

Abstract
Anophthalmia and microphthalmia (A/M) represent severe developmental ocular malformations. Currently, mutations in known genes explain less than 40% of A/M cases. We performed whole genome copy number variation analysis in sixty patients affected with isolated or syndromic A/M. Pathogenic deletions of 3q26 (SOX2) were identified in four independent patients with syndromic microphthalmia. Other variants of interest included regions with a known role in human disease (likely pathogenic) as well as novel rearrangements (uncertain significance). A 2.2-Mb duplication of 3q29 in a patient with nonsyndromic anophthalmia and an 877-kb duplication of 11p13 (PAX6) and a 1.4-Mb deletion of 17q11.2 (NF1) in two independent probands with syndromic microphthalmia and other ocular defects were identified; while ocular anomalies have been previously associated with 3q29 duplications, PAX6 duplications, and NF1 mutations in some cases, the ocular phenotypes observed here are more severe than previously reported. Three novel regions of possible interest included a 2q14.2 duplication which cosegregated with microphthalmia/microcornea and congenital cataracts in one family, and 2q21 and 15q26 duplications in two additional cases; each of these regions contains genes that are active during vertebrate ocular development. Overall, this study identified causative copy number mutations and regions with a possible role in ocular disease in 17% of A/M cases.
Keywords: anophthalmia, microphthalmia, copy number variation, NF1, PAX6, SOX2, 2q14, 3q29
INTRODUCTION
Anophthalmia/microphthalmia (A/M) is characterized by small (microphthalmia) or absent (anophthalmia) eye globes; anophthalmia or microphthalmia is reported in one out of ten pediatric patients with visual impairment with an overall frequency of 1–3/10,000 births (1–3). A/M is associated with additional non-ocular anomalies in approximately one-third of cases; both isolated and syndromic cases can be caused by genetic factors, including chromosomal anomalies and single gene disruptions (4,5). Mutations in SOX2, OTX2 and FOXE3 transcription factors were shown to play major roles in A/M phenotypes with some contribution from CHX10, RAX, PITX3, PAX6, GDF6, STRA6, FRAS1, CHD7, BMP4 and other factors (4,5). At this point, mutations in known genes explain less than 40% of this condition (5); therefore, additional causative factors need to be discovered.
Analyses of chromosomal abnormalities such as translocations or deletions/duplications of chromosomal material have been widely utilized to discover genomic regions associated with human disorders and to subsequently identify causative gene(s). This approach resulted in the identification of numerous human ocular disease genes including PITX2 for Axenfeld-Rieger syndrome (6); SOX2 for Anophthalmia syndrome (7); B3GALTL for Peters Plus syndrome (8); and CHD7 for CHARGE syndrome (9). Copy number variation (CNV) analysis or array comparative genomic hybridization (array-CGH) allows high resolution screening for structural alterations in human genomes (10). CNVs are deletions or duplications of various sizes that can be inherited or occur as dominant de novo mutations. Recessive disease causing mutations can be clinically observed if a deleted allele happens to be combined with a mutation-carrying allele (8,11).
In order to better understand the mechanisms of these debilitating human phenotypes, identification of novel genetic factors involved in human ocular syndromes will be essential. In this manuscript, we report results of the array-CGH analysis of sixty patients affected with anophthalmia/microphthalmia that identified pathogenic CNVs as well as several candidate regions/genes that can now be further investigated.
MATERIALS AND METHODS
Human samples
This human study was approved by the Institutional Review Boards of Children’s Hospital of Wisconsin and Albert Einstein Healthcare Network with written informed consent obtained for every subject. Genomic DNA was extracted using standard procedures from blood or buccal samples. Previously reported patients were excluded (12,13). Sixty individuals with anophthalmia/microphthalmia were included in this study; 35 patients had syndromic A/M and the remaining 25 had isolated ocular defects. Previous screening excluded mutations in SOX2, OTX2, PITX2, PITX3, FOXE3, and BMP4 (12,14–17).
Copy number variation analysis
Patients were screened for structural genomic variation using the Affymetrix Genome-Wide Human SNP Array 6.0 (Santa Clara, CA, USA) including over 1.8 million genetic markers as previously described (18). The copy number analysis was performed using the Canary Algorithm and Affymetrix Genotyping Console 4.1.1 software, protocols and workflows. Standard settings for intensity QC were utilized with a median absolute pairwise difference (MAPD) greater than 0.4 and criteria that a CNV region of interest must contain consistent information from a minimum of five markers and/or be at least 150-kb in size. Copy number probe data were subjected to custom region analysis to obtain information for all RefSeq (NCBI build GRCh37/hg19) genes. In addition, a second custom region analysis of 203 genes known to be involved in developmental ocular disorders was performed and included all exonic/intronic regions as well as 200-kb of surrounding sequence. Regions identified as polymorphic/benign (>1% frequency) when compared to the Database of Genomic Variants (DGV; http://projects.tcag.ca/variation/) were excluded from further analysis (19). The remaining copy number variants were classified as ‘pathogenic’ if the CNV was repeatedly associated with the ocular findings in prior reports, as ‘uncertain clinical significance; likely pathogenic’ if the CNV involved regions with a known role in human disease but not clearly associated with the specific ocular findings, and as ‘uncertain clinical significance’ if the CNV was novel or rarely seen (<1%) in the DGV but not associated with human disease in the literature (19).
SOX2 deletions less than 100-kb were validated by TaqMan Copy Number assay using the Hs02675353_cn probe (Applied Biosystems, Carlsbad, CA) located in the exon of SOX2; available family members were tested with the same probe. TaqMan analysis of family members for the PAX6 duplication was completed using the Hs00240871_cn probe (Applied Biosystems, Carlsbad, CA).
RESULTS
Pathogenic copy number mutations
Deletions of 3q26.33 including the SOX2 gene were identified in four independent patients (Patients 1–4; Table 1; Figure 1). The deletion size ranged from 3.6-Mb (Patient 1) to ~30-kb (Patient 4) (Figure 1). All four patients were affected with bilateral anophthalmia/severe microphthalmia, mental retardation/developmental delay, and growth deficiency. Three patients had an ataxic gait (Patients 1, 2 and 3) and one (Patient 4) was reported to have an unsteady gait. Two patients (Patients 1 and 2) had micropenis and another (Patient 4) had hypoplastic uterus and ovaries. Parents were unaffected in all cases; the SOX2 deletions in Patients 1 and 3 were determined to be de novo (parental samples were not available for testing in remaining cases). Deletion of this region is not present in control populations reported in the Database of Genomic Variants (DGV).
Table 1.
Pathogenic copy number mutations.
| Patient | Ocular Phenotype | Non-ocular phenotype | Region | Copy state | Position | Size (Mb)a | Genes in region | Phenotype associated with gene/region |
|---|---|---|---|---|---|---|---|---|
| Patient 1 | B anophthalmia | Severe mental retardation, short stature, growth hormone deficiency, micropenis, ataxic gait | 3q26.32-q26.33 | Loss | 178008903–181595830 | 3.6 (2075) | KCNMB2, ZMAT3, PIK3CA, KCNMB3, ZNF639, MFN1, GNB4, ACTL6A, MRPL47, NDUFB5, USP13, PEX5L, TTC14, CCDC39, FXR1, DNAJC19, SOX2-OT, SOX2 | SOX2 anophthalmia syndrome: ocular, brain, pituitary, genitourinary, and gastroesophageal anomalies |
| Patient 2 | B anophthalmia, cryptophthalmia | Mental retardation, short stature, cryptorchidism, micropenis, ataxic gait | 3q26.33 | Loss | 181363922–182842838 | 1.5 (761) | SOX2-OT, SOX2, FLJ46066, ATP11B, DCUN1D1, MCCC1, LAMP3 | SOX2 anophthalmia syndrome: see above |
| Patient 3 | B anophthalmia | Developmental delays, short stature, ataxic gait | 3q26.33 | Loss | 181381394–181472140 | 0.09 (29) | SOX2-OT, SOX2 | SOX2 anophthalmia syndrome: see above |
| Patient 4 | B anophthalmia | Severe mental retardation, short stature, hypoplastic uterus and ovaries, unsteady gait | 3q26.33 | Loss | 181441700–181472140 | 0.03 (16) | SOX2-OT, SOX2 | SOX2 anophthalmia syndrome: see above |
Number of probes indicative of haploid state is specified in parentheses
Figure 1.

Pathogenic copy number mutations. (a–d) Affymetrix Genome-Wide Human SNP Array 6.0 data for Patients 1–4 with 3q26 deletions including SOX2; breakpoints are indicated with arrows; an enlargement of the deleted region is shown for Patients 3 and 4. (e) The UCSC Genome Browser (http://genome.ucsc.edu) view of the deleted region indicating the positions of genes (GRCh37/hg19 assembly); the rectangular bars below indicate the extent of the deletion in each patient.
Copy number variants of uncertain significance (including likely pathogenic)
Six additional CNVs of uncertain significance with a possible role in ocular disease were identified (Table 2; Figure 2); these CNVs were either not seen or rarely seen (<1%) in control populations reported in the DGV. Three of the CNVs affect regions with a known role in human disease and were considered to be likely pathogenic and the remaining three represent potential novel loci.
Table 2.
Copy number variants of uncertain significance (including likely pathogenic).
| Patient | Ocular Phenotype | Non-ocular phenotype | Region | Copy state | Position | Size (Mb)a | Genes in region | Phenotype for gene/region | Significance for ocular disease |
|---|---|---|---|---|---|---|---|---|---|
| Patient 5 | B severe microphthalmia | None | 3q29 | Gain | 195383130–197550799 | 2.2 (1018) | SDHAP2, MUC20, MUC4, MIR570, TNK2, SDHAP1, TFRC, LOC401109, ZDHHC19, SLC51A, PCYT1A, TCTEX1D2, TM4SF19-TCTEX1D2, TM4SF19, UBXN7, RNF168, C3orf43, WDR53, FBXO45, LRRC33, CEP19, PIGX, PAK2, SENP5, NCBP2, LOC152217, PIGZ, MFI2-AS1, MFI2, DLG1, MIR4797, DLG1-AS1, BDH1, LOC220729, KIAA0226, MIR922, FYTTD1, LRCH3 | 3q29 duplication syndrome: developmental delay, obesity, heart defects, microcephaly, variable other features | Ocular anomalies (A/M, anterior segment dysgenesis) occasionally reported |
| Patient 6b | B microphthalmia, congenital cataract, glaucoma, retinal detachment, optic nerve hypoplasia | Mild microcephaly, hypotonia, history of gross motor delay, pituitary hypoplasia, ADHD | 11p13 | Gain | 31609664–32486962 | 0.88 (707) | ELP4, PAX6, DKFZp686K1684, RCN1, WT1, WT1-AS | Variable ocular defects (typically mild), facial dysmorphism, other features | Mice with Pax6 overexpression show embryonic eye defects |
| Patient 7b | R microphthalmia, iris coloboma, corneal leukoma | Neurofibromatosis type 1; B clinodactyly, redundant periumbilical skin | 17q11.2 | Loss | 28997779–30420528 | 1.4 (621) | SUZ12P1, CRLF3, CORPS, ATAD5, TEFM, ADAP2, RNF135, DPRXP4, MIR4733, NF1, OMG, EVI2B, EVI2A, RAB11FIP4, MIR4724, MIR193A, MIR4725, MIR365B, UTP6, SUZ12, LRRC37B, SH3GL1P1 | Neurofibromatosis type 1 | Ocular anomalies in some patients; Nf1 mutant mice have embryonic eye defects |
| Patient 8b | B microphthalmia, microcornea, congenital cataract | Microcephaly, developmental delays, short stature | 2q14.2 | Gain | 120078243–120623545 | 0.55 (298) | C2orf76, DBI, TMEM37, SCTR, PCDP1, TMEM177, PTPN4 | None | Nanophthalmos 3 locus; TMEM37, ptpn4a, dbi are expressed in lens |
| Patient 9 | B anophthalmia | Choanal atresia, broad nasal bridge, indented nasal tip | 2q21.1 | Gain | 130783696–131157859 | 0.37 (111) | POTEF CCDC74B-AS1, CCDC74B, SMPD4, MZT2B, TUBA3E CCDC115, IMP4, PTPN18 | None | SMPD4 enriched in the lens in mouse embryos |
| Patient 10 | R severe microphthalmia | None | 15q26.3 | Gain | 102025982–102311246 | 0.29 (183) | PCSK6, TM2D3, TARSL2 | Levy-Shanske (tetrasomy): facial dysmorphism, craniosynostosis and other defects | PCSK6 is enriched in the lens in mouse embryos |
number of probes indicative of CNV is specified in parentheses;
familial cases (details are provided in the text); regions/genes associated with likely pathogenic variants are shown in bold
Figure 2.

Likely pathogenic copy number variants involving regions with a known role in human disease. (a–c) Affymetrix Genome-Wide Human SNP Array 6.0 data for Patients 5–7; breakpoints are indicated with arrows. The UCSC Genome Browser (http://genome.ucsc.edu) view of the affected region indicating the positions of genes for each region (GRCh37/hg19 assembly). (a) 3q29 duplication in Patient 5; genes located within the minimum critical internal (~1.58 Mb) for 3q29 microduplication syndrome as defined by Goobie et al. 2008 (19) are indicated in bold font. (b) 11p13 duplication including PAX6 in Patient 6. (c) 17q11 deletion including NF1 in Patient 7.
Patient 5 is an 18-year-old male with isolated bilateral severe microphthalmia and normal development. CNV analysis identified a 2.2-Mb duplication of 3q29 (Figure 2a) which is not seen in the general population (DGV). This CNV is consistent with 3q29 microduplication, a syndrome which includes A/M or anterior segment ocular anomalies in some cases, typically in association with developmental delay/mental retardation and variable other anomalies (20,21).
Patient 6 is a 6-year-old female with bilateral microphthalmia, congenital cataract, glaucoma diagnosed at age 2 months, abnormal pupil (failure to dilate), optic nerve hypoplasia, and retinal detachment. Non-ocular anomalies include borderline microcephaly, hypotonia with history of gross motor delays (resolved), pituitary hypoplasia, and ADHD. The patient was found to carry an 877-kb duplication of 11p13 including the PAX6 gene (Figure 2b); this duplication was not observed in the general population (DGV). The patient’s mother was found to carry the same duplication and is affected with bilateral congenital cataract, glaucoma diagnosed at age 11 years, myopia, abnormal pupil (failure to dilate), and right corneal opacity; non-ocular anomalies include unilateral sensorineural hearing loss and short stature. The patient’s unaffected father and sister were found to have normal copy number at this region. There is a strong family history of congenital cataracts with or without glaucoma: the patient’s maternal uncle, maternal first cousin, maternal grandmother, three maternal great-aunts and uncles, the maternal great-grandmother, and maternal great-great-grandmother are all affected but were unavailable for testing.
Patient 7 is a 10-month-old male with Neurofibromatosis type 1 along with right microphthalmia, iris coloboma, and corneal leukoma as well as bilateral clinodactyly and redundant periumbilical skin. His mother and sister are also affected with Neurofibromatosis type 1 with no structural ocular anomalies. He was found to have a 1.4-Mb deletion of 17q11.2 including the NF1 gene (Figure 2c) that was not observed in the general population (DGV).
Patient 8 is a 2-year-old female with bilateral microphthalmia, microcornea, congenital cataract, and nystagmus along with microcephaly, developmental delays, mild hyperextensibility, and short stature. She was found to have a 545-kb duplication of 2q14.2 (Figure 3a). The full duplication is not seen in the general population (DGV); one case of a partial duplication of PCDP1 was noted (DGV). There is a strong family history with the patient’s brother affected with microphthalmia, microcornea, congenital cataracts, and nystagmus along with microcephaly and speech delay while the patient’s mother has microcornea, congenital cataract, nystagmus, glaucoma diagnosed at 16 years of age, and 2 missing lateral incisors; both the brother and mother carry the same 2q14.2 duplication (Figure 3a). There is no established phenotype associated with 2q14.2 duplication.
Figure 3.
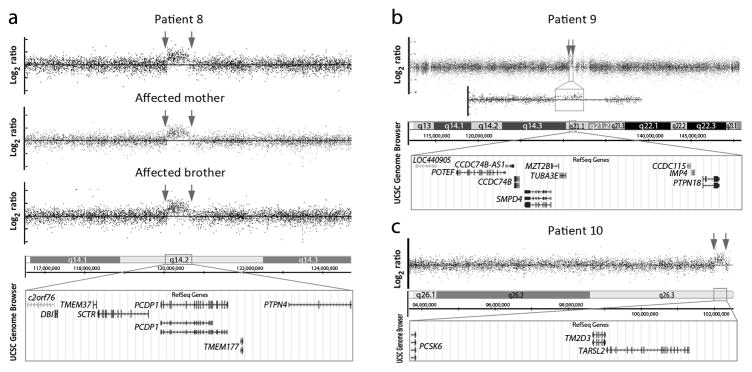
Copy number variants of uncertain significance with a possible role in human ocular disease. (a–c) Affymetrix Genome-Wide Human SNP Array 6.0 data for Patients 5–10; breakpoints are indicated with arrows. The UCSC Genome Browser (http://genome.ucsc.edu) view of the affected region indicating the position of genes for each region (GRCh37/hg19 assembly). (a) 2q14.2 duplication in Patient 8, her affected mother, and her affected brother. (b) 2q21.1 duplication in Patient 9; an enlargement of the duplicated region is also shown. (c) 15q26.3 duplication in Patient 10.
Patient 9 is a 1-week-old female with bilateral anophthalmia, choanal atresia, broad nasal bridge, and indented nasal tip. She was found to have a 374-kb duplication of 2q21.1 (Figure 3b), which is seen in <1% of the general population (DGV); copy number variation of this region is not well-characterized.
Patient 10 is a 40-year-old female with isolated right severe microphthalmia. She was found to carry a 285-kb duplication of 15q26.3 (Figure 3c), seen in <1% of the general population (DGV). Duplication and tetrasomy for the q26.1-q26.3 region is previously reported, associated with overgrowth, craniofacial dysmorphism, and severe developmental delay, but ocular anomalies have not been noted.
DISCUSSION
Our results are consistent with previous copy number variation studies of ocular developmental anomalies (including A/M, coloboma, congenital cataract, and anterior segment dysgenesis) which identified causative CNVs in 3–15% (22–24). Two of these studies also reported novel candidate regions; there was no overlap between the candidate regions presented in these studies and those identified here (22,23).
SOX2 disruption is the most common cause of A/M, accounting for 10–20% of cases (14), so the high frequency of SOX2 deletions (4/60; 7%) in our study is not surprising. The phenotypes reported here are typical for SOX2 anophthalmia syndrome (MIM:206900) (14); while hypoplastic uterus/ovaries are not commonly noted, this phenotype has been reported previously (25) and may be under recognized. Similar to previous studies, there is no correlation between the severity of the disease and deletion size, suggesting that other genes in the region are not contributing to the phenotype. Review of the other genes included in the deletions in Patient 1 and 2 support this conclusion. Only two genes have been associated with human disease in heterozygous form, neither of which are due to haploinsufficiency: heterozygous germline gain-of-function mutations in PIK3CA are associated with Cowden syndrome and somatic mosaic missense mutations in this gene are seen in other conditions (26) while heterozygous dominant-negative mutations in GNB4 cause Charcot-Marie-Tooth (27). Three genes are associated with recessive metabolic or ciliary dyskinesia phenotypes (CCDC39, DNAJC19, MCCC1) (28–30); the remaining genes have no reported human phenotype (http://omim.org/).
Ocular defects, including microphthlamia and anterior segment defects, have been occasionally reported in patients with 3q29 microduplications (MIM:611936), but most patients also have mental retardation/developmental delay and variable other anomalies (20,21). The duplication seen in Patient 5 overlaps the critical region defined by Goobie et al. 2008 (20) extending from TFRC to BDH1; while the full duplication was not seen in controls, the centromeric (TFRC) and telomeric (BDH1) portions of this region do appear to be polymorphic (DGV). Patient 5 reported here is the first case with isolated ocular anomalies and provides further evidence for the importance of this region in ocular development. DLG1 has been proposed as a likely candidate for the ocular findings (20); DLG1 is expressed in the developing lens and retinal pigment epithelium (31). Mutant mice with Dlg1 deficiency were found to have increased proliferation in the lens, along with growth deficiency, craniofacial abnormalities, kidney defects, and neonatal lethality (31,32).
Copy number mutations identified in Patients 6 and 7 that involve PAX6 and NF1, respectively, are likely to contribute to these patients’ phenotypes. PAX6 duplications (as seen in Patient 6) have been previously reported in association with eye anomalies, but not congenital cataracts and glaucoma as observed in this family. Most previous reports involve larger duplications identified cytogenetically including bands 11p12 and/or 11p14 in addition to 11p13. Ocular findings in these patients are highly variable with strabismus and nystagmus most commonly reported; microphthalmia, iris hypoplasia, eccentric pupils, and retinal defects have occasionally been reported as well (33). More recently, a patient with a 166-kb duplication of PAX6 and the first exon of ELP4 was reported; she was affected with seizures, developmental delay, microcephaly, ADHD and autistic features, and only minor ocular features, consisting of slight hypopigmentation of the fundi, accommodative esotropia with high hyperopia, astigmatism, amblyopia, and ptosis (34). Significant overexpression of PAX6 in a mouse model (5–7 and 10–45 copies) resulted in ocular defects ranging from iris and corneal defects to severe microphthalmia (35).
Deletion of NF1 causes Neurofibromatosis type 1 (NF1; MIM 162200), which was seen in Patient 7 in addition to the ocular phenotype (unilateral microphthalmia, iris coloboma, and corneal leukoma). The patient’s mother and sister are also affected with NF1 but without ocular anomalies. Deletion of NF1 is seen in ~5% of patients with NF1; the 1.4Mb deletion seen here is entirely consistent with the most commonly observed deletion (type-1). Patients with this deletion have been well-characterized with no report of structural ocular defects (36). At the same time, while structural eye defects are not typical of NF1 (37), one patient with NF1 with anterior segment anomalies (bilateral posterior embryotoxon and left Peters anomaly) and cerebral artery stenosis was reported; the authors postulated that the ocular defects may have been caused by vascular disruption during embryonic development (38). In addition, gonioscopic evaluation of nine NF1 patients by ultrasonic biomicroscopy suggested that almost half (8/19 eyes) had an abnormal (occludable) anterior chamber angle (39). Finally, homozygous deletion of Nf1 in mice results in lens dysgenesis; eye phenotypes included normal eyes (19%), anophthalmia or microphthalmia (28%), and absent lens with intact retina (53%); study of embryonic development suggested that Nf1 is required for lens vesicle formation (40). None of the other genes in the region are known to play a role in ocular development (http://omim.org/). Therefore, it is possible that deletion of the NF1 gene is not sufficient to produce an ocular phenotype unless combined with other ocular risk alleles, possibly inherited from the patient’s father in this case. Alternatively, the patient’s ocular phenotype may be due to a separate genetic (or environmental) cause unrelated to the NF1 deletion.
The 2q14.2 duplication seen in Patient 8 and two affected family members is not currently associated with an established phenotype but shows evidence of being causative for the phenotype in the family. The region overlaps with the Nanophthalmos 3 locus (MIM 611897), 2q11-q14, mapped in a family with simple microphthalmia, small corneas, and high hyperopia (41); there were no variations identified in the general population for this region. Several genes within the region are of potential interest: TMEM37 is enriched in the lens (42), ptpn4a in zebrafish showed expression in the retina during development (43), and dbi in zebrafish showed embryonic ocular expression (44). Further study is needed to determine the contribution of the 2q14.2 duplication to the ocular phenotype seen in the family. The final two variants of uncertain significance, 2q21.2 and 15q26.3 duplications, are occasionally seen in control populations (<1%) and thus may represent rare variants or potential susceptibility factors. Both regions contain genes enriched in the lens: SMPD4 in the 2q21.1 duplication and PCSK6 in the 15q26.3 duplication (42).
While interpretation of the clinical significance of copy number mutations that affect regions/genes of known ocular phenotypes is usually straightforward, the clinical relevance of novel structural genomic changes, or even of variants previously described in patients with a somewhat different ocular manifestation, is often uncertain without further studies. Analysis of additional family members may provide helpful information but needs to be considered with caution since many pathogenic CNVs were found to display variable expressivity or incomplete penetrance (20,45,46). Additional investigation of the regions reported in this paper in human patient populations with similar phenotypes and/or animal models will allow for better understanding of their potential role in human disease and may lead to the identification of novel developmental ocular factors.
Acknowledgments
The authors are thankful to Rebecca C. Tyler for executing TaqMan assays on select regions and Rachel Lorier, Stephen Hall, Katie Felhofer and Andrea Lenarduzzie for assistance with Affymetrix array CNV analysis. The authors also gratefully acknowledge the patients and their families for their participation in research studies. This work was supported by the National Institutes of Health awards R01EY015518 (EVS), R21DC010912 (EVS) and funds provided by the Children’s Hospital of Wisconsin (EVS), along with 1UL1RR031973 from the Clinical and Translational Science Award (CTSA) program.
Footnotes
THE AUTHORS DECLARE NO CONFLICT OF INTEREST.
References
- 1.Clementi M, Tenconi R, Bianchi F, Botto L, Calabro A, Calzolari E, Cianciulli D, Mammi I, Mastroiacovo P, Meli P, Spagnolo A, Turolla L, Volpato S. Congenital eye malformations: a descriptive epidemiologic study in about one million newborns in Italy. Birth Defects Orig Artic Ser. 1996;30 (1):413–424. [PubMed] [Google Scholar]
- 2.Kallen B, Robert E, Harris J. The descriptive epidemiology of anophthalmia and microphthalmia. Int J Epidemiol. 1996;25 (5):1009–1016. doi: 10.1093/ije/25.5.1009. [DOI] [PubMed] [Google Scholar]
- 3.Forrester MB, Merz RD. Descriptive epidemiology of anophthalmia and microphthalmia, Hawaii, 1986–2001. Birth Defects Res A Clin Mol Teratol. 2006;76 (3):187–192. doi: 10.1002/bdra.20237. [DOI] [PubMed] [Google Scholar]
- 4.Verma AS, Fitzpatrick DR. Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007;2:47. doi: 10.1186/1750-1172-2-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bardakjian TM, Schneider A. The genetics of anophthalmia and microphthalmia. Curr Opin Ophthalmol. 2011;22 (5):309–313. doi: 10.1097/ICU.0b013e328349b004. [DOI] [PubMed] [Google Scholar]
- 6.Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14 (4):392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- 7.Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR. Mutations in SOX2 cause anophthalmia. Nat Genet. 2003;33 (4):461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- 8.Lesnik Oberstein SA, Kriek M, White SJ, Kalf ME, Szuhai K, den Dunnen JT, Breuning MH, Hennekam RC. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am J Hum Genet. 2006;79 (3):562–566. doi: 10.1086/507567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36 (9):955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 10.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7 (2):85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 11.Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007;39 (7 Suppl):S48–54. doi: 10.1038/ng2092. [DOI] [PubMed] [Google Scholar]
- 12.Reis LM, Tyler RC, Schilter KF, Abdul-Rahman O, Innis JW, Kozel BA, Schneider AS, Bardakjian TM, Lose EJ, Martin DM, Broeckel U, Semina EV. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum Genet. 2011;130 (4):495–504. doi: 10.1007/s00439-011-0968-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bardakjian TM, Kwok S, Slavotinek AM, Schneider AS. Clinical report of microphthalmia and optic nerve coloboma associated with a de novo microdeletion of chromosome 16p11. 2. Am J Med Genet A. 2010;152A (12):3120–3123. doi: 10.1002/ajmg.a.33492. [DOI] [PubMed] [Google Scholar]
- 14.Schneider A, Bardakjian T, Reis LM, Tyler RC, Semina EV. Novel SOX2 mutations and genotype-phenotype correlation in anophthalmia and microphthalmia. Am J Med Genet A. 2009;149A (12):2706–2715. doi: 10.1002/ajmg.a.33098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schilter KF, Schneider A, Bardakjian T, Soucy JF, Tyler RC, Reis LM, Semina EV. OTX2 microphthalmia syndrome: four novel mutations and delineation of a phenotype. Clin Genet. 2011;79 (2):158–168. doi: 10.1111/j.1399-0004.2010.01450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reis LM, Tyler RC, Volkmann Kloss BA, Schilter KF, Levin AV, Lowry RB, Zwijnenburg PJ, Stroh E, Broeckel U, Murray JC, Semina EV. PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur J Hum Genet. 2012;20 (12):1224–1233. doi: 10.1038/ejhg.2012.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reis LM, Tyler RC, Schneider A, Bardakjian T, Stoler JM, Melancon SB, Semina EV. FOXE3 plays a significant role in autosomal recessive microphthalmia. Am J Med Genet A. 2010;152A (3):582–590. doi: 10.1002/ajmg.a.33257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, Hessner MJ, Verbsky J. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360 (23):2438–2444. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13 (7):680–685. doi: 10.1097/GIM.0b013e3182217a3a. [DOI] [PubMed] [Google Scholar]
- 20.Goobie S, Knijnenburg J, Fitzpatrick D, Sharkey FH, Lionel AC, Marshall CR, Azam T, Shago M, Chong K, Mendoza-Londono R, den Hollander NS, Ruivenkamp C, Maher E, Tanke HJ, Szuhai K, Wintle RF, Scherer SW. Molecular and clinical characterization of de novo and familial cases with microduplication 3q29: guidelines for copy number variation case reporting. Cytogenet Genome Res. 2008;123 (1–4):65–78. doi: 10.1159/000184693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ballif BC, Theisen A, Coppinger J, Gowans GC, Hersh JH, Madan-Khetarpal S, Schmidt KR, Tervo R, Escobar LF, Friedrich CA, McDonald M, Campbell L, Ming JE, Zackai EH, Bejjani BA, Shaffer LG. Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal microduplication. Mol Cytogenet. 2008;1:8-8166-1-8. doi: 10.1186/1755-8166-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raca G, Jackson CA, Kucinskas L, Warman B, Shieh JT, Schneider A, Bardakjian TM, Schimmenti LA. Array comparative genomic hybridization analysis in patients with anophthalmia, microphthalmia, and coloboma. Genet Med. 2011;13 (5):437–442. doi: 10.1097/GIM.0b013e318204cfd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delahaye A, Bitoun P, Drunat S, Gerard-Blanluet M, Chassaing N, Toutain A, Verloes A, Gatelais F, Legendre M, Faivre L, Passemard S, Aboura A, Kaltenbach S, Quentin S, Dupont C, Tabet AC, Amselem S, Elion J, Gressens P, Pipiras E, Benzacken B. Genomic imbalances detected by array-CGH in patients with syndromal ocular developmental anomalies. Eur J Hum Genet. 2012;20 (5):527–533. doi: 10.1038/ejhg.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balikova I, de Ravel T, Ayuso C, Thienpont B, Casteels I, Villaverde C, Devriendt K, Fryns JP, Vermeesch JR. High Frequency Of Submicroscopic Chromosomal Deletions in Patients with Idiopathic Congenital Eye Malformations. Am J Ophthalmol. 2011;151 (6):1087–1094. e45. doi: 10.1016/j.ajo.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 25.Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson IC, Dattani MT. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest. 2006;116 (9):2442–2455. doi: 10.1172/JCI28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orloff MS, He X, Peterson C, Chen F, Chen JL, Mester JL, Eng C. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am J Hum Genet. 2013;92 (1):76–80. doi: 10.1016/j.ajhg.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soong BW, Huang YH, Tsai PC, Huang CC, Pan HC, Lu YC, Chien HJ, Liu TT, Chang MH, Lin KP, Tu PH, Kao LS, Lee YC. Exome Sequencing Identifies GNB4 Mutations as a Cause of Dominant Intermediate Charcot-Marie-Tooth Disease. Am J Hum Genet. 2013;92 (3):422–430. doi: 10.1016/j.ajhg.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merveille AC, Davis EE, Becker-Heck A, Legendre M, Amirav I, Bataille G, Belmont J, Beydon N, Billen F, Clement A, Clercx C, Coste A, Crosbie R, de Blic J, Deleuze S, Duquesnoy P, Escalier D, Escudier E, Fliegauf M, Horvath J, Hill K, Jorissen M, Just J, Kispert A, Lathrop M, Loges NT, Marthin JK, Momozawa Y, Montantin G, Nielsen KG, Olbrich H, Papon JF, Rayet I, Roger G, Schmidts M, Tenreiro H, Towbin JA, Zelenika D, Zentgraf H, Georges M, Lequarre AS, Katsanis N, Omran H, Amselem S. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat Genet. 2011;43 (1):72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davey KM, Parboosingh JS, McLeod DR, Chan A, Casey R, Ferreira P, Snyder FF, Bridge PJ, Bernier FP. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J Med Genet. 2006;43 (5):385–393. doi: 10.1136/jmg.2005.036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gallardo ME, Desviat LR, Rodriguez JM, Esparza-Gordillo J, Perez-Cerda C, Perez B, Rodriguez-Pombo P, Criado O, Sanz R, Morton DH, Gibson KM, Le TP, Ribes A, de Cordoba SR, Ugarte M, Penalva MA. The molecular basis of 3-methylcrotonylglycinuria, a disorder of leucine catabolism. Am J Hum Genet. 2001;68 (2):334–346. doi: 10.1086/318202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen MM, Nguyen ML, Caruana G, Bernstein A, Lambert PF, Griep AE. Requirement of PDZ-containing proteins for cell cycle regulation and differentiation in the mouse lens epithelium. Mol Cell Biol. 2003;23 (24):8970–8981. doi: 10.1128/MCB.23.24.8970-8981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahoney ZX, Sammut B, Xavier RJ, Cunningham J, Go G, Brim KL, Stappenbeck TS, Miner JH, Swat W. Discs-large homolog 1 regulates smooth muscle orientation in the mouse ureter. Proc Natl Acad Sci U S A. 2006;103 (52):19872–19877. doi: 10.1073/pnas.0609326103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aalfs CM, Fantes JA, Wenniger-Prick LJ, Sluijter S, Hennekam RC, van Heyningen V, Hoovers JM. Tandem duplication of 11p12-p13 in a child with borderline development delay and eye abnormalities: dose effect of the PAX6 gene product? Am J Med Genet. 1997;73 (3):267–271. doi: 10.1002/(sici)1096-8628(19971219)73:3<267::aid-ajmg7>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 34.Aradhya S, Smaoui N, Marble M, Lacassie Y. De novo duplication 11p13 involving the PAX6 gene in a patient with neonatal seizures, hypotonia, microcephaly, developmental disability and minor ocular manifestations. Am J Med Genet A. 2011;155 (2):442–444. doi: 10.1002/ajmg.a.33814. [DOI] [PubMed] [Google Scholar]
- 35.Schedl A, Ross A, Lee M, Engelkamp D, Rashbass P, van Heyningen V, Hastie ND. Influence of PAX6 gene dosage on development: overexpression causes severe eye abnormalities. Cell. 1996;86 (1):71–82. doi: 10.1016/s0092-8674(00)80078-1. [DOI] [PubMed] [Google Scholar]
- 36.Mautner VF, Kluwe L, Friedrich RE, Roehl AC, Bammert S, Hogel J, Spori H, Cooper DN, Kehrer-Sawatzki H. Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1. 4 Mb type-1 NF1 deletions. J Med Genet. 2010;47 (9):623–630. doi: 10.1136/jmg.2009.075937. [DOI] [PubMed] [Google Scholar]
- 37.Schnur RE. Type I neurofibromatosis: a geno-oculo-dermatologic update. Curr Opin Ophthalmol. 2012;23 (5):364–372. doi: 10.1097/ICU.0b013e3283570127. [DOI] [PubMed] [Google Scholar]
- 38.Smith M, Heran MK, Connolly MB, Heran HK, Friedman JM, Jett K, Lyons CJ, Steinbok P, Armstrong L. Cerebrovasculopathy in NF1 associated with ocular and scalp defects. Am J Med Genet A. 2011;155A (2):380–385. doi: 10.1002/ajmg.a.33788. [DOI] [PubMed] [Google Scholar]
- 39.Emre S, Palamar M, Ulusoy MO, Gencoglan G. Ciliary body cysts in neurofibromatosis: a new coexistence? Graefes Arch Clin Exp Ophthalmol. 2012;250 (6):857–861. doi: 10.1007/s00417-011-1830-6. [DOI] [PubMed] [Google Scholar]
- 40.Carbe C, Zhang X. Lens induction requires attenuation of ERK signaling by Nf1. Hum Mol Genet. 2011;20 (7):1315–1323. doi: 10.1093/hmg/ddr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Wang JX, Wang CY, Yu P, Zhou Q, Chen YG, Zhao LH, Zhang YP. Localization of a novel gene for congenital nonsyndromic simple microphthalmia to chromosome 2q11-14. Hum Genet. 2008;122 (6):589–593. doi: 10.1007/s00439-007-0435-y. [DOI] [PubMed] [Google Scholar]
- 42.Lachke SA, Ho JW, Kryukov GV, O’Connell DJ, Aboukhalil A, Bulyk ML, Park PJ, Maas RL. iSyTE: integrated Systems Tool for Eye gene discovery. Invest Ophthalmol Vis Sci. 2012;53 (3):1617–1627. doi: 10.1167/iovs.11-8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Eekelen M, Overvoorde J, van Rooijen C, den Hertog J. Identification and expression of the family of classical protein-tyrosine phosphatases in zebrafish. PLoS One. 2010;5 (9):e12573. doi: 10.1371/journal.pone.0012573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thisse B, Thisse C. Fast Release Clones: A High Throughput Expression Analysis. ZFIN Direct Data Submission. 2004 ( http://zfin.org)
- 45.Delahaye A, Pipiras E, Benzacken B. Chromosomal microarray analysis in ocular developmental anomalies. Expert Rev Mol Diagn. 2012;12 (5):425–427. doi: 10.1586/erm.12.41. [DOI] [PubMed] [Google Scholar]
- 46.Hoppman-Chaney N, Wain K, Seger P, Superneau D, Hodge J. Identification of single gene deletions at 15q13.3: further evidence that CHRNA7 causes the 15q13.3 microdeletion syndrome phenotype. Clin Genet. 2012 doi: 10.1111/j.1399-0004.2012.01925.x. [DOI] [PubMed] [Google Scholar]