

Abstract
Study Objectives:
The hypocretins (HCRTs) are two hypothalamic peptides predominantly localized to neurons in the perifornical, dorsomedial, and lateral hypothalamic area (PF-LHA). Evidence suggests that HCRT signaling is critical for the promotion and stabilization of active-arousal and its loss or malfunction leads to symptoms of narcolepsy. In the PF-LHA, HCRT neurons are intermingled with glutamate-expressing neurons and also co-express glutamate. Evidence suggests that HCRT-glutamate interactions within the PF-LHA may play a critical role in maintaining behavioral arousal. However, the relative contributions of the glutamate and HCRT in sleep-wake regulation are not known.
Design:
We determined whether a lack of HCRT signaling in the prepro-orexin-knockout (HCRT-KO) mouse attenuates/compromises the wake-promoting ability of glutamatergic activation of the PF-LHA region. We used reverse microdialysis to deliver N-methyl-D-aspartate (NMDA) into the HCRT zone of the PF-LHA in HCRT-KO and wild-type (WT) mice to evaluate the contributions of glutamatergic vs. HCRT signaling in sleep-wake regulation.
Measurements and Results:
As compared to respective controls, local perfusion of NMDA into the PF-LHA, dose-dependently increased active-waking with concomitant reductions in nonREM and REM sleep in spontaneously sleeping WT as well as HCRT-KO mice. However, compared to WT, the NMDA-induced behavioral changes in HCRT-KO mice were significantly attenuated, as evidenced by the higher dose of NMDA needed and lower magnitude of changes induced in sleep-wake parameters. Although not observed in WT mice, the number of cataplectic events increased significantly during NMDA-induced behavioral arousal in HCRT-KO mice.
Conclusions:
The findings of this study are consistent with a hypothesis that synergistic interactions between hypocretin and glutamatergic mechanisms within the perifornical, dorsomedial, and lateral hypothalamic area are critical for maintaining behavioral arousal, especially arousal involving elevated muscle tone.
Citation:
Kostin A, Siegel JM, Alam MN. Lack of hypocretin attenuates behavioral changes produced by glutamatergic activation of the perifornical-lateral hypothalamic area. SLEEP 2014;37(5):1011-1020.
Keywords: orexin, hypocretin, perifornical area, prepro-orexin knockout, sleep, dorsomedial hypothalamus, lateral hypothalamus, N-methyl-D-aspartate
INTRODUCTION
The hypocretins (HCRT-1 and 2), also called orexins, are unique hypothalamic peptides that have been implicated in many physiological functions including sleep-wake regulation.1–4 In the brain, HCRT-expressing neurons are predominantly localized in the perifornical, dorsomedial, and lateral hypothalamic area (PF-LHA).5–8 Much evidence suggests that HCRT system promotes and stabilizes waking with concomitant suppression of non-rapid eye movement (nonREM) as well as REM sleep and associated muscle atonia. HCRT neurons are active during waking, especially during active arousal, and are quiescent during nonREM and REM sleep.9–11 The in vivo selective stimulation of HCRT neurons genetically targeted with channelrhodopsin-2, a light-activated cation channel, induces awakenings during both nonREM and REM sleep, whereas optogenetic or pharmacogenetic silencing of HCRT neurons suppress waking and induce sleep.12,13 A loss of HCRT signaling is linked with the pathogenesis of narcolepsy including fragmentation of sleep-wakefulness and cataplexy-like state in human and animals.8,14–18 HCRT neurons project widely to brain structures implicated in sleep-wake regulation, especially to those regions that are involved in arousal and motor control, where HCRTs exert excitatory effects via two G-protein coupled receptors, viz., HCRT-1 and HCRT-2 receptors, to promote waking.1,4,8,19–21
In the PF-LHA, HCRT neurons are intermingled with various other neuronal groups including neurons expressing glutamate, a major excitatory neurotransmitter.22 In addition, PF-LHA also receives glutamatergic afferents from other brain areas including the basal forebrain and several hypothalamic and brainstem regions.19,23–25 Evidence suggests that glutamatergic neurons within the PF-LHA are also involved in the promotion and/or maintenance of waking and that this glutamatergic action, in part, may be mediated via mutually excitatory interactions between HCRT and glutamatergic neurons. For example: (a) local glutamatergic activation of the HCRT field by microinjections of L-glutamic acid or N-methyl-D-aspartate (NMDA) elicit arousal or arousal-related behaviors including locomotion, rearing, feeding and drinking and suppress nonREM/REM sleep26,27; (b) moderate to high levels of glutamate receptor genes as well as proteins and vesicular glutamate transporter-2 immunoreactivity have been reported in the PF-LHA28–30; (c) majority of HCRT neurons express mRNA for the vesicular glutamate transporters or co-express glutamate31 and glutamate is also co-released from the terminals of HCRT neurons30,32; and (d) glutamate release in the PF-LHA increases rapidly with waking onset.33 In vitro pharmacologic evidence suggests that glutamatergic synaptic input to HCRT neurons, at least partly, contributes to the HCRT neuronal excitability,34 which in the intact animal potentially activates other wake-promoting regions leading to the promotion of waking and suppression of nonREM/REM sleep. While the HCRT-glutamatergic interactions potentially play a critical role in maintaining a high level of HCRT neuronal excitability during waking, especially active waking, the contributions of this interaction to sleep-wake regulation in freely behaving animals are not known.
In this study, we determined whether a lack of HCRT signaling in prepro-orexin knockout (HCRT-KO) mice attenuates or compromises the wake-promoting ability of the PF-LHA glutamatergic activation, thus affecting sleep-wake regulation. We used reverse microdialysis to deliver NMDA into the PF-LHA of HCRT-KO and wild-type (WT) mice to evaluate the contributions of glutamatergic versus HCRT signaling in sleep-wake regulation. Our findings indicate that, while PF-LHA glutamatergic activation in both WT and HCRT-KO mice induced prolonged behavioral arousal with concomitant decreases in nonREM and REM sleep, the magnitude of effects were significantly attenuated in HCRT-KO mice. In HCRT-KO mice, the frequency of behavioral arrest or cataplectic events increased significantly during NMDA-induced behavioral arousal, whereas no such event was observed in WT mice.
EXPERIMENTAL PROCEDURES
Experiments were conducted on 2 groups of male mice; prepro-hypocretin mutated C57BL/6J-129/SvEv (HCRT-KO) and their wild-type (WT) littermates. The C57BL/6J-129/SvEv mice were generated as reported previously,35 were maintained as heterozygotes, and crossed to obtain null mutants (KO). The genotype of HCRT-KO mice was confirmed by polymerase chain reaction using the previously described method.36 The mice were maintained on 12:12h light: dark cycle (lights on at 08:00), an ambient temperature of 24 ± 1°C, and with food and water available ad libitum. All experiments were conducted in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Research Committee of the Veterans Affairs Greater Los Angeles Healthcare System. Mice used in this study were 3-4 months old at the beginning of the procedures.
Surgical Implantation
Under surgical anesthesia (ketamine + xylazine: 80:10 mg/ kg; i.p.) and aseptic conditions, electroencephalogram (EEG) and electromyogram (EMG) electrodes were implanted for polygraphic monitoring of sleep-waking states in 12 WT and 8 HCRT-KO mice. A microdialysis guide cannula (CMA-7, CMA/Microdialysis, USA) was stereotaxically implanted such that its tip rested 2 mm dorsal to the PF-LHA (AP −1.5 to −1.8 mm; L 0.8 mm; H 6.0 mm)37 and was blocked with a stylet. Mice were allowed to recover from the surgical procedure in their home cages for 7-10 days and then connected to the recording cables to acclimatize them to the recording procedure for additional 7 days before the start of the experiments.
Experimental Protocol
At least 12 h before the experiments, the stylet of the micro-dialysis guide cannula was removed and a microdialysis probe (CMA/7, semipermeable cuprophane membrane tip length, 1 mm; outer diameter, 0.24 mm; molecular cutoff size, 6 kDa; CMA/Microdialysis, USA) was inserted through the cannula into the PF-LHA, secured, and flushed with artificial cerebrospinal fluid (aCSF; composition in mM, 145 NaCl, 2.7 KCl, 1.3 MgSO4, 1.2 CaCl2, and 2 Na2HPO4; pH, 7.2) at a flow rate of 1μL/min. The probe was implanted such that the estimated diffusion area of the semi-permeable membrane was within or predominantly included the HCRT neuronal field. The time taken by the aCSF solution to travel from the reservoir to the tip of the probe was precisely calculated.
Data Acquisition and Drug Delivery
Studies suggest that within the PF-LHA, HCRT and glutamatergic neurons exhibit minimal activation during lights-on phase when rats spent significantly more time in sleep.38–40 Therefore, the effects of glutamatergic activation in the PF-LHA on sleep-wakefulness in HCRT-KO versus WT mice were studied during lights-on phase to maximize the salience of the effects.
The EEG and EMG activities of each animal were recorded continuously, starting 1 h after lights-on, for 7 h either with aCSF perfusion (control) or during perfusion of aCSF for 2 h as a baseline, followed by 250 or 500 μM of N-methyl-D-aspartate (NMDA) for 3 h, and followed again by aCSF perfusion for 2 h as recovery. Each animal was subjected to all 3 experimental conditions. NMDA was dissolved in distilled water at 100 mM concentration and then the stock solution was diluted to 250 or 500 μM concentrations in aCSF for perfusion. The sequence of aCSF or NMDA treatments was switched among animals. Amplified and filtered EEG and EMG signals were continuously digitized and stored on the hard disk of the host computer with the use of an integrated computer interface device (Cambridge Electronic Design 1401; supporting software, Spike 2; London).
Histology
At the end of the experiment, under deep anesthesia (100 mg/kg, i.p., pentobarbital) mice were perfused transcardially with 10-15 mL of 0.1M phosphate buffer (pH 7.2) followed by 50 mL of 4% paraformaldehyde in phosphate buffer. The brains were removed and equilibrated in 10%, 20%, and finally 30% sucrose until they sank. Horizontal sections through the PF-LHA were freeze-cut at 30 μm thickness and alternate sections from the series of sections spanning the probe tract were immunostained to reveal HCRT-1 proteins.40,41
For HCRT-1 immunostaining, first, sections were washed in Tris-buffered saline (TBS) and then incubated in rabbit anti-HCRT-1 (orexin-A) antibody (1:1000, Calibiochem, USA) for 40-48 h at 4°C. The sections were then incubated in biotinylated goat anti-rabbit IgG (1:500 Vector Laboratories, USA) for 2 h, followed by incubation in ABC (1:250, Vector Laboratories, USA) for 2 h, and then developed with 3,3'-diaminobenzidine producing a brown reaction product for visualization. Two sections from each brain were treated as above, except for the omission of HCRT-1 primary antibody to control for nonspecific staining. Finally sections were rinsed in Tris (3 ×10 min) followed by TBS. The sections were mounted on gelatin-coated slides, air dried, dehydrated, and cover-slipped. Experiments were run in groups of WT and HCRT-KO mice, and brains obtained from WT and HCRT-KO mice were processed together for immunostaining to avoid any staining-related discrepancy. Locations of microdialysis probes were histologically confirmed (Figure 1)
Figure 1.

Localization of the microdialysis probes. Locations of the microdialysis probes, reconstructed on coronal sections through the PF-LHA from the atlas of Paxinos and Franklin (2001). Numbers in the lower right portion of each coronal section indicate the distance (in mm) from the bregma. Photomicrographs of horizontal sections through the PF-LHA from HCRT-KO and WT mice are shown on the top. The circular outlines marked by stars indicate the microdialysis probe tracts in the HCRT zone of the PF-LHA as evident from clusters of HCRT neurons (marked by arrowheads) in the WT and its absence in the HCRT-KO mice. Bars represent actual reconstructed probe lengths and its dorso-ventral locations; gray bars are those cases that were not considered to be within HCRT zone; blank and solid bars represent probe locations in HCRT-KO and WT mice, respectively, that were considered to be in HCRT zone.
Data Analyses
Sleep-wake profiles of animals were scored manually in 10-s epochs in terms of active waking (AW), quiet waking (QW), nonREM, and REM sleep using standard criteria.42 Cataplexy was scored based on the criteria established by the International Working Group on Rodent Models of Narcolepsy43 and included atonia with EEG theta activity that was immediately preceded and followed by active waking. The sleep-wake data during the 3 h of aCSF/NMDA treatment were compared with the data obtained during 2 h of baseline and 2 h of recovery period for WT and HCRT-KO mice. The level of significance among different treatment conditions was determined by using one-way repeated measures ANOVA followed by pairwise multiple comparisons using Student-Newman-Keuls method. We found that 500 μM of NMDA was effective in both WT and HCRT-KO mice. Therefore, we compared changes in sleep-wake parameters in response to 500 μM of NMDA in HCRT-KO versus WT mice using one-way ANOVA.
RESULTS
Site of Drug Delivery
The locations of the microdialysis probes along with the outlines of membrane that were used for perfusing aCSF/ NMDA are shown in Figure 1. These probes were localized mostly in the PF-LHA and adjoining areas between AP −1.5 to −1.8,37 areas where clusters of HCRT neurons, as evident from the HCRT-1 immunostaining in WT mice, are localized. In earlier studies we found that perfusion of bicuculline or serotonin at 2 μL/min (double the rate used in this study) into the PF-LHA40,41 affected Fos-IR in 500-750 μm radius around the probe. Therefore, it is likely that the diffusion field in this study was relatively smaller (∼250-400 μm area); and based on the probe locations, it is likely that the affected areas perfused by NMDA included the perifornical area, portions of dorsomedial and ventromedial hypothalamic area, lateral hypothalamic area, and ventral zona incerta.
Effects of NMDA on Sleep-Wakefulness in WT Mice
The effect of 2 doses of NMDA vs. aCSF into the PF-LHA on sleep-wakefulness was studied in a group of 12 mice, in which each animal was subjected to all 3 treatments. Of these mice, in 5 cases probe locations were either not within the HCRT zone or included minimal portions of the HCRT zone of the PF-LHA; therefore, these mice were used as a control for site specificity. The sleep-wake profiles of mice perfused with aCSF (n = 7) and NMDA (250 μM, n = 6 [one mouse exhibited unusually high waking during baseline and was not included in analysis] and 500 μM, n = 7) into the PF-LHA on percent time (mean ± SEM) spent in waking, nonREM, and REM sleep during the 7-h recording period including 2 h of baseline, 3 h of treatment, and 2 h of post-treatment conditions are shown in Figures 2 and 3. Overall, aCSF treated mice spent a significant portion of the recording time in sleep (nonREM sleep, 66% ± 2% and REM sleep, 9% ± 1%) during the 7-h recording period with comparable amounts of waking, nonREM, and REM sleep during pre-treatment (baseline), treatment, and post-treatment time-slots assigned for NMDA experiments. The sleep-wake profiles during pre- and post-treatment periods in both aCSF and NMDA treated groups were comparable. However, during NMDA perfusion, mice spent significantly more time in AW (F = 35.370; P < 0.001) and less time in nonREM (F = 43.896; P < 0.001) and REM sleep (F = 12.939; P < 0.001). While both doses of NMDA were effective, behavioral changes, i.e., increases in AW with concomitant decreases in nonREM and REM sleep, induced by 500 μM NMDA were significantly greater than that induced by 250 μM of NMDA treatment. As compared to aCSF or baseline control, neither dose of NMDA affected QW. The sleep-wake profiles of animals where NMDA perfusion was not in the HCRT zone of the PF-LHA exhibited minimal changes during NMDA treatments and were comparable to those observed in aCSF treated animals across the experiment (Figure 3).
Figure 2.

Effects of NMDA on sleep-wakefulness in HCRT-KO and WT mice. Nearly 8 h of continuous EEG and EMG recording (2 h of baseline, 3 h of NMDA treatment, and ∼3 h of recovery period) showing the sleep-wake profiles (hypnogram) of HCRT-KO (A) and WT (B) mice in response to aCSF and 500 μM of NMDA into the PF-LHA during lights-on phase. The arrival and washout of NMDA are shown by solid and blank arrows, respectively. As a control, aCSF was continuously perfused into the PF-LHA. NMDA produced stronger behavioral arousal in WT and a relatively muted effect in HCRT-KO mice. In this case, WT mouse practically spent the entire perfusion time in AW but did not exhibit any cataplectic event. On the other hand, although the effect was relatively muted, cataplectic event or atonia can be seen during behavioral arousal in HCRT-KO mouse (also see Figure 4). EEG, electroencephalogram; EMG, electromyogram.
Figure 3.

Effects of NMDA on sleep-wakefulness in HCRT-KO and WT mice. Mean (± SEM) percentages of AW, QW, nonREM, and REM sleep in HCRTKO and WT mice during baseline (2 h with aCSF), during aCSF or NMDA (250 or 500 μM, 3 h) perfusion, and during recovery period (2 h with aCSF). NMDA induced a dose-dependent increase in AW and suppression of nonREM and REM sleep in both HCRT-KO and WT mice. In those cases where the microdialysis probes were not localized in the PF-LHA, even 500 μM of NMDA was not effective in inducing any noticeable change in sleep-wake profiles of animals (shown in Figure). ++, =, **, < 0.01; +, -, *, < 0.05 level of significance (one-way repeated measure ANOVA followed by Student-Newman-Keuls method), +, increase as compared to the baseline; -, decrease as compared to the baseline.
During NMDA perfusion into the PF-LHA, mice exhibited significantly longer bouts of AW (52.0 ± 8.2 s [aCSF] vs. 266 ± 116 s [250 μM NMDA] and 720 ± 201 s [500 μM NMDA]; F = 7.712; P < 0.001) with concomitant shorter bouts of nonREM (156 ± 16 s vs. 89 ± 13 s and 39 ± 12 s; F = 6.498; P < 0.001) and REM sleep (65 ± 6 s vs. 39 ± 15 s and 16 ± 7 s; F = 7.815; P < 0.001). Although the frequency of AW episodes during NMDA treatment were comparable to that observed during aCSF treatment (12 ± 1 vs. 17 ± 3 and 13 ± 6; P = 0.463), the number of nonREM (18 ± 2 vs. 11 ± 3 and 2 ± 1; F = 7.649; P < 0.001) and REM sleep episodes (5.6 ± 0.4 vs. 1.2 ± 0.5 and 0.5 ± 0.3; F = 10.965; P < 0.001) decreased significantly.
Effects of NMDA on Sleep-Wakefulness in HCRT-KO Mice
The effects of 250 μM and 500 μM of NMDA into the PF-LHA on sleep-wakefulness were studied in 8 HCRT KO mice. Of these mice, probe locations in 3 cases were predominantly not within the HCRT zone of the PF-LHA, and thus these animals served as control for site specificity. The sleep-wake profiles of mice perfused with aCSF vs. 250 μM and 500 μM of NMDA into the PF-LHA on percent time (mean ± SEM) spent in waking, nonREM, and REM sleep during the 2 h of baseline, 3 h of treatment, and 2 h of post-treatment conditions are shown in Figures 2 and 3. Overall, mice perfused with aCSF spent significantly less time in waking, especially AW (20% ± 2%) and more time in sleep (nonREM sleep, 63% ± 1.70%, and REM sleep, 7% ± 1%) during the 7-h recording period with comparable amounts of waking, nonREM, and REM sleep during pre-treatment, treatment, and post-treatment time-slots assigned for NMDA experiments. While the sleep-wake profiles of both aCSF and NMDA treated mice were comparable during pre-treatment and post-treatment phases, during NMDA perfusion into the PF-LHA, HCRT-KO mice spent significantly more time in AW (F = 16.519; P < 0.001) and less time in nonREM (F = 15.859; P < 0.001) and REM sleep (F = 3.316; P = 0.007; Figure 3). Of the 2 doses used, 500 μM NMDA-induced AW with concomitant decreases in nonREM and REM sleep, were significantly greater compared to those induced by 250 μM of NMDA treatment; 250 μM NMDA only marginally increased AW and suppressed nonREM and REM sleep. As compared to aCSF or baseline control, both doses of NMDA did not affect QW. The sleep-wake profiles of animals where NMDA perfusion was not in the HCRT zone of the PF-LHA were comparable to those observed in aCSF-treated animals across the experiment.
During NMDA perfusion into the PF-LHA, HCRT-KO mice also exhibited significantly longer bouts of AW (46 ± 9 s [aCSF] vs. 100 ± 24 s [250 μM NMDA] and 225 ± 62 s [500 μM NMDA]; F = 6.029; P < 0.001) with concomitant shorter bouts of nonREM (98 ± 6 s vs. 94 ± 13 s and 54 ± 7s; F = 6.235; P < 0.001) and REM sleep (75 ± 10 s vs. 64 ± 13 s and 32 ± 15 s; F = 1.568; P = 0.174). While the number of AW episodes during NMDA treatment was comparable to that observed during aCSF treatment (15 ± 1 vs. 17 ± 3 and 14 ± 2; P = 0.649), the number of nonREM (25 ± 3 vs. 21 ± 2.0 and 10 ± 3; F = 4.643; P < 0.001) and REM sleep episodes (4.8 ± 0.6 vs. 3.4 ± 0.6 and 1.6 ± 0.6; F = 3.433; P = 0.006) decreased significantly.
Effects of NMDA on Cataplexy in WT and HCRT-KO Mice
In WT mice, no behavioral arrest or cataplectic event was observed during baseline or aCSF perfusion. No such event was observed even during NMDA perfusion, when WT mice spent significantly longer time in AW with longer AW episodes (Figure 2).
The HCRT-KO mice, on the other hand, exhibited cataplexy, although rarely during baseline and more frequently during NMDA perfusion. An example of a cataplectic event and the number of times mice exhibited such events during the 2 h of baseline, 3 h of treatment with aCSF vs. 250 μM and 500 μM of NMDA, and 2 h of post-treatment duration are shown in Figure 4. Overall, during aCSF perfusion, mice rarely exhibited behavioral arrests during the 7-h recording period with comparable number of events during pre-treatment, treatment, and post-treatment time-slots assigned for NMDA experiments. While the numbers of cataplectic events in HCRT-KO mice treated with both aCSF and NMDA were comparable during pre- and post-treatment phases, during NMDA perfusion into the PF-LHA, these mice exhibited significantly higher frequency of such events (F = 6.397; P < 0.001) in a dose-dependent manner. The number of cataplectic events in HCRT-KO mice, where NMDA perfusion was not in the HCRT zone of the PF-LHA was comparable to that observed in aCSF treated animals across the experiment (Figure 4).
Figure 4.
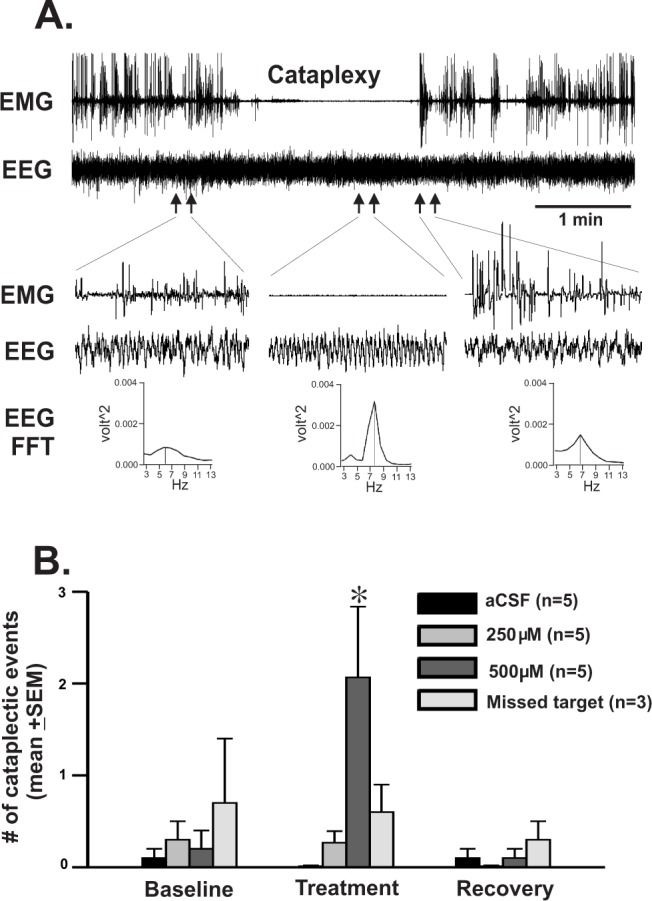
Effects of NMDA on cataplexy in HCRT-KO mice. (A) A typical cataplectic event, which occurred during behavioral arousal induced by 500 μM of NMDA perfusion. An event that included atonia with EEG theta activity and which was immediately preceded and followed by AW was considered as a cataplectic event. (B) Number of cataplectic events (mean ± SEM) encountered during baseline, during two doses of NMDA treatment and during recovery period. In those cases where NMDA perfusion was not in the HCRT zone, only the effect of 500 μM of NMDA is shown. Such events were rare during lights-on phase in spontaneously sleeping and awake animals and even in those animals where NMDA was not in the HCRT zone. However, the number of cataplectic event increased significantly during NMDA perfusion. No cataplectic event was observed in WT mice either during baseline or during NMDA-induced AW (not shown). *, < 0.05 level of significance (one-way ANOVA followed by Student-Newman-Keuls method).
Differential Effects of NMDA in WT vs. HCRT-KO Mice
As noted above, while both 250 μM and 500 μM of NMDA induced significant behavioral changes in WT mice, only 500 μM of NMDA was effective in significantly influencing behavioral states in HCRT-KO mice (Figure 3). A comparison of the sleep-wake profiles of HCRT-KO vs. WT mice in response to 500 μM of NMDA into the HCRT zone of the PF-LHA is shown in Figure 5. While the sleep-wake profiles of HCRT-KO and WT mice during baseline and post-treatment period were comparable, 500 μM of NMDA produced differential effects in HCRT-KO and WT mice.
Figure 5.

Effects of 500 μM of NMDA on sleep-wakefulness in HCRT-KO vs. WT mice. Effects of 500 μM of NMDA on mean (± SEM) percentages of AW, QW, nonREM, and REM sleep (A) and on mean episode duration of AW, QW, nonREM, and REM sleep (B) in HCRT-KO versus WT mice. ++, =, **, < 0.01; +, -, *, < 0.05 level of significance (one-way ANOVA followed by Student-Newman-Keuls method), +, increase as compared to the baseline; -, decrease as compared to the baseline, *, as compared to the WT.
NMDA-induced AW was significantly higher in WT mice than in HCRT-KO mice. Although the number of AW episodes was comparable, the mean duration of AW episodes in WT mice was also significantly longer than that induced in HCRT-KO mice. Concomitantly, NMDA-induced suppression of nonREM sleep was significantly stronger in WT mice than that observed in HCRT-KO mice. There was a significant shortening of nonREM sleep bouts or episode duration in both groups; however, the magnitude of this effect was relatively small in HCRT-KO mice as compared to the WT mice (−50 ± 4 vs.−71 ± 11; as compared to respective baselines; NS). There were significant reductions in REM sleep amount as well as the duration of REM sleep episodes in both HCRT-KO and WT mice. However, although not significant, in general HCRT-KO mice exhibited relatively smaller magnitude of suppression in REM sleep as well as the shortening of REM sleep episodes (−40% ± 24% vs. −79% ± 9%, as compared to respective baselines; NS; Figure 5). HCRT-KO mice also exhibited more episodes of nonREM (20.5 ± 1.8 [baseline] vs. 9.7 ± 2.9 [NMDA] in HCRT-KO versus 15.6 ± 1.4 [baseline] vs. 2.3 ± 0.7 [NMDA] in WT mice; NS) and REM sleep (4.6 ± 1.1 [baseline] vs.1.6 ± 0.6 [NMDA] in HCRT-KO versus 4.1 ± 0.6 [baseline] vs. 0.5 ± 0.3 [NMDA] in WT mice; NS) during NMDA perfusion. QW did not change significantly in either HCRT-KO or WT mice.
During NMDA-induced AW, while WT mice did not exhibit any behavioral arrests, the frequency of cataplectic events increased significantly in HCRT-KO mice (Figure 4).
DISCUSSION
This study demonstrates that the activation of glutamatergic transmission in the PF-LHA, produced by local perfusion of an NMDA receptor agonist via reverse microdialysis, increased AW with concomitant suppression of nonREM and REM sleep in spontaneously sleeping and undisturbed WT as well as HCRT-KO mice. However, as compared to the WT, NMDA-induced behavioral changes were significantly attenuated in HCRT-KO mice. In HCRT-KO mice, a higher dose of NMDA was needed to produce a significant behavioral response, as the magnitude of changes induced in sleep-wake parameters by NMDA were significantly lower. The frequency of behavioral arrests or cataplexy also increased significantly during NMDA treatment in HCRT-KO mice. The findings of this study suggest that synergic interactions between HCRT and glutamatergic mechanisms within the PF-LHA is critical for maintaining behavioral arousal and associated muscle tone.
In this study, microdialysis probes were largely localized in the HCRT zone of the PF-LHA. Therefore, it is likely that NMDA-induced differential behavioral changes as observed comparing WT versus HCRT-KO mice were caused by the activation of neurons in the diffusion field of the microdialysis probe including HCRT neurons in the PF-LHA. It is also likely that the observed effects were physiological response to the NMDA because: (a) aCSF perfusion at the same site and in the same animal was not effective in inducing behavioral changes; (b) the effects were reversible; (c) the effect was site specific, for NMDA was maximally effective in HCRT zone and produced minimal effects if its perfusion was not within the PF-LHA; and (d) the effects were dose-dependent. We further note that unlike microinjection, drug delivery via reverse microdialysis provided a better control of drug concentration and its delivery in undisturbed animals and also reduced the likelihood that treatment effects could be due to mechanical or inflammatory responses.44
The findings that NMDA stimulation in the HCRT zone of the PF-LHA produced behavioral arousal or AW and suppressed nonREM/REM sleep is consistent with and complementary to a previous report in the rat, where local microinjection of glutamic acid into the PF-LHA produced arousal and suppressed spontaneous nonREM/REM sleep.26 PF-LHA contains a mixture of neuronal populations, including wake-active non-HCRT neurons as well as MCH and GABAergic neurons that have been implicated in the regulation of sleep.45–49 The relative distribution of NMDA receptor on various neuronal subgroups, in particular on GABAergic and MCH vs. HCRT neurons, is not well characterized. However, given the findings of this study that the net physiological output of the HCRT PF-LHA activation was AW-promoting, it is likely that behavioral changes observed in this study were predominantly caused by the activation of wake-active HCRT and perhaps by other neurons of unknown neurotransmitter phenotypes. NMDA potentially activated a glutamatergic-HCRT-glutamatergic/HCRT positive feedback loop in the diffusion field of the probe leading to further activation/recruitment of other wake-promoting neurons locally as well as at HCRT projections to major arousal systems including basal forebrain, locus coeruleus (LC), dorsal raphe nucleus (DRN), and tuberomammillary nucleus (TMN), resulting in the manifestation of stronger behavioral arousal (see Figure 2) and consequent suppression of non-REM/REM sleep. That PF-LHA neurons including HCRT neurons are under glutamatergic influences is supported by the findings that glutamate is co-localized with HCRT neurons, and the excitability of the HCRT neurons, in part, is modulated by local glutamatergic synaptic inputs (see Introduction). Furthermore, we found that whereas the frequency of AW episodes, potentially reflective of state initiations remained unaffected, the episode duration increased significantly. This suggests that HCRT neurons are potentially critical for maintaining sustained behavioral arousal.
Evidence suggests that glutamatergic system plays a role in the promotion/maintenance of wakefulness (see introduction). In this study a comparison of the sleep-wake response profiles of HCRT-KO vs. WT mice to the PF-LHA glutamatergic activation suggest synergistic and complementary roles of the two systems in the control of behavioral arousal. A key finding of this study is that the ability of NMDA to induce AW and suppressing nonREM/REM sleep was significantly compromised in HCRT-KO mice. It is not surprising given the large body of evidence implicating HCRT neurons in the regulation of behavioral arousal and associated muscle tone with concomitant suppression of nonREM/REM sleep. For example: (a) HCRT neurons exhibit maximal discharge during AW (behavioral arousal), particularly in association with muscle movement, that decrease dramatically (4∼5 fold) during QW/drowsy state and decrease further during nonNREM and REM sleep9–11; (b) consistent with the discharge activity, Fos-IR in HCRT neurons or levels of HCRT in cerebrospinal fluid are higher during active-arousal38,50; (c) a loss or malfunction of HCRT system leads to cataplexy in human and animal models1,4,8; and (d) administrations of HCRT-peptide, either centrally or locally at projection sites, suppress cataplexy or behavioral arrests and increase wakefulness.1,4,8,51 These findings indicate that HCRT neurons are central to behavioral arousal consisting of higher muscle tone/locomotion. And therefore, a compromised response to glutamatergic activation might be a consequence of inadequate activation, due to the lack of HCRT production, of local and well as other wake-promoting systems in the HCRT circuitry.
Interestingly, in this study the frequency of behavioral arrests or cataplectic events increased in NMDA-induced arousal in HCRT-KO mice, while no such event was observed in its WT counterpart. It is plausible that a normal function of HCRT is to maintain muscle tone and that with HCRT cell activation, the concurrent activation of non-HCRT cells (or perhaps of “HCRT cells” lacking HCRT in the HCRT-KO mice) in the PF-LHA suppress muscle tone. Therefore, in the intact animal, a balance is maintained during behavioral arousal. However, with the activation of this area in the absence of HCRT signaling, an imbalance, leading to occasional loss of muscle tone occurs during behavioral arousal, as observed in this study.
In this study, glutamatergic activation of the HCRT zone was effective in promoting behavioral arousal and suppressing nonREM and REM sleep even in HCRT-KO mice, although to a lesser extent. This could be due to the fact that in the PF-LHA, HCRT neurons constitute only a subset of neuronal population, which is active during AW and quiescent during nonREM or nonREM-REM sleep.48,52 Therefore, it is likely that NMDA-induced behavioral changes in HCRT-KO mice were caused by the activation of those wake-active non-HCRT neuronal populations. It is also plausible that in HCRT-KO mice, while HCRT production is compromised, the neuronal network as well as other neurotransmitters signaling molecules that co-localize with HCRT neurons including dynorphin, glutamate, and Narp remains intact.53,54 Fibers containing such co-transmitters are found intermingled with HCRT terminals in many wake-promoting areas including aminergic cell groups, namely, TMN, LC, and DRN, and thus may complement HCRT actions.32 For example, evidence suggests that dynorphin increases the excitability of TMN neurons by suppressing its GABAergic input.55 Therefore, it is likely that some of those co-peptides and/or co-transmitters released by glutamatergic activation of HCRT neurons contributed to behavioral changes by acting locally as well as at HCRT projection sites.
In conclusion, the results of this study are consistent with earlier findings that HCRT system within the PF-LHA is critically involved in the regulation of behavioral arousal and associated muscle tone. However, the effectiveness of glutamatergic stimulation in the PF-LHA to promote behavioral arousal even in HCRT-KO mice suggests that other non-HCRT neuronal groups or other neurotransmitters/neuropeptides that colocalize with HCRT neurons contribute to the regulation of behavioral arousal as well. The neurotransmitter phenotype of these neurons and their relative contributions or the role of co-transmitters in influencing HCRT circuitry remains to be fully characterized. This study indicates that glutamatergic system within the PF-LHA is one of such player. It is likely that synergistic interactions between HCRT and glutamatergic systems within the PF-LHA rapidly activate multiple arousal systems via fast-acting glutamatergic mechanism leading to wake onset, whereas a relatively slower and stable release of HCRT peptide stabilizes the activation of such neurons to maintain behavioral arousal, especially that involving elevated muscle tone.
DISCLOSURE STATEMENT
This was not an industry supported study. This work was supported by the Medical Research Service, US Department of Veterans Affairs and MH064109. The authors have indicated no financial conflicts of interest.
REFERENCES
- 1.Alam MN, Szymusiak R, McGinty D. Hypocretinergic system: role in REM-sleep regulation. In: Mallick BN, Pandi-Perumal SR, McCarley R, Morrison AR, editors. Rapid eye movement sleep. Cambridge University Press; 2011. pp. 234–46. [Google Scholar]
- 2.Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. Control of sleep and wakefulness. Physiol Rev. 2012;92:1087–187. doi: 10.1152/physrev.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Datta S. Cellular and chemical neuroscience of mammalian sleep. Sleep Med. 2010;11:431–40. doi: 10.1016/j.sleep.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel JM. The neurobiology of sleep. Semin Neurol. 2009;29:277–96. doi: 10.1055/s-0029-1237118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Lecea L, Kilduff TS, Peyron C, et al. The hypocretins: hypothalamusspecific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–7. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peyron C, Tighe DK, van den Pol AN, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–85. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 8.Ohno K, Sakurai T. Orexin neuronal circuitry: role in the regulation of sleep and wakefulness. Front Neuroendocrinol. 2008;29:70–87. doi: 10.1016/j.yfrne.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi K, Lin JS, Sakai K. Neuronal activity of orexin and nonorexin waking-active neurons during wake-sleep states in the mouse. Neuroscience. 2008;153:860–70. doi: 10.1016/j.neuroscience.2008.02.058. [DOI] [PubMed] [Google Scholar]
- 10.Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–98. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005;25:6716–20. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–4. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsunematsu T, Kilduff TS, Boyden ES, Takahashi S, Tominaga M, Yamanaka A. Acute optogenetic silencing of orexin/hypocretin neurons induces slow-wave sleep in mice. J Neurosci. 2011;31:10529–39. doi: 10.1523/JNEUROSCI.0784-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–7. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- 15.Thannickal TC, Moore RY, Nienhuis R, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–74. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin L, Faraco J, Li R, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–76. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- 17.Nishino S. The hypothalamic peptidergic system, hypocretin/orexin and vigilance control. Neuropeptides. 2007;41:117–33. doi: 10.1016/j.npep.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Anaclet C, Parmentier R, Ouk K, et al. Orexin/hypocretin and histamine: distinct roles in the control of wakefulness demonstrated using knock-out mouse models. J Neurosci. 2009;29:14423–38. doi: 10.1523/JNEUROSCI.2604-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida K, McCormack S, Espana RA, Crocker A, Scammell TE. Afferents to the orexin neurons of the rat brain. J Comp Neurol. 2006;494:845–61. doi: 10.1002/cne.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Methippara MM, Alam MN, Szymusiak R, McGinty D. Effects of lateral preoptic area application of orexin-A on sleep-wakefulness. Neuroreport. 2000;11:3423–6. doi: 10.1097/00001756-200011090-00004. [DOI] [PubMed] [Google Scholar]
- 21.Thakkar MM, Ramesh V, Strecker RE, McCarley RW. Microdialysis perfusion of orexin-A in the basal forebrain increases wakefulness in freely behaving rats. Arch Ital Biol. 2001;139:313–28. [PubMed] [Google Scholar]
- 22.Abrahamson EE, Moore RY. The posterior hypothalamic area: chemoarchitecture and afferent connections. Brain Res. 2001;889:1–22. doi: 10.1016/s0006-8993(00)03015-8. [DOI] [PubMed] [Google Scholar]
- 23.Henny P, Jones BE. Innervation of orexin/hypocretin neurons by GABAergic, glutamatergic or cholinergic basal forebrain terminals evidenced by immunostaining for presynaptic vesicular transporter and postsynaptic scaffolding proteins. J Comp Neurol. 2006;499:645–61. doi: 10.1002/cne.21131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niu JG, Yokota S, Tsumori T, Qin Y, Yasui Y. Glutamatergic lateral parabrachial neurons innervate orexin-containing hypothalamic neurons in the rat. Brain Res. 1358:110–22. doi: 10.1016/j.brainres.2010.08.056. [DOI] [PubMed] [Google Scholar]
- 25.Chou TC, Scammell TE, Gooley JJ, Gaus SE, Saper CB, Lu J. Critical role of dorsomedial hypothalamic nucleus in a wide range of behavioral circadian rhythms. J Neurosci. 2003;23:10691–702. doi: 10.1523/JNEUROSCI.23-33-10691.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alam MA, Mallick BN. Glutamic acid stimulation of the perifornical-lateral hypothalamic area promotes arousal and inhibits non-REM/REM sleep. Neurosci Lett. 2008;439:281–6. doi: 10.1016/j.neulet.2008.05.042. [DOI] [PubMed] [Google Scholar]
- 27.Li FW, Deurveilher S, Semba K. Behavioural and neuronal activation after microinjections of AMPA and NMDA into the perifornical lateral hypothalamus in rats. Behav Brain Res. 2011;224:376–86. doi: 10.1016/j.bbr.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 28.Khan AM, Curras MC, Dao J, et al. Lateral hypothalamic NMDA receptor subunits NR2A and/or NR2B mediate eating: immunochemical/ behavioral evidence. Am J Physiol. 1999;276:R880–91. doi: 10.1152/ajpregu.1999.276.3.R880. [DOI] [PubMed] [Google Scholar]
- 29.Eyigor O, Centers A, Jennes L. Distribution of ionotropic glutamate receptor subunit mRNAs in the rat hypothalamus. J Comp Neurol. 2001;434:101–24. doi: 10.1002/cne.1167. [DOI] [PubMed] [Google Scholar]
- 30.Henny P, Brischoux F, Mainville L, Stroh T, Jones BE. Immunohisto-chemical evidence for synaptic release of glutamate from orexin terminals in the locus coeruleus. Neuroscience. 2010;169:1150–7. doi: 10.1016/j.neuroscience.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosin DL, Weston MC, Sevigny CP, Stornetta RL, Guyenet PG. Hypothalamic orexin (hypocretin) neurons express vesicular glutamate transporters VGLUT1 or VGLUT2. J Comp Neurol. 2003;465:593–603. doi: 10.1002/cne.10860. [DOI] [PubMed] [Google Scholar]
- 32.Torrealba F, Yanagisawa M, Saper CB. Colocalization of orexin a and glutamate immunoreactivity in axon terminals in the tuberomammillary nucleus in rats. Neuroscience. 2003;119:1033–44. doi: 10.1016/s0306-4522(03)00238-0. [DOI] [PubMed] [Google Scholar]
- 33.John J, Ramanathan L, Siegel JM. Rapid changes in glutamate levels in the posterior hypothalamus across sleep-wake states in freely behaving rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R2041–9. doi: 10.1152/ajpregu.90541.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Gao XB, Sakurai T, van den Pol AN. Hypocretin/Orexin excites hypocretin neurons via a local glutamate neuron-A potential mechanism for orchestrating the hypothalamic arousal system. Neuron. 2002;36:1169–81. doi: 10.1016/s0896-6273(02)01132-7. [DOI] [PubMed] [Google Scholar]
- 35.Chemelli RM, Willie JT, Sinton CM, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–51. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 36.Kayaba Y, Nakamura A, Kasuya Y, et al. Attenuated defense response and low basal blood pressure in orexin knockout mice. Am J Physiol Regul Integr Comp Physiol. 2003;285:R581–93. doi: 10.1152/ajpregu.00671.2002. [DOI] [PubMed] [Google Scholar]
- 37.Paxinos G, Franklin KJ. The mouse brain in stereotaxic coordinates. Second ed. San Diego: Academic Press; 2001. [Google Scholar]
- 38.Espana RA, Valentino RJ, Berridge CW. Fos immunoreactivity in hypocretin-synthesizing and hypocretin-1 receptor-expressing neurons: effects of diurnal and nocturnal spontaneous waking, stress and hypocretin-1 administration. Neuroscience. 2003;121:201–17. doi: 10.1016/s0306-4522(03)00334-8. [DOI] [PubMed] [Google Scholar]
- 39.Estabrooke IV, McCarthy MT, Ko E, et al. Fos expression in orexin neurons varies with behavioral state. J Neurosci. 2001;21:1656–62. doi: 10.1523/JNEUROSCI.21-05-01656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar S, Szymusiak R, Bashir T, Rai S, McGinty D, Alam MN. Effects of serotonin on perifornical-lateral hypothalamic area neurons in rat. Eur J Neurosci. 2007;25:201–12. doi: 10.1111/j.1460-9568.2006.05268.x. [DOI] [PubMed] [Google Scholar]
- 41.Alam MN, Kumar S, Bashir T, et al. GABA-mediated control of hypocretin- but not melanin-concentrating hormone-immunoreactive neurones during sleep in rats. J Physiol. 2005;563:569–82. doi: 10.1113/jphysiol.2004.076927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franken P, Malafosse A, Tafti M. Genetic determinants of sleep regulation in inbred mice. Sleep. 1999;22:155–69. [PubMed] [Google Scholar]
- 43.Scammell TE, Willie JT, Guilleminault C, Siegel JM. A consensus definition of cataplexy in mouse models of narcolepsy. Sleep. 2009;32:111–6. [PMC free article] [PubMed] [Google Scholar]
- 44.Quan N, Blatteis CM. Microdialysis: a system for localized drug delivery into the brain. Brain Res Bull. 1989;22:621–5. doi: 10.1016/0361-9230(89)90080-4. [DOI] [PubMed] [Google Scholar]
- 45.Hassani OK, Henny P, Lee MG, Jones BE. GABAergic neurons intermingled with orexin and MCH neurons in the lateral hypothalamus discharge maximally during sleep. Eur J Neurosci. 2010;32:448–57. doi: 10.1111/j.1460-9568.2010.07295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hassani OK, Lee MG, Jones BE. Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle. Proc Natl Acad Sci U S A. 2009;106:2418–22. doi: 10.1073/pnas.0811400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konadhode RR, Pelluru D, Blanco-Centurion C, et al. Optogenetic stimulation of MCH neurons increases sleep. J Neurosci. 2013;33:10257–63. doi: 10.1523/JNEUROSCI.1225-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alam MN, Gong H, Alam T, Jaganath R, McGinty D, Szymusiak R. Sleep-waking discharge patterns of neurons recorded in the rat perifornical lateral hypothalamic area. J Physiol. 2002;538:619–31. doi: 10.1113/jphysiol.2001.012888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luppi PH, Clement O, Fort P. Paradoxical (REM) sleep genesis by the brainstem is under hypothalamic control. Curr Opin Neurobiol. 2013;23:786–92. doi: 10.1016/j.conb.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 50.Martins PJ, D'Almeida V, Pedrazzoli M, Lin L, Mignot E, Tufik S. Increased hypocretin-1 (orexin-a) levels in cerebrospinal fluid of rats after short-term forced activity. Regul Pept. 2004;117:155–8. doi: 10.1016/j.regpep.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 51.Mieda M, Willie JT, Hara J, Sinton CM, Sakurai T, Yanagisawa M. Orexin peptides prevent cataplexy and improve wakefulness in an orexin neuron-ablated model of narcolepsy in mice. Proc Natl Acad Sci U S A. 2004;101:4649–54. doi: 10.1073/pnas.0400590101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rai S, Kumar S, Alam MA, Szymusiak R, McGinty D, Alam MN. A1 receptor mediated adenosinergic regulation of perifornical-lateral hypothalamic area neurons in freely behaving rats. Neuroscience. 2010;167:40–8. doi: 10.1016/j.neuroscience.2010.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chou TC, Lee CE, Lu J, et al. Orexin (hypocretin) neurons contain dynorphin. J Neurosci. 2001;21:RC168. doi: 10.1523/JNEUROSCI.21-19-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blouin AM, Thannickal TC, Worley PF, Baraban JM, Reti IM, Siegel JM. Narp immunostaining of human hypocretin (orexin) neurons: loss in narcolepsy. Neurology. 2005;65:1189–92. doi: 10.1212/01.wnl.0000175219.01544.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eriksson KS, Sergeeva OA, Selbach O, Haas HL. Orexin (hypocretin)/ dynorphin neurons control GABAergic inputs to tuberomammillary neurons. Eur J Neurosci. 2004;19:1278–84. doi: 10.1111/j.1460-9568.2004.03243.x. [DOI] [PubMed] [Google Scholar]