puche AC, Elnabawi A, Sieghart W, Aurelian L, June HL., Sr Binge Drinking: In Search of its Molecular Target via the GABA(A) Receptor. Front Neurosci. 2011;5:123. doi: 10.3389/fnins.2011.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AR, Yi HS, Warnock KT, Mamczarz J, June HL, Jr, Mallick N, Krieter PA, Tonelli L, Skolnick P, Basile AS, June HL., Sr Effects of the triple monoamine uptake inhibitor DOV 102,677 on alcohol-motivated responding and antidepressant activity in alcohol-preferring (P) rats. Alcohol Clin Exp Res. 2012;36(5):863–73. doi: 10.1111/j.1530-0277.2011.01671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.