

RSV infection induces IL-25 that contributes to the pathogenesis of pulmonary disease during primary infection and asthma-like exacerbations.
Keywords: mucus, Th2, cytokines
Abstract
One of the most severe pathologic responses of RSV infection is associated with overproduction of cytokines and inflammation, leading to mucus hypersecretion. This study investigated the role of IL-25 in the development of RSV-associated immunopathology. IL-25 and its receptor IL-17RB were increased following RSV infection, and IL-25 blockade using neutralizing antibodies reduced RSV-associated pathology, AHR, and type 2 cytokine production. Likewise, IL-17RB−/− mice demonstrated a modified inflammatory response during RSV infection characterized by decreased Th2 and increased Th17 cytokine production. Additionally, the IL-17RB−/− mice demonstrated significantly reduced inflammation and cytokine production in a model of RSV-driven asthma exacerbation. These results indicate that IL-25 regulates the inflammatory response to RSV infection and that its inhibition may enable a reduction in the severity of RSV-associated pulmonary inflammation, including during viral-induced asthma exacerbation.
Introduction
RSV is a ubiquitous pathogen and the most common cause of hospitalization in the first year of life [1–3]. Upwards of 95% of children have been exposed to the virus by age 2, and although subsequent infections are generally less severe, RSV represents a difficult target for the adaptive immune system in that T and B cell memory is not fully protective and does not prevent reinfection from occurring throughout life [4]. Opportunistic infections with RSV can have significant impacts on elderly and immunocompromised populations and present with variable symptoms ranging from asymptomatic illness to bronchiolitis, requiring hospitalization, and in the most severe cases, death, as a result of respiratory failure [5–7]. Initial attempts to generate a vaccine against RSV ended in failure when individuals inoculated with an inactivated virus became hyper-responsive to subsequent RSV infection, causing profound type 2 inflammation, mucus hypersecretion, and pulmonary eosinophilia [8]. The clinical importance of RSV and the absence of an effective vaccine make understanding immune regulation of RSV infection an important area of research.
IL-25 (IL-17E) is an IL-17 family member that regulates multiple aspects of mucosal immunity by promoting type 2 inflammation and production of IL-4, IL-5, and IL-13 [9]. Previous studies have suggested that IL-25 can have a significant role in RSV-induced immune responses [10]. We investigated the role of IL-25 in the pathogenesis of RSV, specifically, how inhibition of IL-25-mediated signals may alter the development of RSV-associated immunopathology. Here, we report that IL-25 is up-regulated following primary RSV infection and that peak cytokine expression corresponds with the intensity of inflammation. Our findings indicate that IL-25 and its receptor, IL-17RB, play an important role in the establishment of type 2 immunopathology in RSV infection. Furthermore, using a model of RSV-associated exacerbation of allergic airways disease in the absence of IL-17RB, a reduction in type 2 cytokine production and pathology was observed. Thus, IL-25 (IL-17E) and its receptor may be relevant targets for RSV-induced disease and provide a therapeutic and diagnostic cytokine to follow during disease for a predictive measurement of severity.
MATERIALS AND METHODS
Animals
Female mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). IL-17RB−/− mice were from Amgen (Thousand Oaks, CA, USA) and bred at the University of Michigan. Animal experiments were approved by the University of Michigan University Committee on Care.
RSV
Our laboratory used two clinical isolates of the antigenic subgroup A strain of RSV, Lines 19 [11] and A2-20 [12]. Both have been demonstrated in animal models to mimic human infection by stimulating mucus production, promoting AHR, and increasing IL-13 [12, 13]. Line 19 virus was used for BALBc mice, whereas the A2-20 strain was used for C57BL/6J mice, including IL-17RB−/− animals.
Viral exacerbation model
To simulate RSV exacerbation of allergic airway disease, C57BL/6J and IL-17RB−/− mice were sensitized to allergen on Days 0 and 14 and then inoculated with RSV on Day 17 of allergen sensitization. Mice then received additional allergen challenges on Days 21 and 23, with harvest on Day 24, as described [14].
Measurement of AHR
AHR was measured using mouse plethysmography, which is designed specifically for the low tidal volumes as a direct measure of pulmonary function, as described [13]. Airway resistance was measured in a closed plethysmograph by directly assessing tracheal pressure and comparing the level with box pressure changes using direct ventilation methodology. Once baseline levels had stabilized, a methacholine challenge was given via tail vein. The response was monitored, and the peak airway resistance was recorded as a measure of AHR.
Lung histology
Mouse lungs were perfused with 10% formalin (Sigma-Aldrich, St. Louis, MO, USA), instilled directly into the trachea, then immersed in 10% formalin solution overnight at room temperature, and stained with H&E or PAS.
LN restimulation
Draining mediastinal LNs from mice were isolated, and single-cell suspensions were obtained, as described above for in vitro experiments. LN cells (4×105/well in triplicate) were restimulated with 10 μl mL−1 allergen, 10 ng mL−1 IL-25, or both. RNA was isolated after 2 h in culture; supernatants were analyzed after 48 h in culture.
Flow cytometry
Analyses of pulmonary cell populations were assessed using standard techniques. Data were collected on a LSR II flow cytometer (BD Biosciences, San Jose, CA, USA) and on a FACSAria (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR, USA). The following mAb were obtained from BD Biosciences: CD8a-PE, CD11c-PE, CD125-PE, CD80-PE, CD86-PE, Nk1.1-PE, CD11b-PerCP-Cy5.5, MHC II(I-Ad)-APC, and Ly-6G-PeCy7; from BioLegend (San Diego, CA, USA): CD4-FITC, CD8a-FITC, B220-FITC, CD8a-PE, Ly-6C-PE, CD3-PeCy7, ScaI-Pe-Cy7, CCR3-AF647, Gr-1-AF700, IL-7Rα- APC-Cy7, and streptavidin-APC-Cy7; from eBioscience (San Diego, CA, USA): CD4-PeCy7, CD11c-APC-Cy7, Gr-1-PB, CD49b-PE, F4/80-PE, and FcϵR1a-AF647. Mouse anti-mouse IL-17RB was provided by A. L. Budelsky (Amgen, Seattle, WA, USA) and biotinylated for use in flow cytometry. The cell-surface markers of spleen cell controls were not altered by the collagenase treatment compared with untreated cells.
Quantitative PCR
Studies to quantitate mRNA were assessed using Real-Time PCR and reagents (Life Technologies, Grand Island, NY, USA), as described previously, with quantitation of fold increase with the 2−ΔΔ comparative threshold method [15].
Statistical analysis
Data were evaluated by one-way ANOVA and where appropriate, evaluated further with the parametric Student-Newman-Keuls test for multiple comparisons or the nonparametric Mann-Whitney rank-sum test.
RESULTS AND DISCUSSION
IL-25 and its receptor IL-17RB are expressed and participate in RSV-induced airway disease
Previous studies have identified that IL-25 participates in Th2-associated responses linked to changes in pathophysiology in the lung [15–17]. The results of the present study indicate that IL-25 represents a target associated with pathogenesis of severe lower respiratory tract infections. Type 2 cytokines—IL-13, in particular—act as primary mediators of mucus hypersecretion, and the ability of IL-25 to promote Th2 cytokine production may therefore contribute to airway obstruction associated with poor clinical outcomes. In our initial experiments, we sought to characterize whether IL-25 was expressed during the inflammatory response to RSV infection temporal expression of Il-25 (Fig. 1A) and IL-17RB (Fig. 1B) lung transcripts (Fig. 1). Transcripts of both genes increased following infection, peaking between Days 4 and 8 and returning to baseline levels by Day 12. These responses correlated well with other potentially pathogenic cytokines produced within the lungs of RSV-infected mice, including IL-13 and IFN-γ (Fig. 1C). To understand further the relationship of IL-25 and T cell activation, lung draining LNs from infected mice were stimulated with IL-25, RSV, or the combination. The data in Fig. 1D illustrate that IL-25 enhanced not only Th2 cytokines IL-5 and IL-13 but also Th1-associated IFN-γ. When we examined further the level of activation of lung draining LN T cells, we found that those CD4+ T cells that were IL-17RB+ had increased expression of two critical transcription factors associated with cytokine production—GATA3 and T-bet (Fig. 1E). Interestingly, previous studies have identified that IL-17RB was expressed primarily on effector/memory T cells [18, 19]. In contrast, not only were there fewer IL-17RB+ CD8 T cells, but also, they did not express higher transcription factors (Fig. 1E). Thus, whereas IL-25 has been shown to up-regulate GATA3 expression, its receptor may be expressed generally on activated CD4+ T cells and once expressed, enhance the production of cytokines.
Figure 1. Expression of IL-25 and its receptor IL-17RB in lungs of RSV-infected Balb/c mice is associated with cytokine responses and T cell cytokine expression.
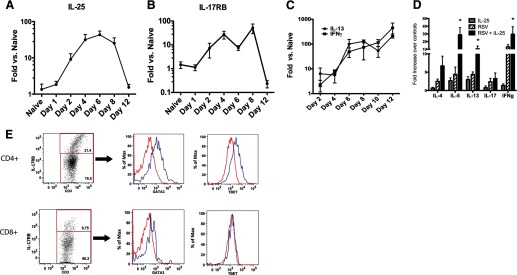
(A) IL-25 and (B) IL-17RB mRNA expression are both up-regulated during RSV infection in the lungs and (C) correlates to increased expression of RSV-induced cytokines, IFN-γ, and IL-13. (D) Quantitative PCR analysis of RSV-restimulated LN cells from 8-day-infected animals, with or without IL-25. *P<0.05. (E) Flow cytometry of reactivated T cells from RSV-infected mice demonstrates that CD4+ IL-17RB+ cells (blue line) preferentially express increased transcription factors, GATA3 and T-bet, whereas CD8+ T cells did not. Data represents mean ± se from five mice.
To investigate further the role of IL-25 in the inflammatory response to RSV infection, mice infected with RSV were treated with a polyclonal antibody to IL-25. The effects of anti-IL-25 therapy in mice were assessed on Day 10 postinfection, corresponding to peak pathophysiology in our model. RSV-infected mice, treated with anti-IL-25 antibody, had a significant reduction in AHR (Fig. 2A), as well as in the mucus-associated gene gob5 (Fig. 2B). Histologic analysis confirmed that inhibiting IL-25 reduced airway pathology and mucus (Fig. 2C); however, it did not affect specific subsets of inflammatory cells in the lung (Fig. 2D). Next, the draining LNs of RSV-infected animals were collected and restimulated in vitro to assess the effects of IL-25 blockade on antigen-specific cytokine production (Fig. 2E). RSV-restimulated LN cells from mice treated with control IgG produced type 2 cytokines in response to RSV, whereas LN cells from animals that had been treated with anti-IL-25 antibody showed dramatic reductions in production of type 2 cytokines. In contrast, the expression of IL-17a and IFN-γ was increased significantly in those animals treated with anti-IL-25. Therefore, these results indicate that IL-25 inhibition altered the activation of RSV-specific T lymphocytes and skewed the response to a less-pathogenic phenotype. Numerous studies have demonstrated that IL-25 regulates Th1 and Th17 immune responses [20–22], and most recently, the expression of IL-25 in the gut mucosa can regulate inflammatory bowel disease via IL-10 [23]. Interestingly, our studies did not observe any effect on viral clearance with anti-IL-25 treatment (data not shown). The expression of IL-17RB may provide a regulatory effect to inflammatory responses that can help protect tissue at mucosal surfaces. However, in the case of RSV, IL-25 overexpression may be linked to immunopathogenesis, especially in susceptible patient populations, including those with underlying pulmonary diseases, such as asthma.
Figure 2. Blockade of IL-25 during RSV infection in Balb/c mice attenuates goblet cell-associated pathology and mucus production.
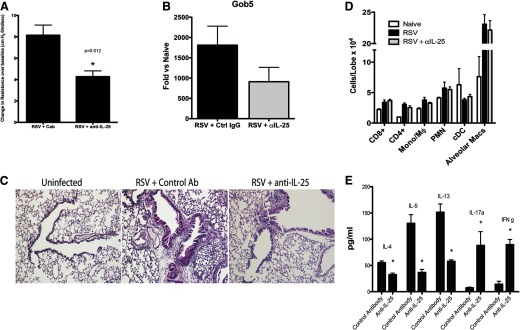
Naive Balb/c mice were infected with RSV (1×105 PFU/mouse), treated with control or anti-IL-25 polyclonal rabbit anti-mouse antibody on Days 0, 2, 4, and 6, and harvested on Day 8. (A) AHR and (B) mucus-associated pulmonary gob5 expression after 8 days of infection. Cab, control antibody; Ctrl, control. (C) Histologic assessment of PAS-stained lung sections illustrates a reduction in mucus-stained airways (pink). (D) Flow cytometric assessment of individual immune cell populations in lungs of infected mice depicts no reduction in cell accumulation. Mono, Monocyte; Mϕ, macrophage; cDC, conventional dendritic cell; Macs, macrophages. (E) RSV-restimulated lung draining LN culture supernatants were assayed by Bioplex for cytokine levels. Data represent mean ± se from five mice/group. *P < 0.05 and is representative of three repeat studies.
RSV infection of IL-17RB−/− mice demonstrates reduced pathogenic responses
To lend genetic proof to support our findings, IL-17RB−/− mice, which lack the ability to respond to IL-25, were infected with RSV. After 8 days of infection, IL-17RB−/− mice showed diminished pathology following RSV infection, including a reduction in airway inflammation and mucus hypersecretion (Fig. 3A). The reduced pathologic severity was accompanied by increased clearance of the virus, as depicted by expression of RSV G protein mRNA expression in the lungs of IL-17RB−/− mice compared with WT mice (Fig. 3B). There was a reduction in pulmonary cytokine transcripts, including IL-5, IL-13, and IFN-γ (Fig. 3C). Furthermore, cytokine production in draining LN cultures demonstrated the importance of IL-25 in regulating the antigen-specific response to RSV infection (Fig. 3D). Similar to the anti-IL-25-treated animals, there was a reduction in Th2 cytokines with a striking and significant increase in IL-17 production. When we examined transcription factor expression from the restimulated LNs of infected mice, we observed a decrease in GATA3 and T-bet in IL-17RB−/− mice (Fig. 3E). Examination of IL-17RB−/− T cells demonstrated that in the absence of IL-25 signaling, T cells are capable of responding to antigen and of producing cytokine (as evidenced by IL-17A in response to RSV). Taken together, these findings indicate that T cells may require activation before becoming IL-25-responsive and that activation itself can occur in an IL-25-independent manner. However, as described previously [24–26], IL-25 may act as an additional polarizing signal for antigen-dependent responses, enabling T cells to differentiate and mount a Th2 response in situations where IL-25 is being produced, such as allergic asthma and RSV infection. The alteration of immune responses and shift in the phenotype and reduced lung pathology are consistent with the findings with anti-IL-25 treatment and the reduction in the cytokine, and transcription factor levels may be reflective of more-efficient viral clearance. Interestingly, in studies from our lab and others, IL-17 has been suggested to contribute to lung pathology, in particular, to mucus hypersecretion. However, in the case of IL-25 neutralization or in IL-17RB−/− mice, when Th2 cytokines are reduced, the increased IL-17 does not lead to more pathogenesis. We have observed this aspect previously in studies with RSV vaccine development [27], where high levels of IL-17 in the absence of Th2 cytokines do not promote enhanced disease, suggesting that the most-severe disease occurs when a combined set of mediators is present.
Figure 3. Infection of IL-17RB−/− C5BL/6 mice demonstrates a reduction in disease pathology and alteration of cytokine responses.
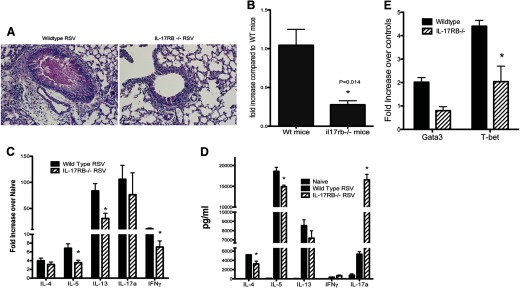
(A) Examination of lung pathology, 8 days post-RSV infection, demonstrated a reduction in inflammation and PAS-stained mucus production. (B) Quantitative PCR of RSV G protein in the lungs of 8-day-infected mice. The cytokine mRNA expression in the lungs (C) and protein in RSV-rechallenge draining LN cells (D) displayed a different profile in IL-17RB−/− mice. (E) The mRNA expression of GATA3 and T-bet transcription factors was reduced in IL-17RB−/− mice. Data represent mean ± se from five mice/group and are representative of three repeat studies. *P < 0.05.
Inhibition of IL-25 signaling impacts RSV-induced allergic inflammation significantly
Childhood viral infections have multiple effects on the risk for an eventual asthma diagnosis [28]. For example, individuals with sensitivities to house dust mite are at an up to sevenfold-higher risk for asthma if they experience a severe respiratory infection—RSV or rhinovirus [29]—supporting the fact that genetic predisposition plays an important role [30], as children hospitalized with RSV bronchiolitis before their first birthday are at a fivefold-increased risk of asthma if one parent has a history of allergic disease [31]. Thus, an individual that is susceptible to allergic disease or has underlying allergen sensitivity may be more sensitive to adverse affects of RSV infection. To understand whether IL-25 influences the ability of RSV to exacerbate allergic disease, we used a model of allergic asthma to simulate a viral exacerbation with RSV. WT and IL-17RB−/− mice were sensitized to allergen, as described previously [14], infected with RSV during the active allergic responses, and then challenged with allergen during the course of RSV infection (Day 5 postinfection). Our initial characterization of RSV-induced exacerbation demonstrated a significant increase in IL-25 mRNA expression that corresponded to a significant increase in IL-17RB+ CD11b+, GR1+ granulocytes (Fig. 4A), which we have characterized previously as playing an important role in driving airway pathology [32]. The data also indicate that there was a significant increase in IL-17RB+ CD4 T cells during allergen-induced responses that were not increased further by RSV exacerbation (Fig. 4A). No significant increase in NKT cells was observed in our RSV infection model and no change in number of IL-17RB+ NKT cells during exacerbation (data not shown), in contrast to observations in previous studies [10]. When we examined cellular changes in the total cell subsets in allergic mice exacerbated with RSV, we observed no significant change in any subset, although some reduction in CD4+CD69+ (activated) T cells was observed in the IL-17RB−/− mice (Fig. 4B), which may have been a result of increased viral clearance. As histology indicates, increases in infiltrates were observed during the RSV-induced exacerbation, and the absence of IL-17RB did not subjectively have an obvious decrease in cell infiltration (Fig. 4C). Studies with RSV infection response alone identified that CD4+ T cells were the primary pathogenic targets for IL-25 activation, with few IL-17RB+ granulocytes. However, when examining a RSV-induced allergen exacerbation, the most prominent IL-17RB+ cell population that increased was a granulocytic cell. We have termed this previously subset type 2 myeloid cells that produced IL-4 and IL-13 and were closely related to eosinophils [32]. Multiple subsets of myeloid cells appear to express IL-17RB, including basophils [33]. Interestingly eosinophil numbers, assessed by flow and specific granule stain, were not changed upon RSV exacerbation, matching our earlier observations [14]. Thus, there appear to be multiple subsets of leukocytes that express IL-17RB and have the ability to respond to IL-25 and impact severity of disease. However, the most apparent difference histologically between groups was the reduction in mucus production in the RSV-exacerbated IL-17RB−/− group, with RSV-allergen-exacerbated WT mice displaying more intense staining than the IL-17RB−/− mice, as indicated by the arrows (Fig. 4C). The analysis of lung mRNA expression demonstrated substantial reduction in the level of RSV-exacerbated IL-13 and the mucus associated gene gob5 in the IL-17RB−/− mice, confirming the histologic picture in Fig. 4C (Fig. 4D). Thus, overall, there appeared to be a significant reduction in the RSV-induced allergic airway exacerbation response in the absence of IL-17RB, characterized by an alteration of pathology. The latter aspect may reflect enhanced clearance of RSV that would ultimately lead to decreased immune activation.
Figure 4. RSV-induced exacerbation of allergic airway responses is attenuated in IL-17RB−/− C57BL/6 mice.
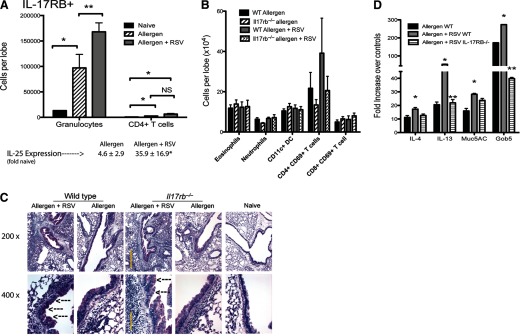
Allergic mice were infected with RSV and challenge two times with allergen (see Materials and Methods) to expose animals simultaneously to the stimuli. (A) Flow cytometry analyses indicated that the primary IL-17RB+ cell population that was increased was granulocytes. (B) Analysis of all leukocyte subsets indicated no significant change in any individual population. (C) Histological examination reflected the flow cytometry analysis in (B) indicating similar inflammatory cell accumulation in IL-17RB−/− mice; however, a reduced intensity in the PAS-postitive staining was evident in the RSV-exacerbated animals compared with WT. Arrows indicate PAS-stained goblet cells. (D) Quantitative PCR analysis of whole lung mRNA indicated that RSV-exacerbated IL-17RB−/− animals demonstrated a reduction in IL-13 and gob5 mucus gene expression. Muc5AC, Mucin 5AC. Data represent mean ± se from five mice/group.*P < 0.05; **P<0.01. This study was repeated twice with similar results.
Other recent studies have also highlighted that IL-17A can provide pathogenic signals that promote an enhanced disease phenotype associated with PMNs and mucus hypersecretion [34–36]. As IL-17A and IL-25 (IL-17E) share a common receptor chain—IL-17RA—and have common signaling properties, including a steroid-resistant phenotype [32, 37], targeting the common receptor may be beneficial. Recent strategies include mAb to the IL-17RA chain that target the binding of IL-17A and IL-25 [38]. This latter strategy may be important, given the cross-regulatory effect that IL-25 and IL-17A appear to have for one another. In the context of RSV infection, where a dysregulated inflammatory response drives disease pathology, the ability to regulate IL-17- and IL-25-induced inflammatory signals could represent a powerful tool for preventing unwanted consequences of viral infection. Our data suggest that significant pathologic components associated with RSV infection and asthma exacerbation may be managed effectively through the targeting of IL-25-mediated activation.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health grants RO1 HL059178 and AI036302.
Footnotes
- AHR
- airway hyper-responsiveness
- APC
- allophycocyanin
- IL-17RB−/−
- IL-17RB deficient
- PAS
- periodic acid-Schiff
- RSV
- respiratory syncytial virus
- T-bet
- T-box expressed in T cells
AUTHORSHIP
B.C.P. contributed to the writing, design, performance, and concept. V.D. contributed to the design and performance. A.R. contributed to the performance. N.W.L. contributed to the writing, design, performance, and concept.
DISCLOSURES
The authors have no conflict of interest with the results presented in this manuscript.
REFERENCES
- 1. Sigurs N. (2002) Clinical perspectives on the association between respiratory syncytial virus and reactive airway disease. Respir. Res. 3 (Suppl. 1), S8–S14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Szabo S. M., Levy A. R., Gooch K. L., Bradt P., Wijaya H., Mitchell I. (2013) Elevated risk of asthma after hospitalization for respiratory syncytial virus infection in infancy. Paediatr. Respir. Rev. 13 (Suppl. 2), S9–S15 [DOI] [PubMed] [Google Scholar]
- 3. Hall C. B. (2012) The burgeoning burden of respiratory syncytial virus among children. Infect. Disord. Drug Targets 12, 92–97 [DOI] [PubMed] [Google Scholar]
- 4. Habibi M. S., Openshaw P. J. (2012) Benefit and harm from immunity to respiratory syncytial virus: implications for treatment. Curr. Opin. Infect. Dis. 25, 687–694 [DOI] [PubMed] [Google Scholar]
- 5. Elliot A. J., Fleming D. M. (2008) Influenza and respiratory syncytial virus in the elderly. Expert Rev. Vaccines 7, 249–258 [DOI] [PubMed] [Google Scholar]
- 6. Falsey A. R. (2007) Respiratory syncytial virus infection in adults. Semin. Respir. Crit. Care Med. 28, 171–181 [DOI] [PubMed] [Google Scholar]
- 7. Falsey A. R., Walsh E. E. (2005) Respiratory syncytial virus infection in elderly adults. Drugs Aging 22, 577–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Graham B. S. (2011) Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol. Rev. 239, 149–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paul W. E., Zhu J. (2010) How are T(H)2-type immune responses initiated and amplified? Nat. Rev. Immunol. 10, 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaiko G. E., Phipps S., Angkasekwinai P., Dong C., Foster P. S. (2010) NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J. Immunol. 185, 4681–4690 [DOI] [PubMed] [Google Scholar]
- 11. Herlocher M. L., Ewasyshyn M., Sambhara S., Gharaee-Kermani M., Cho D., Lai J., Klein M., Maassab H. F. (1999) Immunological properties of plaque purified strains of live attenuated respiratory syncytial virus (RSV) for human vaccine. Vaccine 17, 172–181 [DOI] [PubMed] [Google Scholar]
- 12. Stokes K. L., Chi M. H., Sakamoto K., Newcomb D.C., Currier M. G., Huckabee M. M., Lee S., Goleniewska K., Pretto C., Williams J. V., Hotard A., Sherrill T. P., Peebles R. S., Jr., Moore M. L. (2011) Differential pathogenesis of respiratory syncytial virus clinical isolates in BALB/c mice. J. Virol. 85, 5782–5793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lukacs N. W., Moore M. L., Rudd B. D., Berlin A. A., Collins R. D., Olson S. J., Ho S. B., Peebles R. S., Jr., (2006) Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus. Am. J. Pathol. 169, 977–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schaller M. A., Kallal L. E., Lukacs N. W. (2008) A key role for CC chemokine receptor 1 in T-cell-mediated respiratory inflammation. Am. J. Pathol. 172, 386–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dolgachev V., Petersen B. C., Budelsky A. L., Berlin A. A., Lukacs N. W. (2009) Pulmonary IL-17E (IL-25) production and IL-17RB+ myeloid cell-derived Th2 cytokine production are dependent upon stem cell factor-induced responses during chronic allergic pulmonary disease. J. Immunol. 183, 5705–5715 [DOI] [PubMed] [Google Scholar]
- 16. Tamachi T., Maezawa Y., Ikeda K., Kagami S., Hatano M., Seto Y., Suto A., Suzuki K., Watanabe N., Saito Y., Tokuhisa T., Iwamoto I., Nakajima H. (2006) IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. J. Allergy Clin. Immunol. 118, 606–614 [DOI] [PubMed] [Google Scholar]
- 17. Sharkhuu T., Matthaei K. I., Forbes E., Mahalingam S., Hogan S. P., Hansbro P. M., Foster P. S. (2006) Mechanism of interleukin-25 (IL-17E)-induced pulmonary inflammation and airways hyper-reactivity. Clin. Exp. Allergy 36, 1575–1583 [DOI] [PubMed] [Google Scholar]
- 18. Terrier B., Bieche, Maisonobe I. T., Laurendeau I., Rosenzwajg M., Kahn J. E, Diemert M. C, Musset L., Vidaud M., Sene D., Costedoat-Chalumeau N., Le Thi-Huong D., Amoura Z., Klatzmann D., Cacoub P., Saadoun D. (2010) Interleukin-25: a cytokine linking eosinophils and adaptive immunity in Churg-Strauss syndrome. Blood 116, 4523–4531 [DOI] [PubMed] [Google Scholar]
- 19. Wang Y. H., Liu Y. J. (2009) Thymic stromal lymphopoietin, OX40-ligand, and interleukin-25 in allergic responses. Clin. Exp. Allergy 39, 798–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishii A., Oboki K., Nambu A., Morita H., Ohno T., Kajiwara N., Arae K., Sudo H., Okumura K., Saito H., Nakae S. (2010) Development of IL-17-mediated delayed-type hypersensitivity is not affected by down-regulation of IL-25 expression. Allergol. Int. 59, 399–408 [DOI] [PubMed] [Google Scholar]
- 21. Zaph C., Du Y., Saenz S. A., Nair M. G., Perrigoue J. G., Taylor B. C., Troy A. E., Kobuley D. E., Kastelein R. A., Cua D. J., Yu Y., Artis D. (2008) Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J. Exp. Med. 205, 2191–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kleinschek M. A., Owyang A. M., Joyce-Shaikh B., Langrish C. L., Chen Y., Gorman D. M., Blumenschein M. W., McClanahan T., Brombacher F., Hurst S. D., Kastelein R. A., Cua D. J. (2007) IL-25 regulates Th17 function in autoimmune inflammation. J. Exp. Med. 204, 161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Su J., Chen T., Ji X. Y., Liu C., Yadav P. K., Wu R., Yang P., Liu Z. (2013) IL-25 downregulates Th1/Th17 immune response in an IL-10-dependent manner in inflammatory bowel disease. Inflamm. Bowel Dis. 19, 720–728 [DOI] [PubMed] [Google Scholar]
- 24. Wang Y. H., Angkasekwinai P., Lu N., Voo K. S., Arima K., Hanabuchi S., Hippe A., Corrigan C. J., Dong C., Homey B., Yao Z., Ying S., Huston D. P., Liu Y. J. (2007) IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J. Exp. Med. 204, 1837–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Angkasekwinai P., Park H., Wang Y. H., Chang S. H., Corry D. B., Liu Y. J., Zhu Z., Dong C. (2007) Interleukin 25 promotes the initiation of proallergic type 2 responses. J. Exp. Med. 204, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fort M. M., Cheung J., Yen D., Li J., Zurawski S. M., Lo S., Menon S., Clifford T., Hunte B., Lesley R., Muchamuel T., Hurst S. D., Zurawski G., Leach M. W., Gorman D. M., Rennick D. M. (2001) IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 15, 985–995 [DOI] [PubMed] [Google Scholar]
- 27. Lindell D. M., Morris S. B., White M. P., Kallal L. E., Lundy P. K., Hamouda T., Baker J. R., Jr., Lukacs N. W. (2011) A novel inactivated intranasal respiratory syncytial virus vaccine promotes viral clearance without Th2 associated vaccine-enhanced disease. PLoS One 6, e21823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stein R. T., Sherrill D. W., Morgan J., Holberg C. J., Halonen M., Taussig L. M., Wright A. L., Martinez F. D. (1999) Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet 354, 541–545 [DOI] [PubMed] [Google Scholar]
- 29. Holt P. G., Upham J. W., Sly P. D. (2005) Contemporaneous maturation of immunologic and respiratory functions during early childhood: implications for development of asthma prevention strategies. J. Allergy Clin. Immunol. 116, 16–24 [DOI] [PubMed] [Google Scholar]
- 30. Drysdale S. B., Milner A. D., Greenough A. (2012) Respiratory syncytial virus infection and chronic respiratory morbidity—is there a functional or genetic predisposition? Acta Paediatr. 101, 1114–1120 [DOI] [PubMed] [Google Scholar]
- 31. Sigurs N., Aljassim F., Kjellman B., Robinson P. D., Sigurbergsson F., Bjarnason R., Gustafsson P. M. (2010) Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 65, 1045–1052 [DOI] [PubMed] [Google Scholar]
- 32. Petersen B. C., Budelsky A. L., Baptist A. P., Schaller M. A., Lukacs N. W. (2012) Interleukin-25 induces type 2 cytokine production in a steroid-resistant interleukin-17RB+ myeloid population that exacerbates asthmatic pathology. Nat. Med. 18, 751–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang H., Mobini R., Fang Y., Barrenas F., Zhang H., Xiang Z., Benson M. (2010) Allergen challenge of peripheral blood mononuclear cells from patients with seasonal allergic rhinitis increases IL-17RB, which regulates basophil apoptosis and degranulation. Clin. Exp. Allergy 40, 1194–1202 [DOI] [PubMed] [Google Scholar]
- 34. Ono N., Kusunoki T., Ikeda K. (2012) Relationships between IL-17A and macrophages or MUC5AC in eosinophilic chronic rhinosinusitis and proposed pathological significance. Allergy Rhinol. (Providence) 3, e50–e54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wakashin H., Hirose K., Iwamoto I., Nakajima H. (2009) Role of IL-23-Th17 cell axis in allergic airway inflammation. Int. Arch. Allergy Immunol. 149 (Suppl. 1), 108–112 [DOI] [PubMed] [Google Scholar]
- 36. Newcomb D. C., Boswell M. G., Sherrill T. P., Polosukhin V. V., Boyd K. L., Goleniewska K., Brody S. L., Kolls J. K., Adler K. B., Peebles R. S., Jr., (2013) IL-17A induces signal transducers and activators of transcription-6-independent airway mucous cell metaplasia. Am. J. Respir. Cell Mol. Biol. 48, 711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McKinley L., Alcorn J. F., Peterson A., Dupont R. B., Kapadia S., Logar A., Henry A., Irvin C., G., Piganelli J. D., Ray A., Kolls J. K. (2008) TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 181, 4089–4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barlow J. L., McKenzie A. N. (2009) IL-25: a key requirement for the regulation of type-2 immunity. Biofactors 35, 178–182 [DOI] [PubMed] [Google Scholar]