

How do we learn? More specifically, what biochemical and structural changes occur in the brain during the processes of learning and memory? The truth is that, despite decades of research, we still do not have a satisfactory answer to this question. One of the leading theories, the Hebbian learning paradigm (1), proposes that memories are encoded by altering the strength of individual synaptic connections in response to certain stimuli. This process, termed “synaptic plasticity”, could allow a neuron to respond preferentially to some of its presynaptic partners while reducing its response to others, and in this way the neuron learns to respond to only particular stimuli.
One of the best-studied forms of synaptic plasticity is long-term potentiation (LTP) at excitatory glutamateric synapses (2). LTP can be induced at these synapses by strong, high-frequency stimulation, and results in an increased postsynaptic response to subsequent basal stimulation (2). The potentiated synapses have increased numbers of glutamate receptors, typically AMPA receptors, and have enlarged dendritic spines with larger synapses (3). This is a long-lasting, potentially permanent change in the synapse which then persists in the absence of any additional stimulation.
A useful analogy here is to a light switch. A light switch in the off-position will remain off indefinitely, but a strong enough stimulus (your finger) can move the switch to the on-position. Once there, it will remain in the on-position even after your finger is removed. In the same way, the strength of the synapse does not change unless it is given an appropriate stimulus, but once changed the stimulus is no longer required to maintain the synapse in the potentiated state. Permanent changes like this are the basis of information storage and can act as a memory: the light switch stores the fact that it was once pushed up, while the synapse stores the memory of a previous encounter with a particular excitation.
Whereas it is easy to design a light switch, it is not as obvious how to design a biochemical switch that would be stable under stochastic fluctuations and protein turnover. In a now classic article, Lisman (4) showed how such a switch could be constructed by coupling an activatable, autophosphorylating kinase, K, with a phosphatase, P. The reaction diagram for this system is shown in Fig. 1 A. Lisman showed that the activation curve of such a kinase could have a region of hysteresis, as shown in Fig. 1 B. To see how this can act like a switch, imagine that the basal stimulation level is S1 and the system is initially in a low-activity state at point A. Small fluctuations in stimulation level are unable to drive the system to high activity, which protects the system against accidental stochastic activation. However, a large stimulation, S2, can activate the system, moving it to point B. When the stimulation returns to its basal level, the system remains in the high activity state at point C. Note that the basal stimulation level plays an important role here; if, for example, the basal stimulation level was lower, S0, so that the resting activity level was at point D, then the system would not act like a switch despite the region of hysteresis.
Figure 1.
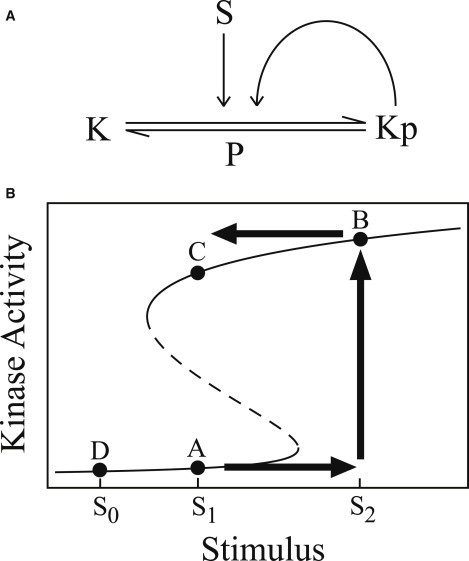
An example of a biochemical switch. (A) Schematic reaction diagram of an autophosphorylating kinase. The kinase, K, can be phosphorylated and activated by a stimulus, S. Activated kinase, Kp, can phosphorylate inactive kinase. The kinase is dephosphorylated by a phosphatase, P. (B) A plot of kinase activity as a function of stimulus intensity. At low and high stimulus levels, there is a single steady state, but at intermediate stimulus levels there are two stable steady states (and an unstable steady state, shown as a dashed line). The labeled points are described in the text.
A strong candidate for the autophosphorylating kinase of Lisman (4) is calcium/calmodulin-dependent protein kinase II (CaMKII), a highly expressed, synaptically localized kinase that is known to have important roles in learning and memory (5). CaMKII is activated by synaptic calcium transients, and once activated it can autophosphorylate on T286 (5). Autophosphorylation at this site maintains the kinase in a partially active state even in the absence of calcium. This is site-dephosphorylated by protein phosphatase 1 (PP1), which is also synaptically localized, and thus the CaMKII-PP1 system might function as the bistable synaptic switch underlying LTP (5). CaMKII is more complicated than Lisman’s original proposed kinase because it exists as a 12-subunit holoenzyme and T286 phosphorylation is an intersubunit reaction (5), but early models that included these details suggested that CaMKII-PP1 could form a bistable switch over a wide range of conditions (6). However, other, later experiments suggest that CaMKII is only transiently activated following LTP induction (7), and an in vitro experiment showed no hysteresis in the CaMKII-PP1 system (8). Thus, despite its theoretical appeal, it appeared CaMKII could not exhibit bistability.
However, in this issue of the Biophysical Journal, Urakubo et al. (9) use an in vitro system to demonstrate that the CaMKII-PP1 system becomes bistable in the presence of a peptide composed of the CaMKII-binding region of the NMDA receptor (NMDAR) GluN2B subunit. To my knowledge, this is the first definitive demonstration of bistability in a synaptic protein system. GluN2B is a major CaMKII binding partner in the postsynaptic density, and recently the CaMKII-NMDAR complex was shown to be essential in the maintenance of LTP (10). Once established, LTP could be eliminated by a peptide that blocks the CaMKII-NMDAR interaction. The importance of this interaction could be structural, but the results of Urakubo et al. (9) suggest that this interaction could be essential to maintain kinase bistability.
Urakubo et al. (9) provide two additional results that serve to suggest the in vitro observation of bistability could have physiological relevance:
-
1.
They show that bistability occurs at the level of individual holoenzymes. This is an important observation, because CaMKII holoenzymes only interact in vivo under pathological conditions.
-
2.
They show that mutations which change the CaMKII-GluN2B binding affinity change the size of the region of bistability but do not eliminate hysteresis. The binding affinity is known to be affected by phosphorylation and other CaMKII binding partners at the synapse, but the robustness displayed here makes it more likely this mechanism could operate under the variable conditions in vivo.
However, Urakubo et al. (9) also found that bistability required calcium concentrations above 200 nM, whereas in vivo calcium levels are in the 80–150 nM range. This observation is in agreement with the most recent model of the CaMKII-PP1 system, which suggested that bistable regions will always be above resting calcium levels for any reasonable variations of kinase and phosphatase activity (11). In this case, the in vivo activity curve would resemble Fig. 1 B, with point D as the resting point; hysteresis is present in the activity curve but the system is unable to act like a switch. Of course, it is entirely possible that in vivo interactions with other proteins alter the system in such as way as to extend the region of bistability down to basal calcium levels.
Clearly, additional research is needed. It will be of particular importance to reproduce these results in cellular systems. Nevertheless, the results of Urakubo et al. (9) provide important and surprising new information about the behavior of the CaMKII system, and represent a major milestone in the near-30-year quest to find bistability in a memory-related protein.
Acknowledgments
This work was supported by National Institutes of Health postdoctoral fellowship No. 1F32 NS077751-01 and grant No. P41 RR013186.
References
- 1.Brown T.H., Kairiss E.W., Keenan C.L. Hebbian synapses: biophysical mechanisms and algorithms. Annu. Rev. Neurosci. 1990;13:475–511. doi: 10.1146/annurev.ne.13.030190.002355. [DOI] [PubMed] [Google Scholar]
- 2.Malenka R.C., Nicoll R.A. Long-term potentiation—a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 3.Murakoshi H., Yasuda R. Postsynaptic signaling during plasticity of dendritic spines. Trends Neurosci. 2012;35:135–143. doi: 10.1016/j.tins.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lisman J.E. A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc. Natl. Acad. Sci. USA. 1985;82:3055–3057. doi: 10.1073/pnas.82.9.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lisman J., Schulman H., Cline H. The molecular basis of CaMKII function in synaptic and behavioral memory. Nat. Rev. Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 6.Zhabotinsky A.M. Bistability in the Ca2+/calmodulin-dependent protein kinase-phosphatase system. Biophys. J. 2000;79:2211–2221. doi: 10.1016/S0006-3495(00)76469-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lengyel I., Voss K., Bliss T.V.P. Autonomous activity of CaMKII is only transiently increased following the induction of long-term potentiation in the rat hippocampus. Eur. J. Neurosci. 2004;20:3063–3072. doi: 10.1111/j.1460-9568.2004.03748.x. [DOI] [PubMed] [Google Scholar]
- 8.Bradshaw J.M., Kubota Y., Schulman H. An ultrasensitive Ca2+/calmodulin-dependent protein kinase II-protein phosphatase 1 switch facilitates specificity in postsynaptic calcium signaling. Proc. Natl. Acad. Sci. USA. 2003;100:10512–10517. doi: 10.1073/pnas.1932759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urakubo H., Sato M., Kuroda S. In vitro reconstitution of a memory switch of CaMKII by NMDA receptor-derived peptide. Biophys. J. 2014;106:1414–1420. doi: 10.1016/j.bpj.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanhueza M., Lisman J. The CaMKII/NMDAR complex as a molecular memory. Mol. Brain. 2013;6:10. doi: 10.1186/1756-6606-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michalski P.J. The delicate bistability of CaMKII. Biophys. J. 2013;105:794–806. doi: 10.1016/j.bpj.2013.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]