

Abstract
Phosphate plays essential biological roles and its plasma level in humans requires tight control to avoid bone loss (insufficiency) or vascular calcification (excess). Intestinal absorption and renal reabsorption of phosphate are mediated by members of the SLC34 family of sodium-coupled transporters (NaPi-IIa,b,c) whose membrane expression is regulated by various hormones, circulating proteins, and phosphate itself. Consequently, NaPi-II proteins are also potentially important pharmaceutical targets for controlling phosphate levels. Their crucial role in Pi homeostasis is underscored by pathologies resulting from naturally occurring SLC34 mutations and SLC34 knockout animals. SLC34 isoforms have been extensively studied with respect to transport mechanism and structure-function relationships; however, the three-dimensional structure is unknown. All SLC34 transporters share a duplicated motif comprising a glutamine followed by a stretch of threonine or serine residues, suggesting the presence of structural repeats as found in other transporter families. Nevertheless, standard bioinformatic approaches fail to clearly identify a suitable template for molecular modeling. Here, we used hydrophobicity profiles and hidden Markov models to define a structural repeat common to all SLC34 isoforms. Similar approaches identify a relationship with the core regions in a crystal structure of Vibrio cholerae Na+-dicarboxylate transporter VcINDY, from which we generated a homology model of human NaPi-IIa. The aforementioned SLC34 motifs in each repeat localize to the center of the model, and were predicted to form Na+ and Pi coordination sites. Functional relevance of key amino acids was confirmed by biochemical and electrophysiological analysis of expressed, mutated transporters. Moreover, the validity of the predicted architecture is corroborated by extensive published structure-function studies. The model provides key information for elucidating the transport mechanism and predicts candidate substrate binding sites.
Introduction
Cells depend on inorganic phosphate (Pi) to ensure growth and structural integrity, maintain energy balance, and communicate with their environment. In mammals, Pi transport across the cell membrane is mediated by secondary active transporter proteins that use the free energy from the Na+ gradient and transmembrane (TM) electrical potential to catalyze uphill Pi transport. Multicellular organisms, especially vertebrates, face a particular challenge in maintaining whole body Pi homeostasis: the solubility of Pi is limited in the presence of divalent cations, especially calcium, and excess of Pi in bodily fluids promotes Ca-Pi precipitation. This is of particular concern in patients with chronic kidney disease who tend to retain excessive levels of Pi (hyperphosphatemia), which leads to vascular calcification (1).
A family of Na+-coupled Pi transporters belonging to the solute carrier family SLC34 (NaPi-II), is central to maintaining whole body Pi homeostasis (2). All vertebrates express up to three isoforms of NaPi-II in organs that contribute to maintaining Pi balance. Mammals express NaPi-IIb (SLC34A2) in the intestine to mediate dietary Pi absorption and express NaPi-IIa (SLC34A1) together with NaPi-IIc (SLC34A3) in the renal proximal tubule to mediate reabsorption of Pi from the glomerular filtrate (2–4). The central role of NaPi-II proteins emerged when hormones and metabolic factors known to influence body Pi levels, such as parathyroid hormone, calcitonin, growth hormones, or Pi availability, were found to regulate NaPi-II membrane expression (reviewed in (5,6)). Their homeostatic role was further corroborated by NaPi-II knockout mice, which displayed organ-specific perturbations of Pi handling (7). Of importance, in humans, dysfunction in renal Pi handling can be attributed to naturally occurring mutations in NaPi-IIa (8,9); NaPi-IIc (10), and to Pi-related pathologies for NaPi-IIb in lung and testes (11), further underscoring the importance of these proteins.
Functional analyses of wild-type (WT) and mutant transporters expressed in Xenopus laevis oocytes have allowed a detailed characterization of each isoform. The NaPi-II proteins transport one divalent Pi ion with an apparent affinity (K0.5Pi) of 10–70 μM, together with two (NaPi-IIc) or three Na+ ions (NaPi-IIa and NaPi-IIb) with an apparent affinity (K0.5Na) of 25–50 mM (reviewed in (12)). The experimental evidence supports the notion that NaPi-II proteins mediate transport according to the canonical alternating access mechanism (13–15). In the normal transport cycle (Fig. 1 A), substrate binding at the external side is ordered, with 2 Na+ ions binding before Pi. These cation interactions, together with intrinsic mobile charges that are postulated to alter the empty carrier orientation between inward and outward facing conformations (states 0 and 1, Fig. 1 A), effectively couple membrane potential to the NaPi-IIa/b transport cycle to act as a transport driving force (12). In contrast, the electroneutral NaPi-IIc cycle is insensitive to membrane voltage and only one of the Na+ ions preceding Pi binding is translocated. However, for all three isoforms, binding of the last Na+ is a rate-determining partial reaction (Fig. 1 A, transition 4 → 5) (16,17) and moreover, the similarity of apparent substrate affinities suggests that they share the same substrate recognition and translocation structural unit.
Figure 1.
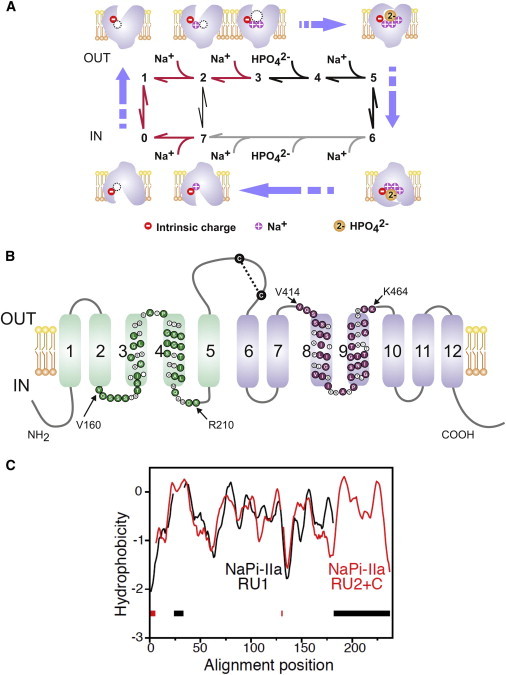
Kinetic and structural features of NaPi-IIa. (A) The transport cycle is depicted as a sequence of partial reactions between conformational states, numbered 0–7 (reviewed in (12)). Cartoons illustrate the ordered nature of protein-substrate interaction based on experimental evidence. Partial reactions on the cytosolic side have not been explicitly identified except the last Na+ release (transition 7 → 0) (68). Red arrows: electrogenic partial reactions (involving charge movement); black arrows: electroneutral partial reactions. In the absence of external Pi, Na+ ions can also translocate via a leak transition (2 ↔ 7) (66). For the electroneutral cycle (of NaPi-IIc), all transitions are electroneutral and the first Na+ ion to bind (transition 1 → 2) is hypothesized not to translocate (58). (B) Secondary topology of NaPi-IIa based on previous experimental evidence and bioinformatic predictions (12). The assumed boundaries of repeat regions (rat sequence) are indicated (52); colored symbols indicate identical or conserved residues in each repeat. A disulfide bridge links the two halves of the protein in the large extracellular loop. (C) Hydropathy plots of the regions predicted to contain the structural repeats in NaPi-II, averaged over a set of sequence homologs and aligned using AlignMe. Region 1, containing residues 86–256 of the human NaPi-IIa (RU1, black) is aligned to region 2 plus C-terminus consisting of residues 335–564 (RU2+C, red). Gaps in the alignment are indicated by dashes along the base of the plot. To see this figure in color, go online.
All vertebrate NaPi-II homologs identified and studied to date are assumed to have identical TM topologies (12) (Fig. 1 B). Specifically, the protein contains two sets of TM helices, each containing a copy of a conserved sequence motif, separated by a large extracellular loop. Epitope tagging and cysteine scanning mutagenesis experiments suggest that these repeated units have inverted TM topologies and that both termini are cytosolic (12,18). However, a three-dimensional structural model of NaPi-II proteins that integrates these functional and structural data is still lacking, preventing further understanding of the transport mechanism and limiting the interpretation of structure-function data.
The inverted-topology repeat architecture represents a common structural leitmotif displayed by a number of secondary active transporters for which crystal structures have been determined recently (see e.g. (13,19,20).). Such repeats provide a pseudosymmetry to the fold that has a number of important implications for function. For example, these transporters frequently harbor a central substrate binding site at the axis of pseudosymmetry. This site is then accessed alternately via two pseudosymmetric pathways, each constructed from equivalent elements from the two repeats (21,22). Interestingly, binding sites for sodium ions have also been identified by studying pseudosymmetric positions in structures of a sodium-coupled transporter, BetP (23). Thus, although many transporters have evolved significant breaks in this pseudosymmetry, presumably as a means to introduce specificity and variety, consideration of the repeats and their symmetry can be a useful starting point for structure-function studies of secondary transporters.
The recognition of the conserved inverted-topology sequence repeat in NaPi-II prompted us to search for a suitable template among the crystal structures of solute transporters reported to date, from which a homology model could be generated, compared with a wealth of experimental evidence, and used to predict putative binding site regions.
Methods
Computational methods
Full details of the computational procedures are provided in the Supporting Material. A brief description follows. Sequences of NaPi-II transporters were identified using HMMER (24) from the National Center for Biotechnology Information (NCBI) nonredundant (nr) database, clustered using UCLUST (25), and aligned with MSAProbs (26). Segments of the multiple sequence alignments were converted to averaged hydropathy profiles and aligned with the AlignMe web server (27). Hidden Markov-models (HMMs) of the same segments were constructed after identifying homologs from the NCBI nr20 database using HHblits (28). HMMs were aligned using HHalign (29).
Template structures were searched for using PSI-BLAST (30), Phyre2 (31), COMA (32), and HHpred (29). Template-target alignments of either full-length proteins or individual repeats were generated with either Profile+Secondary-Structure (AlignMePS) or +Transmembrane (AlignMePST) modes on the AlignMe server (27), or with HHalign.
A homology model was constructed using Modeler (33) based on the structure of VcINDY (PDB identifier 4F35). After refinement and guided by conservation patterns from the ConSurf server (34), the percentage of identical residues between VcINDY and NaPi-II was ∼15% in repeat unit 1 (RU1) and 11% in repeat unit 2 (RU2). A single model was selected after 2000 Modeler attempts, with two sodium ions and a phosphate molecule modeled in putative binding sites. The model was evaluated using ProQM (35) and PROCHECK (36).
Functional expression in oocytes and transport assays
Reagents and solutions
Oocytes were incubated in Modified Barth’s solution that contained (in mM): 88 NaCl, 1 KCl, 0.41 CaCl2, 0.82 MgSO4, 2.5 NaHCO3, 2 Ca(NO3)2, 7.5 HEPES, adjusted to pH 7.5 with TRIS and supplemented with antibiotics doxycyclin and gentamicin (5 mg/l). The solution compositions were as follows: Control superfusate (100 Na) (in mM): 100 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, adjusted to pH 7.4 using TRIS. Choline solution (100 Ch): as for 100 Na with isosmotic substitution of 100 mM choline chloride. Pi was added to the required substrate concentration from 1 M K2HPO4 and KH2PO4 stocks premixed to give the required pH (7.4).
Molecular biology and single point mutations
cDNA encoding WT human NaPi-IIa was subcloned into a vector containing the 5′ and 3′ UTRs from Xenopus β-globin to improve its expression in oocytes. Mutations were introduced using the Quickchange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The sequence was verified by sequencing (Microsynth, AG, Balgach, Switzerland), plasmids were linearized with XbaI and cRNA was synthesized in presence of Cap analog using the T3 Message Machine kit (Ambion, Austin, TX).
Functional expression and transport assays
X. laevis frogs were purchased from Xenopus Express France (Vernassal, France). Ovarian lobes were surgically removed and the oocytes isolated following standard protocols (e.g. (37)). Animal procedures were performed according to Swiss Cantonal and Federal legislation. Injected oocytes (50 nl of cRNA (0.2 μg/μl)) were incubated for 3–5 days in modified Barth’s solution. Radioisotope uptake assays were performed using standard procedures as described elsewhere (e.g. (38)) using 100 Na solution and 1 mM cold Pi to which 32Pi (specific activity 10 mCi/mmol Pi) was added. Standard two-electrode voltage clamp hardware was used for electrophysiology (GeneClamp, model 500, Molecular Devices, Sunnyvale, CA) and controlled using pClamp 8-9 software (Molecular Devices). Clampfit (Molecular Devices) was used for postacquisition analysis.
Immunostaining
After 3 days of expression, oocytes were fixed according to standard procedures (e.g. (39)). Further details are given in the Supporting Material.
Results
Identifying the repeated elements and peripheral TM helices in NaPi-IIa
Modeling the structure of a protein is aided by knowledge of the conserved and repeated structural elements within its fold. Although it was clear from earlier studies, and from the presence of a repeated conserved motif (Fig. S1), that NaPi-II transporters contain a structural repeat with inverted topology, it was not obvious where the boundaries of the repeat units lay, or was it clear whether their overall fold had an equivalent already reported. Aligning family-averaged hydropathy profiles of different fragments of the sequence clearly shows that the structure comprises two repeat units (RU1, RU2) plus a C-terminal extension of two additional TM helices (Fig. 1 C). These segments correspond to TMs 1–5, TMs 6–10 and TMs 11–12, respectively, of the published topology (Fig. 1 B).
A similar result was obtained by aligning HHalign hidden HMMs of region 1 with region 2 or with region 2+C (see Methods; Fig. S1, Fig. S2). This confirmed that regions 1 and 2 have similar secondary structures and further indicated that they have ∼27% identical residues. We concluded that RU1 and RU2 in NaPi-II comprise residues 86–256 and 335–489, respectively, and residues 504–564 form peripheral TM helices that are not part of the core fold.
Identifying a structural homolog of NaPi-IIa
To identify whether any available structure would be a suitable template for NaPi-IIa, several approaches were attempted (see Methods), of which HHpred provided the most promising clue. Specifically, among the top five hits was a Na+-coupled dicarboxylate transporter from Vibrio cholera (VcINDY) (40), also known as NaDC, a member of the Divalent Anion:Na+ Symporter (DASS, 2.A.47) (41) or SLC13 family (42), which also includes Na+-coupled inorganic anion (sulfate and phosphate) transporters (41). The VcINDY structural fold belongs to the ST[3] class (43) and contains an inverted-topology repeat fold (40). The E-values of the top 10 hits were rather large (>2 × 102), indicative of low confidence levels. Among those top 10 were three aquaporins, a GlpG protease, a KQT potassium channel, and a disulphide reductase, DsbD, as well as three water-soluble proteins. Pairwise alignments of NaPi-IIa with four of the five highest-scoring membrane proteins (aquaporin-4, GlpG protease, AQY1 aquaporin, and VcINDY) using AlignMe in PS mode indicated that the template with the greatest fold similarity among these hits was VcINDY, with 62% sequence coverage and 7% identical residues, compared to ≤40% coverage and ≤4% identity for the other four proteins. (The other HHpred hit in the top five, the KQT potassium channel was excluded because the region matched to NaPi-II comprised unconserved residues.)
The high E-value and small percentage of identical residues in VcINDY and NaPi-II suggested significant differences between their sequences, though these properties do not preclude them from sharing a common core fold given that structure is much more conserved during evolution than sequence. Indeed, the two repeats in inverted-topology membrane proteins can contain <10% identical residues, despite being structurally related (14,40,44). In the aforementioned search, HHpred matched the conserved QSSS NaPi-II motif to a SNT motif that contributes to Na+ and dicarboxylate binding in VcINDY (Fig. S4 B), which suggested that the binding site regions at least are conserved. To assess whether the similarities between VcINDY and NaPi-II extend beyond function, the presence of inverted repeats and hydrophilic binding motifs, we used a number of other bioinformatic approaches (see Methods) culminating in homology modeling and comparison with experimental data.
An important factor complicating the detection of distant sequence relationships between transporter proteins has been the presence of additional structurally-peripheral TM helices that confound the alignment algorithms. A case in point is the BCCT and NSS families (14,20,40,44,45), which contain two additional TM helices either before or after, respectively, the core inverted-topology fold. The structural similarity between these families is clear using hydropathy profile alignments, especially after separating out the repeats of each protein, because that reduces the chances of core helices aligning to peripheral helices (46).
In a family-averaged hydropathy profile alignment of full-length NaPi-IIa and VcINDY, RU1 of NaPi-IIa was aligned with several helices in the first repeat of VcINDY (Fig. S4 A, green dashed box). That segment of the alignment resembled the NaPi-II repeat alignment (Fig. 1 B), suggesting a relationship between the two proteins. However, RU2 was not aligned to the second repeat of VcINDY in this global alignment, due to a shift of one TM segment (Fig. S4 A, orange dashed box). Therefore, to identify putative corresponding helices in both VcINDY and NaPi-II repeats, we separately aligned hydropathy profiles of RU1 or RU2 from the two proteins (Fig. 2 A), as well as HMMs of these repeat units (Fig. S4 C and D). These alignments from both methods, as well as alignments obtained using AlignMe in PST mode, clearly indicated that the first two TMs of each of the repeats of VcINDY (Fig. S3 A) has no equivalent in NaPi-IIa.
Figure 2.
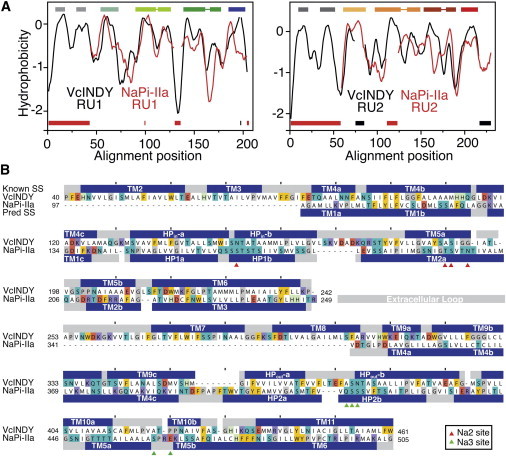
Comparison of repeat units in NaPi-II and the sodium-carboxylate transporter VcINDY suggests that they share a common fold. (A) Hydrophobicity profiles of the two repeat units in NaPi-II (red line) and VcINDY (black line). Hydropathy plots averaged over a set of sequence homologs of human NaPi-IIa or of VcINDY were aligned using AlignMe for repeat unit 1 (left) and repeat unit 2 (right). (B) Sequence alignment between VcINDY and the core region of human NaPi-IIa used for modeling. The helices assigned from the structure of VcINDY (Known SS) and the PSIPRED predictions for helices in NaPi-IIa (Pred SS) are shown as blue bars above and below the sequences, respectively. Residues whose side chains contribute to site Na2 (red triangles) or Na3 (green triangles) are indicated. To see this figure in color, go online.
In VcINDY, these additional helices (TMs 2, 3, 7, and 8) are located at the periphery of the VcINDY monomer (Fig. S3 A), and therefore could be omitted from the NaPi-IIa model without affecting the integrity of the core transport unit. Indeed, in the case of citrate carrier (CitS), which was recently shown to share a similar architecture with VcINDY, substrate-activated structural changes observed in electron microscopy projection maps include no conformational changes in these helices. This indicated that they act as a stator with little or no functional role (47). The fact that both NaPi-IIa repeats align to the equivalent structurally repeated regions in VcINDY (i.e., the C-terminal segment of each repeat, Fig. 2 A) gave support to the proposed relationship between the two proteins. At the same time, the significant number of additional helices in VcINDY compared to NaPi-IIa, before, between, and after the repeats (Fig. S3 A) indicated why the alignments of the full-length sequences were poor and the scores were low.
According to HHalign and AlignMePST alignments of the individual repeats the proposed Na+ and substrate binding site residues of both RU1 and RU2 of VcINDY were aligned to the most conserved regions of NaPi-IIa (Fig. 2 B, Fig. S4 B, and Fig. S4 C). This matching of conserved residues, the consistency of the alignments using three different alignment methodologies, i.e., AlignMePST, HHalign, and hydropathy profile alignment, and the equivalency of the matched helices in both repeats, together provided support for the choice of VcINDY as a template for NaPi-II transporters.
A structural model of human NaPi-IIa
Although the expected accuracy of a model with this level of sequence similarity is modest, it should suffice for comparison with topological and accessibility data even if individual TM segments are misaligned by one or two helical turns (48). An initial model of human NaPi-IIa comprising residues 97–249 (RU1) and 341–505 (RU2) was constructed (Fig. 3 and Fig. 4, A–C), whose topology differs in some aspects from earlier proposals (Fig. 1 B). The earlier, simpler proposal, based on the assumption of membrane-spanning segments, agrees with the present model on the nature of TMs 1 and 3 (previously 1 and 5) and TMs 4 and 6 (previously 6 and 10). In addition, the overall topology, in the sense of the accessibility of the loops, is unchanged. However, in its oversimplicity, the earlier model did not satisfactorily explain the unusual hydropathic nature of the segments between TMs 1 and 3, and between TMs 4 and 6 (Figs. 2 B, 3 A, and 4 C). Because these segments in VcINDY consist of helical hairpins that do not fully span the membrane, as well as long nonhelical elements within TM segments, the model based on VcINDY therefore makes sense of the hydropathy plots. At the same time, the updated topology based on VcINDY agrees well with results from studies using in vitro glycosylation (49) and cysteine scanning mutagenesis and accessibility measurements (SCAM -substituted cysteine accessibility method (50)), e.g. (51–55).), as shown mapped onto the model (Fig. 3 B, Fig. 4 C).
Figure 3.
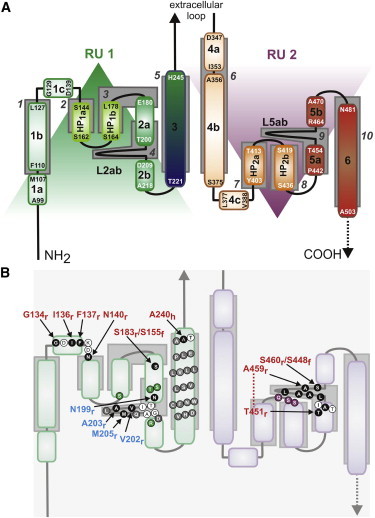
Schematics of refined topology of NaPi-IIa. (A) Topological elements are colored shades of green for RU1 and shades of red for RU2, and the structural repeats (RU1, RU2) are indicated (shaded triangles). Approximate positions of helical domains according to an earlier, simplified topology (gray boxes) are numbered according to the scheme in Fig. 1B for the human NaPi-IIa sequence. (B) Comparison with published experimental data. The position of key residues is indicated. Residues that are accessible based on cysteine scanning (SCAM) studies (black solid circles); residues that are inaccessible based on SCAM (gray solid circles) are highlighted. Sites accessible from the external milieu (red) or accessible only from the internal milieu (blue) are labeled according to the organism: flounder NaPi-IIb (f), human (h) or rat (r) NaPi-IIa. Positions on the topology of residues mutated in this study are also shown (green and violet solid circles for RU1 and RU2, respectively). To see this figure in color, go online.
Figure 4.
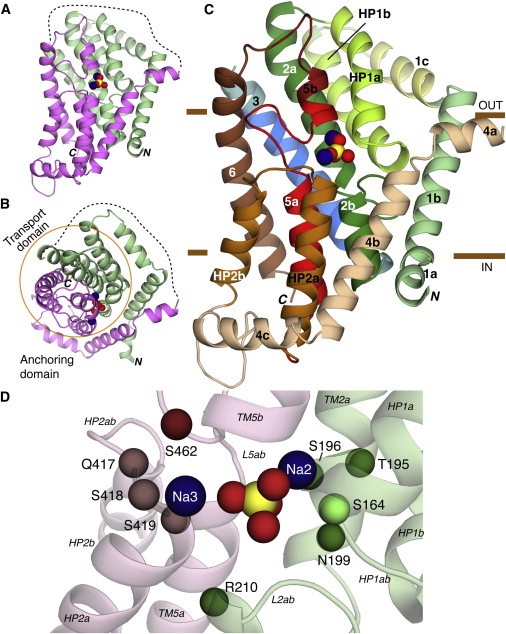
Structural model of human NaPi-IIa based on VcINDY. (A–C) Overview of the predicted fold of the NaPi-IIa model represented by cartoon helices. Two sodium ions (blue) and Pi (yellow, red) are shown as spheres. The model is viewed (A and C) from within the plane of the membrane, or (B) from the extracellular side. (A and B) Location of the structural repeats RU1 (green) and RU2 (pink), and the connecting long extracellular loop (dashed lines). The transport domain is highlighted by an orange circle, in B. In C, individual helices are colored according to Fig. 3A. Regions for which experimental data relating to the topology are highlighted: residues accessible to the extracellular side (yellow) or buried (blue) in helix 1c and 3, respectively; and residues expected to be helical in TM5a, TM5b, and the 5ab loop (red). The approximate extents of the membrane are indicated by brown bars. (D) The predicted binding site region in the model of NaPi-IIa. The Cα-atoms of conserved and polar residues close to the putative binding site are highlighted, as are bound Pi and sodium ions (spheres). Residues N199 and S462 have previously been implicated in substrate binding. Residues S164, T195, S196, R210, Q417, S418, S419 are investigated in this study. To see this figure in color, go online.
First, SCAM in the linker between TM1 and TM2 of RU1 (see Fig. 3 B) established that this region is accessible to the external medium (56), consistent with its position at the exposed external surface of our model (helix 1c, Fig. 4 C).
Second, SCAM in RU2 identified an externally accessible region in loop L5ab between TM5a and TM5b (Fig. 3 B) (53). In our model (Fig. 4 C), this linker is now localized at the same depth as the proposed substrate coordination sites (see below). Nevertheless, it should be accessible to cysteine modifiers via the same aqueous pathway as that of the substrates. Our original topology (Fig. 1 B) also implied that helix 1c should be more accessible from the external medium than loop L5ab, whereas SCAM studies (56) indicate that both regions have similar apparent accessibilities, which is in better agreement with their positioning relative to the external medium depicted in our three-dimensional model (Fig. 4 C). Finally, a cysteine at position S424, which was considered accessible based on the original topology, did not give resolvable fluorescence upon attempts to label the equivalent site in the flounder isoform (57). In better agreement with that data, the updated model (Figs. 3 and 4 C) places this residue deeper in the structure within HP2b.
Third, thiol modification from the external medium of a Cys substituted at S183 in NaPi-IIa or at the equivalent site in flounder NaPi-IIb (S155) at the top of helix 2a in RU1 (Fig. 3 B) is possible, with minimal functional consequences (49,57,58). These findings are consistent with the relatively exposed position of this site, far from the central binding region in the new fold (Fig. 4 C).
Fourth, SCAM revealed four sites that are accessible only from the cytosol: N199 at the end of TM2a, together with V202, A203, and M205 from L2ab (49,52,57,58) (Fig. 3 B). By contrast, the model suggests that these sites are buried. However, it should be noted that our model corresponds to an outward-open state, in which only the extracellular pathway should be accessible. Transporters containing pseudosymmetric repeats typically have pseudosymmetric pathways, which become exposed in a given state, whereas their counterparts in the other repeat are not (13,14). In NaPi-II, residues from TM5a and loop L5ab—the symmetry equivalents of TM2a and L2ab—are accessible in the modeled outward-open state (Fig. 4), and so it is plausible that L2ab and TM2a are accessible from the internal medium in cytosolic-open conformers of the protein. However, we cannot completely rule out an error in the choice of template or sequence alignment used for the modeling.
Fifth, SCAM in TM3 of human NaPi-IIa indicated that this helix was inaccessible from the external medium, with the exception of A240 at its extracellular end (55) (Fig. 3 B). This pattern fits well with our model, wherein TM3 acts as a buried scaffold with A240 exposed at the extracellular side (Fig. 3 C).
We note that the proposed NaPi-IIa model has the opposite TM topology from the core of VcINDY (Fig. S3), with the first helix of RU1 in NaPi-II originating in the cytoplasm (54). As a consequence, our NaPi-IIa model corresponds to an extracellular-facing state.
Predicting substrate coordination sites
As indicated previously, the Na+ and citrate coordinating residues located in the loops of the hairpins (HPin and HPout) in VcINDY have conserved polar counterparts in hairpins HP1 and HP2 of the NaPi-IIa model (Figs. 2 B and 4 C). In addition, after comparing the structures, we observed that the Cα atom of N199 from TM2a of NaPi-IIa (Fig. 4 D) is located ∼5 Å from the Cα atom of the Na+-binding residue N151 in VcINDY (40) even though they originate from different helices. Although the accuracy of the binding-site region in the model (Fig. 4 D) is highly dependent on the correctness of the sequence alignment, we considered the possibility that residue N199 in NaPi-II plays a similar role to N151 in VcINDY; this possibility would be consistent with an earlier finding that Cys substitution of N199 in NaPi-IIa reduces the apparent Na+ affinity 40-fold (52). We therefore reasoned that other residues in this region might coordinate the substrates or cosubstrates. To identify such residues, we tentatively modeled an ion into this region (see Methods), designated the Na2 site. We observed that three other highly conserved hydroxyl-containing residues lay nearby (Cα atoms within 6 Å of the ion), namely S164 from HP1b, and T195 and S196 from TM2a (Fig. 4 D), which could provide suitable sodium coordinating residues.
Assuming direct coupling between the substrate and Na+, as observed in LeuT (20) and in VcINDY (Fig. S3 B), a Pi ion was tentatively placed at a position equivalent to that of bound citrate (Fig. 4 D). The citrate density was observed at the axis of the twofold pseudosymmetry between the structural repeats, a common position for transporter substrate binding sites (13). During the modeling, we imposed coordination of the Pi by S164 and N199, such that they each coordinate the ion at Na2 and the Pi (Fig. 4 D, see also Fig S5 for coordination in the final model). This reflects the observation that the nature of the side chain at site 199 is a critical determinant of the mode of transport: small polar and nonpolar residues do not alter the coupling mode, albeit while causing a large increase in apparent sodium affinity (see above), whereas large polar and charged residues at this position lead to only the uncoupled leak mode being observed (transition 2 ↔ 7, Fig. 1 A) (52). Although we cannot rule out a more indirect role for these mutations, such an ability to affect the mode of coupling would be consistent with a direct coordination of both substrates (Fig. 4 D, Fig. S5).
In the model, residue S164 (at the start of HP1b) has a symmetry equivalent position, namely S419 at the start of HP2b, and so we speculated that S419 may also coordinate Pi. Finally, beneath the binding site we identified a positively charged residue, R210 from TM2b, whose Cα atom is ∼8 Å from the substrate (Fig. 4 D), suitable for Pi coordination by the arginine side chain. R210 was therefore tested as a putative Pi coordinating residue.
A second Na+ site, pseudosymmetric to that described previously, could be hypothesized from our model—this site would also involve direct coordination by the substrate Pi. Assuming this pseudosymmetry predicts that other residues in HP2ab and L4ab might coordinate an ion in that site. In particular, according to the current model, S462 lies at the start of TM5b, which after mutation to Cys in flounder NaPi-IIb (S448) and in NaPi-IIc (S437), can be modified by a thiol reagent and this modification abolishes transport (17,60,61). Under these conditions, Na+ and Pi can still bind (partial reactions 1 ↔ 2, 2 ↔ 3, and 3 ↔ 4, Fig. 1 A) (17,39,58,60), but either the final Na+ binding step or the translocation step has been compromised, consistent with the notion that S462 contributes to the third Na+ binding site, hereafter designated Na3.
Two other positions predicted to be in L5ab, corresponding to A455 and L457 of NaPi-IIa, have been previously mutated to Cys and subjected to thiol modification. Under these conditions, not only was cotransport function compromised, but the uncoupled leak (transition 2 ↔ 7, Fig. 1 A) significantly increased (53), which provided further confirmation of the importance of this loop in substrate translocation and cosubstrate coupling.
Finally, according to their proximity in the model (the Cα atoms are within 6 Å of the ions), we identified residues Q417 and S418 as potentially coordinating this ion in addition to S419 and S462.
Testing predictions by functional studies of NaPi-IIa mutants
As mentioned previously, the minimal sequence identity between VcINDY and NaPi-IIa limits the certainty with which we can assign specific residues to the binding sites. Therefore, to attempt to refine the structure of these sites in atomic detail, for example using molecular dynamics simulations, would not necessarily be informative. However, the predicted involvement of these residues in substrate coordination requires experimental validation. We therefore mutated S164, T195, S196, R210, Q417, S418, and S419 to Ala or Cys, expressed the mutant constructs in Xenopus oocytes, and performed standard radiotracer and voltage clamp assays to compare their functional behavior with that of WT NaPi-IIa (Table 1 and Fig. 5). For constructs that displayed no measurable functional behavior (namely S196C), we confirmed membrane expression by immunohistochemistry (Fig. S6). Furthermore, we used presteady-state charge relaxation measurements to ascertain if the mutagenesis had affected partial reactions involving charge displacement arising from the empty carrier and the movement of Na+ ions to and from their binding sites (Fig. 1 A) (12).
Table 1.
Functional properties of mutants
| Repeat unit | Construct | 32P uptake | Presteady-state |
Activation indexa |
|
|---|---|---|---|---|---|
| 100Na | Pia | Naa | |||
| WT | + | + | 0.61 | 0.63 | |
| RU1 | S164A | − | + | n.a. | n.a. |
| S164C | − | + | n.a. | n.a. | |
| T195C | − | + | n.a. | n.a. | |
| S196C | − | − | n.a. | n.a. | |
| N199Cb | + | n.d. | 0.10 | 0.03 | |
| R210C | + | + | 0.60 | 0.65 | |
| RU2 | Q417C | + | + | 0.46 | 0.39 |
| S418C | − | + | n.a. | n.a. | |
| S419A | − | + | n.a. | n.a. | |
| S419C | − | + | n.a. | n.a. | |
n.a.: not applicable; n.d.: not determined; −: not detected; +: detected.
Presteady-state relaxations suppressed (−) or left unchanged (+) by 1 mM Pi.
Bold entries indicate constructs used for immunohistological confirmation of membrane expression (Fig S6).
Activation indices were defined as follows: the Pi index was the ratio of Pi-induced current with 0.1 mM Pi to that with 1 mM Pi in 100 mM Na+ and the Na+ index was the ratio of the response to 1 mM Pi with 50 mM Na+ to that with 100 mM Na+. Both indices were determined at −50 mV. These ratios give an approximate indication of shifts in the apparent substrate affinity for screening purposes (e.g., (56)).
From (52) for rat NaPi-IIa isoform, equivalent to N199 in human NaPi-IIa.
Figure 5.
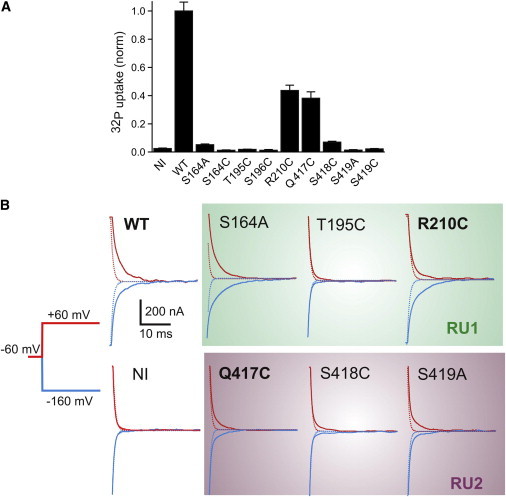
Effect of site-specific mutations on transport and substrate interactions in human NaPi-IIa. (A) Tracer uptake (32P) data normalized to WT for the mutants and noninjected (NI) oocytes as control. Each data set is the mean ± SE of data from two batches of oocytes (4–5 oocytes per batch). (B) Presteady-state relaxations recorded from representative oocytes expressing selected constructs in RU1 and RU2, together with WT and NI as controls superfused in 100 mM Na+. Bold label indicates active Pi transport was also measured. Transient currents shown in response to voltage steps as indicated, from a holding potential = –60 mV to two test potentials: –160 (blue) and +60 mV (red). Traces were baseline corrected and are shown with the same ±500 nA range. Each relaxation was fit with a double exponential function. Dotted traces is the fitted fast component (relaxation time constant <1ms) corresponding to endogenous membrane charging and are comparable to the NI data in all cases. For all mutants shown there was charge movement in excess of the endogenous component. To see this figure in color, go online.
Two constructs (R210C and Q417C) displayed significant 32P uptake (Fig. 5 A), Pi-induced currents (data not shown) and WT-like, resolvable charge relaxations in response to voltage steps in the absence of Pi (Fig. 5 B). However, the uptake levels of these two mutants were significantly smaller than for the WT (Fig. 5 A). Because the uptake assay was performed at only one Pi concentration (1 mM), the lower transport rates could be ascribed to either altered kinetics (apparent substrate affinity, reduced maximum transport rate), or compromised membrane expression, or a combination of both. Given the low transport activity of R210C and Q417C and associated data uncertainties, we did not perform standard dose dependence assays. Instead, to assess whether the mutations had significantly altered the apparent substrate affinities for Na+ and Pi, we determined activation indices defined as the ratio of Pi-induced currents under defined conditions in which either Na+ or Pi concentration was altered (see legend, Table 1) (56). For R210C, the Na+ and Pi activation indices were both close to those of the WT, whereas Q417C showed significantly reduced indices (Table 1). These findings indicated that transport and binding were unaffected by the R210C mutation, but that the Gln-Cys substitution in the predicted Na3 site (Fig. 4 D) reduced the apparent substrate affinities. Nevertheless, for R210C, we noted that the presteady-state relaxations showed a clear asymmetry compared with the WT and other mutants (more charge movement for the hyperpolarizing step compared to the depolarizing step) (Fig. 5 B). This indicated that removal of this charge had affected the voltage-dependent kinetics.
The replacement of S196 in RU1 with Cys did not prevent surface expression (Fig. S6), but was considered lethal, as the oocyte membranes became leaky after 2 days, preventing further functional investigation. Nevertheless, this behavior underscored the potentially critical role played by this residue predicted to be at the Na2 site (Fig. 4 D).
The remaining six constructs (S164A, S164C, T195C, S418C, S419A, and S419C) gave resolvable presteady-state relaxations (Fig. 5 B), but insignificant 32P uptake or Pi-induced current under voltage clamp (Table 1). The presence of presteady-state relaxations that are superimposed on the background linear capacitive charge current of the oocyte membrane (Fig. 5 B) provided strong experimental evidence that, in the absence of Pi, one or more Na+ ions could still interact with these six mutants, in agreement with our kinetic scheme for the WT (Fig. 1 A). However, subsequent partial reactions required for cotransport, such as association of Pi or the last Na+ ion, were compromised by these conservative, and yet apparently deleterious, substitutions.
As described previously, the model predicts that S164 of the first QSSS motif coordinates Na2 and/or Pi and that T195 and S196 of TM2b contribute to the Na2 binding site. The observation of presteady currents, combined with the lack of uptake for both S164A and S164C supports the proposal that S164 is critically required for later steps of transport, consistent with a role in coordinating Na2 and/or Pi.. Similar observations for T195C are consistent with the proposal that it contributes to the Na2 binding site, whereas the lethality of S196C indicates an important, albeit less clearly defined, role for S196.
The model also predicts that Q417, S418, and S419 of the second QSSS motif coordinate Na3, with the latter also possibly coordinating Pi. The ∼50% reduction in Na+ and Pi activation indices of Q417C (Table 1) indicates a reduction in the apparent substrate affinities for both substrates after modification, which is consistent with the proposed role of Q417. Cys substitutions at neighboring sites (S418, S419) resulted in similar properties to the S164 and T195 mutants described above, i.e., the mutants exhibited presteady-state currents while uptake was abolished, consistent with a critical role in later steps in the transport mechanism, most likely in Na+ and Pi coordination. Finally, based on the WT-like behavior of R210C with respect to substrate activation, it is likely that R210 does not contribute to substrate coordination. Nevertheless, the asymmetry of the presteady-state relaxations compared with the WT (Fig. 5 B) suggests that R210 may play a critical role in defining the voltage dependence of the cotransport cycle and therefore may be proximal to the binding sites.
Discussion
The structural repeat has emerged as a common architectural leitmotif in all reported structures of secondary transporters (e.g. (13,22), and in a majority of those, the repeats have inverted TM topologies. The pseudosymmetries introduced by these repeats have functional implications that can include the location of binding sites and the equivalency of repeated elements in defining pathways or gating elements. In NaPi-IIa the repeated sequence of the QSSS motif is particularly prominent and biochemical studies suggested that these were accessible from opposite sides of the membrane, consistent with the repeats exhibiting inverted topologies (52,53). Inspired by the number of new structures available, we used both statistical (using, e.g., HHpred) and qualitative (using hydropathy profile alignments) approaches to search for a putative template. Notably, there is no similarity with the structure of a recently reported fungal proton-dependent Pi transporter (62), which is a member of the major facilitator superfamily of transporters, with its characteristic fold of two lobes, each containing 6 TM helices.
Hydropathy analysis was previously used to predict fold similarity in the ST[3] class to which VcINDY belongs (43). Biochemical topology analysis (63) and two-dimensional crystallography (47) have since confirmed that, e.g., the 2HCT family that includes CitS and the AIT family that includes VcINDY adopt similar architectures, providing support for the use of hydropathy profiles to classify members of this 5TM+hairpin repeat fold.
In NaPi-II, the repeat has fewer TM helices, but that repeat contains a reentrant hairpin before the last helix, as in the ST[3] fold, giving a 3TM+hairpin repeat. The truncated helices contribute to a stator domain (47) and therefore may have minimal effect on transport. Indeed, a variety of lengths are observed in the ST[3] class, including a truncation to a 4-TM helix repeat (63).
The low-resolution electron microscopy-based structure of CitS hints that rearrangements of helices are possible within the context of the same fold (47). We therefore questioned whether a model of NaPi-II could be improved by using CitS as a guiding template. However, comparison of the hydropathy plots of the repeats of CitS with those of NaPi-IIa showed significant differences in the region of the re-entrant hairpins and broken helices (Fig. S7). These were reminiscent of differences between profiles of VcINDY and CitS (Fig. S7). In contrast, the hydropathy profiles of NaPi-IIa and VcINDY share more characteristics (Fig. 2 A). We therefore conclude that, although the three protein families appear to share a common core fold, VcINDY provides the most suitable available template. Moreover, these comparisons (Fig. S7) provide support for the proposal that a homology model based on VcINDY, despite its modest resolution, provides an important step forward in predicting the architecture and approximate binding regions of NaPi-IIa.
We note that hydropathy profile comparisons are statistically poorly defined, therefore the final sequence alignments were made with quantitative techniques in which detailed sequence and secondary structure profiles represent each repeat. The two strategies converged in their matching of TM segments, and placed conserved residues from NaPi-II at the binding region of VcINDY.
The QSSS repeats are hallmarks of all NaPi-II isoforms from bacteria to human (64) and, according to this model, help coordinate Pi, Na2, and Na3. When we mutated residues that contribute to these predicted coordination sites in RU1 (S164, T195, S196) and RU2 (Q417, S418, and S419), even using very conservative substitutions, transport was either markedly affected or abolished completely. These data, combined with previous evidence implicating N199 (52) and S462 (54), provide strong support for their involvement in substrate coordination. The Cys substitution at S462 in RU2 is well tolerated in all NaPi-II isoforms, however when subjected to thiol modification, cotransport function is inhibited, although cation and Pi binding remain (17,54,61). One scenario to explain this would be that binding of the last Na+ ion is restricted by having a bulky MTS moiety at this site. In contrast, Cys substitution at N199 in RU1 leads to a >10-fold reduction in the estimated apparent substrate affinities (52), which is consistent with its proposed proximity to both Pi and Na2. The effect of Cys substitution at Q417 in RU2 was much less dramatic (equivalent to ∼threefold and twofold increases in K0.5Pi and K0.5Na, respectively; see Fig. 5 in (56)). This smaller deviation from the WT behavior may reflect Q417’s proposed proximity to Na3 only (Fig. 4). The possibility of the substrate itself being directly coordinated in part by the cosubstrate, as in the proposed coordination of Na2, Na3, and Pi, and as observed previously for LeuT (20), underscores the notion of strictly coupled transport and offers a molecular basis for the experimentally observed dependence of the apparent affinity constants on the invariant substrate (16,65).
Currently, there is insufficient evidence to locate the binding site of the first Na+ ion, designated Na1, (partial reaction 1 ↔ 2, Fig. 1 A). In the electroneutral isoform this ion is hypothesized to interact allosterically with NaPi-IIc and contribute to the overall cooperativity of Na+ activation, but not to be translocated (17). In contrast, for the electrogenic isoforms (NaPi-IIa and NaPi-IIb) the binding of a Na+ ion to Na1 to the outward facing conformation (state 1) is critical for conferring electrogenicity to the transport cycle; moreover, in the absence of Pi, this ion is assumed to contribute to the uncoupled leak (66). Our model proposes that Pi is tightly coordinated with 2 Na+ ions (at sites Na2 and Na3), and together these would constitute the translocated, and effectively electroneutral, complex for both electroneutral and electrogenic isoforms.
The detection of presteady-state charge movements in the absence of external Pi provides indirect evidence that conformational changes occur in response to changes in membrane potential. For six of the seven mutants whose cotransport function was compromised (Table 1, Fig. 5 A), we were able to resolve such presteady-state relaxations in the presence of 100 mM Na+. The detection of these relaxation currents supported the notion that intrinsic charge movements (analogous to gating currents in ion channels (e.g. (67).) and displacement charge associated with the interaction of a Na+ ion at the postulated Na1 site, most likely still occurred in the mutated constructs. The lack of transport function is therefore consistent with these mutations drastically affecting Na2, Na3, or Pi coordination, events that are common to both electroneutral and electrogenic NaPi-II isoforms. Unfortunately, the small magnitude of the presteady-state relaxations for these dysfunctional mutants precluded more detailed analysis to ascertain whether cation interactions had been altered specifically at Na2 or Na3. Nevertheless, these observations will serve as a basis for more detailed structure-function studies of SLC34 proteins.
In conclusion, we propose a structural model for the architecture of the human, Na+-coupled phosphate transport protein NaPi-II. Because the predictions of the substrate coordination sites (Fig. 4) are based on a homology model at low sequence similarity, they should be considered hypotheses that will help to guide experiments until more detailed structural data for the SLC34 protein family can be obtained. To our knowledge, the strategy used to define equivalent regions in the template by identifying and separating out the structural repeats is novel and may be applied to model other transporters of as yet unknown architecture. As the proposed model is relevant for all members of the SLC34 family it should also provide an important step in structure-function studies of all SLC34 proteins.
Acknowledgments
Financial support was given by The Dunhill Medical Trust (A.W.), Swiss National Science Foundation (I.C.F.), and the Intramural Research Program of the NIH, NINDS (L.R.F.). We thank Eva Hänsenberger for oocyte preparation.
Author contributions: C.F.F., L.R.F.: bioinformatics, modeling, data analysis; M.P.: bioinformatics, molecular biology, transport assays, data analysis; I.C.F.: data analysis; T.K.: oocyte assays, immunohistochemistry; L.R.F., A.W., and I.C.F.: designed experiments and wrote the paper.
Footnotes
Cristina Fenollar-Ferrer and Lucy R. Forrest’s present address is Computational Structural Biology Section, Porter Neuroscience Research Center, National Institutes of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892.
Cristina Fenollar-Ferrer and Monica Patti share equal first authorship.
Contributor Information
Andreas Werner, Email: andreas.werner@newcastle.ac.uk.
Ian C. Forster, Email: iforster@access.uzh.ch.
Lucy R. Forrest, Email: lucy.forrest@nih.gov.
Supporting Material
References
- 1.Shroff R., Long D.A., Shanahan C. Mechanistic insights into vascular calcification in CKD. J. Am. Soc. Nephrol. 2013;24:179–189. doi: 10.1681/ASN.2011121191. [DOI] [PubMed] [Google Scholar]
- 2.Biber J., Hernando N., Forster I. Phosphate transporters and their function. Annu. Rev. Physiol. 2013;75:535–550. doi: 10.1146/annurev-physiol-030212-183748. [DOI] [PubMed] [Google Scholar]
- 3.Forster I.C., Hernando N., Murer H. Phosphate transporters of the SLC20 and SLC34 families. Mol. Aspects Med. 2013;34:386–395. doi: 10.1016/j.mam.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 4.Sabbagh Y., Giral H., Schiavi S.C. Intestinal phosphate transport. Adv. Chronic Kidney Dis. 2011;18:85–90. doi: 10.1053/j.ackd.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biber J., Hernando N., Murer H. Regulation of phosphate transport in proximal tubules. Pflugers Arch. 2009;458:39–52. doi: 10.1007/s00424-008-0580-8. [DOI] [PubMed] [Google Scholar]
- 6.Forster I., Hernando N., Werner A. Phosphate transporters in renal, gastrointestinal, and other tissues. Adv. Chronic Kidney Dis. 2011;18:63–76. doi: 10.1053/j.ackd.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Beck L., Karaplis A.C., Tenenhouse H.S. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc. Natl. Acad. Sci. USA. 1998;95:5372–5377. doi: 10.1073/pnas.95.9.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kenny J., Lees M.M., Bockenhauer D. Sotos syndrome, infantile hypercalcemia, and nephrocalcinosis: a contiguous gene syndrome. Pediatr. Nephrol. 2011;26:1331–1334. doi: 10.1007/s00467-011-1884-z. [DOI] [PubMed] [Google Scholar]
- 9.Magen D., Berger L., Skorecki K. A loss-of-function mutation in NaPi-IIa and renal Fanconi’s syndrome. N. Engl. J. Med. 2010;362:1102–1109. doi: 10.1056/NEJMoa0905647. [DOI] [PubMed] [Google Scholar]
- 10.Bergwitz C., Roslin N.M., Juppner H. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am. J. Hum. Genet. 2006;78:179–192. doi: 10.1086/499409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corut A., Senyigit A., Tolun A. Mutations in SLC34A2 cause pulmonary alveolar microlithiasis and are possibly associated with testicular microlithiasis. Am. J. Hum. Genet. 2006;79:650–656. doi: 10.1086/508263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forster I.C., Hernando N., Murer H. Phosphate transport kinetics and structure-function relationships of SLC34 and SLC20 proteins. Curr. Top. Membr. 2012;70:313–356. doi: 10.1016/B978-0-12-394316-3.00010-7. [DOI] [PubMed] [Google Scholar]
- 13.Forrest L.R., Krämer R., Ziegler C. The structural basis of secondary active transport mechanisms. Biochim. Biophys. Acta. 2011;1807:167–188. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 14.Forrest L.R., Zhang Y.W., Rudnick G. Mechanism for alternating access in neurotransmitter transporters. Proc. Natl. Acad. Sci. USA. 2008;105:10338–10343. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 16.Forster I., Hernando N., Murer H. The voltage dependence of a cloned mammalian renal type II Na+/Pi cotransporter (NaPi-2) J. Gen. Physiol. 1998;112:1–18. doi: 10.1085/jgp.112.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghezzi C., Murer H., Forster I.C. Substrate interactions of the electroneutral Na+-coupled inorganic phosphate cotransporter (NaPi-IIc) J. Physiol. 2009;587:4293–4307. doi: 10.1113/jphysiol.2009.175596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forster I.C., Köhler K., Murer H. Forging the link between structure and function of electrogenic cotransporters: the renal type IIa Na+/Pi cotransporter as a case study. Prog. Biophys. Mol. Biol. 2002;80:69–108. doi: 10.1016/s0079-6107(02)00015-9. [DOI] [PubMed] [Google Scholar]
- 19.Abramson J., Wright E.M. Structure and function of Na(+)-symporters with inverted repeats. Curr. Opin. Struct. Biol. 2009;19:425–432. doi: 10.1016/j.sbi.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamashita A., Singh S.K., Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 21.Forrest L.R. Structural biology. (Pseudo-)symmetrical transport. Science. 2013;339:399–401. doi: 10.1126/science.1228465. [DOI] [PubMed] [Google Scholar]
- 22.Forrest L.R., Rudnick G. The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters. Physiology (Bethesda) 2009;24:377–386. doi: 10.1152/physiol.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khafizov K., Perez C., Forrest L.R. Investigation of the sodium-binding sites in the sodium-coupled betaine transporter BetP. Proc. Natl. Acad. Sci. USA. 2012;109:E3035–E3044. doi: 10.1073/pnas.1209039109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finn R.D., Clements J., Eddy S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server issue):W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y., Schmidt B., Maskell D.L. MSAProbs: multiple sequence alignment based on pair hidden Markov models and partition function posterior probabilities. Bioinformatics. 2010;26:1958–1964. doi: 10.1093/bioinformatics/btq338. [DOI] [PubMed] [Google Scholar]
- 27.Stamm M., Staritzbichler R., Forrest L.R. Alignment of helical membrane protein sequences using AlignMe. PLoS ONE. 2013;8:e57731. doi: 10.1371/journal.pone.0057731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Remmert M., Biegert A., Soding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 2011;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 29.Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 30.Altschul S.F., Madden T.L., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelley L.A., Sternberg M.J. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 32.Margelevicius M., Laganeckas M., Venclovas C. COMA server for protein distant homology search. Bioinformatics. 2010;26:1905–1906. doi: 10.1093/bioinformatics/btq306. [DOI] [PubMed] [Google Scholar]
- 33.Šali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 34.Ashkenazy H., Erez E., Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38(Web Server issue):W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ray A., Lindahl E., Wallner B. Model quality assessment for membrane proteins. Bioinformatics. 2010;26:3067–3074. doi: 10.1093/bioinformatics/btq581. [DOI] [PubMed] [Google Scholar]
- 36.Laskowski R.A., Macarthur M.W., Thornton J.M. PROCHECK - a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 37.Bossi E., Fabbrini M.S., Ceriotti A. Exogenous protein expression in Xenopus oocytes: basic procedures. Methods Mol. Biol. 2007;375:107–131. doi: 10.1007/978-1-59745-388-2_6. [DOI] [PubMed] [Google Scholar]
- 38.Andrini O., Meinild A.K., Forster I.C. Lithium interactions with Na+-coupled inorganic phosphate cotransporters: insights into the mechanism of sequential cation binding. Am. J. Physiol. Cell Physiol. 2012;302:C539–C554. doi: 10.1152/ajpcell.00364.2011. [DOI] [PubMed] [Google Scholar]
- 39.Köhler K., Forster I.C., Murer H. Transport function of the renal type IIa Na+/P(i) cotransporter is codetermined by residues in two opposing linker regions. J. Gen. Physiol. 2002;120:693–705. doi: 10.1085/jgp.20028645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mancusso R., Gregorio G.G., Wang D.N. Structure and mechanism of a bacterial sodium-dependent dicarboxylate transporter. Nature. 2012;491:622–626. doi: 10.1038/nature11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saier M.H., Jr., Paulsen I.T. Whole genome analyses of transporters in spirochetes: Borrelia burgdorferi and Treponema pallidum. J. Mol. Microbiol. Biotechnol. 2000;2:393–399. [PubMed] [Google Scholar]
- 42.Bergeron M.J., Clémençon B., Markovich D. SLC13 family of Na+-coupled di- and tri-carboxylate/sulfate transporters. Mol. Aspects Med. 2013;34:299–312. doi: 10.1016/j.mam.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 43.Lolkema J.S., Slotboom D.J. Classification of 29 families of secondary transport proteins into a single structural class using hydropathy profile analysis. J. Mol. Biol. 2003;327:901–909. doi: 10.1016/s0022-2836(03)00214-6. [DOI] [PubMed] [Google Scholar]
- 44.Crisman T.J., Qu S., Forrest L.R. Inward-facing conformation of glutamate transporters as revealed by their inverted-topology structural repeats. Proc. Natl. Acad. Sci. USA. 2009;106:20752–20757. doi: 10.1073/pnas.0908570106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ressl S., Terwisscha van Scheltinga A.C., Ziegler C. Molecular basis of transport and regulation in the Na(+)/betaine symporter BetP. Nature. 2009;458:47–52. doi: 10.1038/nature07819. [DOI] [PubMed] [Google Scholar]
- 46.Khafizov K., Staritzbichler R., Forrest L.R. A study of the evolution of inverted-topology repeats from LeuT-fold transporters using AlignMe. Biochemistry. 2010;49:10702–10713. doi: 10.1021/bi101256x. [DOI] [PubMed] [Google Scholar]
- 47.Kebbel F., Kurz M., Stahlberg H. Structure and substrate-induced conformational changes of the secondary citrate/sodium symporter CitS revealed by electron crystallography. Structure. 2013;21:1243–1250. doi: 10.1016/j.str.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 48.Faraldo-Gómez J.D., Forrest L.R. Modeling and simulation of ion-coupled and ATP-driven membrane proteins. Curr. Opin. Struct. Biol. 2011;21:173–179. doi: 10.1016/j.sbi.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 49.Radanovic T., Gisler S.M., Murer H. Topology of the type IIa Na+/P(i) cotransporter. J. Membr. Biol. 2006;212:41–49. doi: 10.1007/s00232-006-0033-2. [DOI] [PubMed] [Google Scholar]
- 50.Karlin A., Akabas M.H. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 51.Ehnes C., Forster I.C., Murer H. Structure-function relations of the first and fourth extracellular linkers of the type IIa Na+/Pi cotransporter: II. Substrate interaction and voltage dependency of two functionally important sites. J. Gen. Physiol. 2004;124:489–503. doi: 10.1085/jgp.200409061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Köhler K., Forster I.C., Murer H. Identification of functionally important sites in the first intracellular loop of the NaPi-IIa cotransporter. Am. J. Physiol. Renal Physiol. 2002;282:F687–F696. doi: 10.1152/ajprenal.00282.2001. [DOI] [PubMed] [Google Scholar]
- 53.Lambert G., Forster I.C., Murer H. Cysteine mutagenesis reveals novel structure-function features within the predicted third extracellular loop of the type IIa Na(+)/P(i) cotransporter. J. Gen. Physiol. 2001;117:533–546. doi: 10.1085/jgp.117.6.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lambert G., Traebert M., Murer H. Studies on the topology of the renal type II NaPi-cotransporter. Pflugers Arch. 1999;437:972–978. doi: 10.1007/s004240050869. [DOI] [PubMed] [Google Scholar]
- 55.Virkki L.V., Forster I.C., Murer H. Functionally important residues in the predicted 3(rd) transmembrane domain of the type IIa sodium-phosphate co-transporter (NaPi-IIa) J. Membr. Biol. 2005;206:227–238. doi: 10.1007/s00232-005-0796-x. [DOI] [PubMed] [Google Scholar]
- 56.Ehnes C., Forster I.C., Murer H. Structure-function relations of the first and fourth predicted extracellular linkers of the type IIa Na+/Pi cotransporter: I. Cysteine scanning mutagenesis. J. Gen. Physiol. 2004;124:475–488. doi: 10.1085/jgp.200409060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Virkki L.V., Murer H., Forster I.C. Mapping conformational changes of a type IIb Na+/Pi cotransporter by voltage clamp fluorometry. J. Biol. Chem. 2006;281:28837–28849. doi: 10.1074/jbc.M603861200. [DOI] [PubMed] [Google Scholar]
- 58.Ghezzi C., Meinild A.K., Forster I.C. Voltage- and substrate-dependent interactions between sites in putative re-entrant domains of a Na(+)-coupled phosphate cotransporter. Pflugers Arch. 2011;461:645–663. doi: 10.1007/s00424-011-0948-z. [DOI] [PubMed] [Google Scholar]
- 59.Reference deleted in proof.
- 60.Lambert G., Forster I.C., Murer H. Properties of the mutant Ser-460-Cys implicate this site in a functionally important region of the type IIa Na(+)/P(i) cotransporter protein. J. Gen. Physiol. 1999;114:637–652. doi: 10.1085/jgp.114.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Virkki L.V., Murer H., Forster I.C. Voltage clamp fluorometric measurements on a type II Na+-coupled Pi cotransporter: shedding light on substrate binding order. J. Gen. Physiol. 2006;127:539–555. doi: 10.1085/jgp.200609496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pedersen B.P., Kumar H., Stroud R.M. Crystal structure of a eukaryotic phosphate transporter. Nature. 2013;496:533–536. doi: 10.1038/nature12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.ter Horst R., Lolkema J.S. Membrane topology screen of secondary transport proteins in structural class ST[3] of the MemGen classification. Confirmation and structural diversity. Biochim. Biophys. Acta. 2012;1818:72–81. doi: 10.1016/j.bbamem.2011.09.021. [DOI] [PubMed] [Google Scholar]
- 64.Werner A., Kinne R.K. Evolution of the Na-Pi cotransport systems. Am. J. Physiol. 2001;280:R301–R312. doi: 10.1152/ajpregu.2001.280.2.R301. [DOI] [PubMed] [Google Scholar]
- 65.Virkki L.V., Forster I.C., Murer H. Substrate interactions in the human type IIa sodium-phosphate cotransporter (NaPi-IIa) Am. J. Physiol. Renal Physiol. 2005;288:F969–F981. doi: 10.1152/ajprenal.00293.2004. [DOI] [PubMed] [Google Scholar]
- 66.Andrini O., Ghezzi C., Forster I.C. The leak mode of type II Na(+)-P(i) cotransporters. Channels (Austin) 2008;2:346–357. doi: 10.4161/chan.2.5.6900. [DOI] [PubMed] [Google Scholar]
- 67.Bezanilla F. How membrane proteins sense voltage. Nat. Rev. Mol. Cell Biol. 2008;9:323–332. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- 68.Patti M., Ghezzi C., Forster I.C. Conferring electrogenicity to the electroneutral phosphate cotransporter NaPi-IIc (SLC34A3) reveals an internal cation release step. Pflugers Arch. 2013;465:1261–1279. doi: 10.1007/s00424-013-1261-9. [DOI] [PubMed] [Google Scholar]
- 69.Hessa T., Kim H., von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 70.Custer M., Lotscher M., Kaissling B. Expression of Na-P(i) cotransport in rat kidney: localization by RT-PCR and immunohistochemistry. Am. J. Physiol. 1994;266:F767–F774. doi: 10.1152/ajprenal.1994.266.5.F767. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.