

Abstract
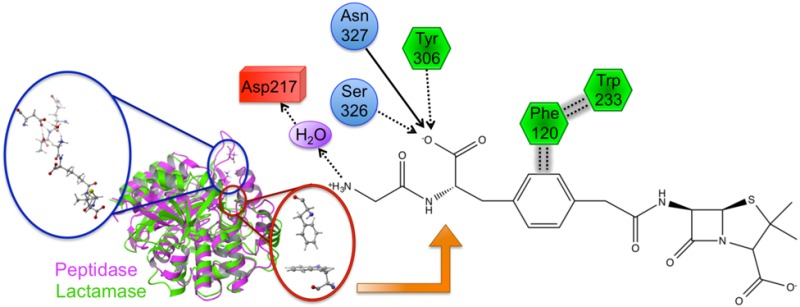
Bacterial resistance to standard (i.e., β-lactam-based) antibiotics has become a global pandemic. Simultaneously, research into the underlying causes of resistance has slowed substantially, although its importance is universally recognized. Key to unraveling critical details is characterization of the noncovalent interactions that govern binding and specificity (DD-peptidases, antibiotic targets, versus β-lactamases, the evolutionarily derived enzymes that play a major role in resistance) and ultimately resistance as a whole. Herein, we describe a detailed investigation that elicits new chemical insights into these underlying intermolecular interactions. Benzylpenicillin and a novel β-lactam peptidomimetic complexed to the Stremptomyces R61 peptidase are examined using an arsenal of computational techniques: MD simulations, QM/MM calculations, charge perturbation analysis, QM/MM orbital analysis, bioinformatics, flexible receptor/flexible ligand docking, and computational ADME predictions. Several key molecular level interactions are identified that not only shed light onto fundamental resistance mechanisms, but also offer explanations for observed specificity. Specifically, an extended π–π network is elucidated that suggests antibacterial resistance has evolved, in part, due to stabilizing aromatic interactions. Additionally, interactions between the protein and peptidomimetic substrate are identified and characterized. Of particular interest is a water-mediated salt bridge between Asp217 and the positively charged N-terminus of the peptidomimetic, revealing an interaction that may significantly contribute to β-lactam specificity. Finally, interaction information is used to suggest modifications to current β-lactam compounds that should both improve binding and specificity in DD-peptidases and their physiochemical properties.
1. Introduction
The bacterial cell wall has fascinated scientists dating back to the early 20th century.1 Its absence in mammalian cells has created an invaluable target for antibiotics research. In 1965, Wise and Park2 and, independently, Tipper and Strominger3 suggested that β-lactam antibiotics, such as penicillin, target d-alanyl-d-alanine transpeptidases (DD-peptidases). Quickly following this proposal, it was demonstrated that these antibiotics prevent the transpeptidation reaction responsible for cross-linking peptidoglycan strands, the polymer that constructs the bacterial cell wall.4 This is accomplished when the β-lactam substrate and the DD-peptidase form a long-lasting acyl enzyme intermediate that can ultimately lead to cell death. (Figure 1)5−8
Figure 1.

The acylation and deacylation reactions are shown for a generalized class of β-lactam antibiotics and a serine protease type enzyme.
β-lactamases, said to predate the antibiotic era, are an evolutionary competitive enzyme class deployed by bacteria to inactivate β-lactam compounds through hydrolysis. β-lactamase enzymes are organized into four classes. Class A, C, and D β-lactamases are serine protease enzymes that confer resistance by structurally similar active sites as compared to DD-peptidases, whereas class B β-lactamases almost always require a divalent zinc ion; hence a different mechanism of β-lactam inactivation is employed for this class.9 Herein, we will focus on the perpetual evolutionary competition between DD-peptidases and β-lactamases.
Penicillin and cephalosporin (i.e., penams and cephams, respectively) derivatives are constituents of the β-lactam antibiotic class. These two classes have a five or six membered sulfur containing ring fused to a four membered β-lactam ring. Varying the framework’s substituents has led to the formation of many broad spectrum antibiotics.10 In their original 1965 work, Tipper and Strominger also suggested that a peptidomimetic substituent attached to a β-lactam framework would increase activity due to its similarity to a DD-peptidase’s natural substrate (Figure 2a).3 However, until recently, this approach had limited success. The breakthrough occurred when structural and kinetic data was reported11,12 examining the effects of a peptidomimetic substrate bound to Streptomyces R61, a low molecular mass bacterial peptidase here after referred to as R61, and Enterobacter cloacae P99 (P99), a class C β-lactamase. The peptidomimetic β-lactam rate of inhibition (ki) is 4 orders of magnitude larger in R61 compared to P99. Furthermore, benzylpenicillin’s (PENG) ki, the previously most effective β-lactam (Figure 2b), is 3 orders of magnitude less compared to the peptidomimetic’s ki. This peptidomimetic β-lactam antibiotic was whimsically named “perfect penicillin” (PPEN) due to its remarkable kinetics (Figure 2c). A “perfect cephalosporin” (PCEPH) was also synthesized (Figure 2d); however, it was not as potent as its penam counterpart. Silvaggi et al.11 cocrystallized this peptidomimetic β-lactam inhibitor with R61.
Figure 2.

(a) Natural substrate for a DD-peptidase. (b) Benzylpenicillin (PENG), also known as penicillin G. (c) Perfect penicillin (PPEN), a substrate that combines a β-lactam/penam framework and the natural substrate. (d) Perfect cephalosporin (PCEPH), a substrate that combines a β-lactam/cepham framework and the natural substrate.
Key structural and functional group features responsible for PPEN’s improved kinetics are characterized in the present work. The perfect penicillin–R61 protein–ligand complex (PPEN–R61) was compared to the benzylpenicillin–R61 protein–ligand complex (PENG–R61) to determine advantages that arise from the peptidomimetic moiety. These complexes were examined with three goals in mind:
-
1.
Identify protein–ligand interactions that govern binding and specificity of β-lactam inhibitors.
-
2.
Elucidate structural differences that contribute to common antibiotic resistance mechanisms.
-
3.
Propose structural modifications of β-lactam inhibitors that take advantage of newly identified protein–ligand information.
To accomplish this, we employ a panoply of computational techniques ranging from virtual screening, molecular dynamics (MD) simulations, hybrid quantum mechanical/molecular mechanical (QM/MM) calculations, charge perturbation analysis (CPA), QM/MM Natural Bond Orbital (NBO) analysis, and bioinformatics. Novel insights into active site flexibility and electrostatics, orbital stabilization, and structure–activity relationships are revealed.
2. Computational Details
2.1. Molecular Dynamics and QM/MM Minimization
The cocrystallized structures of benzylpenicillin and perfect penicillin covalently bound to DD-peptidase Streptomyces R61 (PDB ID: 1PWC and 1PWG, respectively) were used throughout.11 Initial processing of PDB coordinates was done with www.charmming.org.13 Structures were manually back mutated to the noncovalent preacylated forms and classical parameter and topology files for PENG and PPEN were obtained via www.paramchem.org with subsequent manual optimization.14,15 CHARMM16 c37a1 was used to prepare the protein, add hydrogen atoms, and assign protonation states of ionizable residues. His298 was assigned to be protonated in accordance with work by Friesner and co-workers.17 Lys65 was treated as a natural base as determined by PROPKA.18−20 Additionally, a disulfide bridge was added between Cys291 and Cys344. The CHARMM2221 protein and CHARMM3622 generalized force fields (C22 and CGenFF) were used. For the 11 ns MD simulations, the domain decomposition (DOMDEC) parallelization package in CHARMM was used. For production runs, the system was solvated in a cubic box and was equilibrated at constant pressure (1 atm) and temperature (310.15 K). See the Supporting Information for all setup and simulation details.
Subsequently, the full system was minimized using QM/MM23 until a 0.005 kcal mol–1 Å–1 rms gradient tolerance was achieved. PENG/PPEN were treated quantum mechanically during optimization at the B3LYP/6-31G* level of theory,24,25 which has been proven to be a reasonable methodology for ground state geometries compared to dispersion corrected methods for QM/MM.26 The remainder of the system was unrestrained and treated using the C22 force field.
2.2. Charge Perturbation and Natural Bond Orbital Analyses
The charge perturbation analysis (CPA) technique27−29 was used to gain insight into the R61 active site. This involves QM/MM single point calculations where a single residue’s charges are scaled to zero. ΔE is computed by taking the difference of the modified (zero-charge residue) and the full QM/MM electronic energy ΔE = EelecZeroChargeRes (QM/MM) – Eelec (QM/MM). The energetic differences between PPEN–R61 and PENG–R61 can also be determined: ΔΔE = ΔEPPEN – ΔEPENG.
Additionally, QM/MM NBO30−32 was performed on all systems to gain qualitative insight into orbital interactions. Definition of QM regions and link atom placement in NBO calculations are listed in Supporting Information. The total orbital stabilization will be reported as a percentage of the total NBO interactions for each respective PENG/PPEN–R61 complex. All computations were carried out with Q-Chem/CHARMM16 + NBO, using CHARMM version c37a1,16 Q-Chem 4.0,33 and NBO 5.0.34
2.3. Bioinformatics and Virtual Screening
ProBiS, a structure based binding site similarity prediction algorithm,35−37 was applied to 1PWG (PPEN) and 1PWC (PENG). The Z-score, a standard deviation based similarity metric35 was used to quantify results with Z > 2.00 signifying the top 2% of all pairwise alignments. A list of all PDB IDs and corresponding Z-scores are provided in Supporting Information. In addition to ProBiS, Clustal Omega38,39 was used for multiple sequence alignment. FASTA sequences from the PDB were uploaded and aligned; alignment gaps were not removed.
The PubChem40 compound database was used in conjunction with Ligprep41 from Schrödinger to create the diversity set used in the virtual screening studies. Penam and cepham pharmacophores (Figure 3) were used (see the Supporting Information for more details). Grid-based ligand docking with energetics (GLIDE) 5.8 program was used for grid generation and ligand docking studies. Ligands were first evaluated by Glide Standard precision (GlideSP)42 followed by rescoring with the Glide Extra precision (GlideXP)43 function to predict approximate relative binding free energies represented by Glide scores (G-scores). Duplicate ligands were removed; namely different protonation states with a lower docking score compared to their respective top scoring pose. Furthermore, active site flexibility was accounted for using the flexible-ligand/flexible-receptor induced fit docking (IFD) protocol. Top scoring GlideXP ligands (G-score < −20 kcal mol–1) were reduced using IFD; see the Supporting Information for full details. Absorption, distribution, metabolism, and excretion (ADME) properties were predicted using QikProp.44 Qikprop determines pharmaceutically relevant information and physical descriptors for organic compounds. The molecule’s properties are compared to recommended ranges, which are determined from 95% of known drugs.
Figure 3.

Pharmacophore used to generate penam (left) and cepham (right) analogs.
3. Results and Discussion
3.1. Demystifying the Peptidomimetic Advantages
One of the present work’s main goals is to identify protein–ligand interactions that govern binding and specificity of β-lactam inhibitors. To accomplish this task, the QM/MM PPEN/PENG–R61 complexes were compared to identify and characterize different stabilizing features. There are two main structural domains of β-lactam antibiotics: the fused ring scaffold and the “tail” section. The fused ring architecture consists of the β-lactam ring fused to a five/six membered ring, whereas the tail region originates at N2 and is terminated at the ammonio group (N4H3+) and the phenyl ring for PPEN and PENG, respectively. The protein–ligand complex variation is primarily derived from the tail region due to PPEN and PENG’s identical fused ring scaffold.
The advantages of the PPEN’s peptidomimetic tail and PENG’s phenyl ring were first explored by CPA. CPA estimates the electrostatic effect of a single active site residue in a protein–ligand complex. This method is employed with a primary objective in mind; to identify stabilizing/destabilizing active site residues in the PPEN– and PENG–R61 complexes. In Figure 4, negative ΔΔE values indicate that a residue is more stabilizing in PPEN–R61, contrastingly positive ΔΔE values denote stabilizations in PENG–R61. Twenty-five active site residues were examined using CPA due to their proximity to the ligands; seven have a ΔΔE ≥ |10 kcal mol–1|, indicating a different structure–activity role between the complexes. Trp233 was more stabilizing in PENG–R61, whereas Phe120, Asp217, Arg285, Tyr306, Ser326, and Asn327 were more stabilizing in PPEN–R61.
Figure 4.
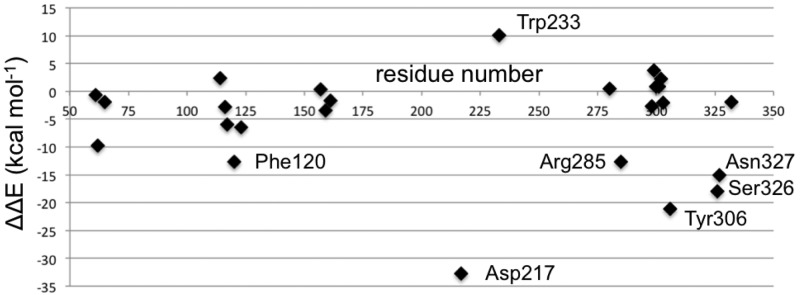
A visual representation of the ΔΔE values in the CPA analysis. This depicts which residues have a greater on impact PPEN or PENG. Negative values indicate the residue is more stabilizing in the PPEN–R61 system, where as positive values show stabilization in PENG–R61.
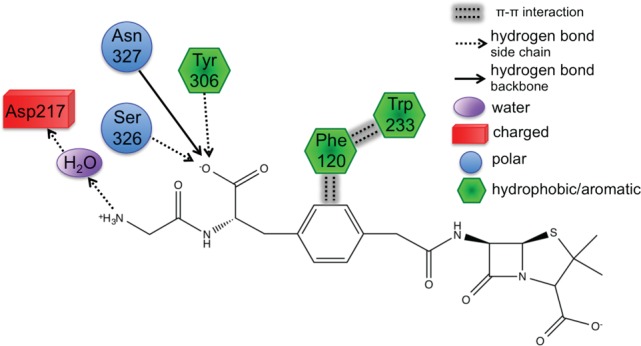
First, Trp233 (the only residue that preferentially stabilizes PENG over PPEN, ΔΔE = 10.0 kcal mol–1) was examined to characterize advantageous protein–ligand interactions. Despite Trp233’s nearly identical location in both proteins, this residue anchors a π–π network that exists solely in PENG–R61. This organization stems from parallel-displaced (standard nomenclature used45−47) π–π stacking between PENG’s phenyl group and the Phe120 side chain (Figure 5, right). The Phe120 then forms an edge-face π–π interaction with Trp233, whereas PPEN lacks these aromatic elements to assist in its stabilization. Although Phe120 plays a critical role in this π–π network, CPA reveals it is more stable in PPEN–R61 than PENG–R61 (ΔΔE = −12.6 kcal mol–1). This is attributed to a weak interaction between Phe120s backbone carbonyl and the CH2 group adjacent to PPEN’s terminal ammonio group (N4H3+). Crystal structures of R61 complexed to a peptide fragment illustrate precedence for this stabilization48,49 with NBO revealing it (Phe120 lone pair (LP) O → σ* C–H) to be approximately half the strength of a gas phase water dimer. To further examine the stability of this network, MD simulations were performed (11 ns). The PENG–Phe120–Trp233 π–π network was maintained over the duration of the simulation with few exceptions (Figure 5, left). The simulation’s measured distances are similar to the QM/MM minimized and crystal structures, which confirms the importance of this network.
Figure 5.
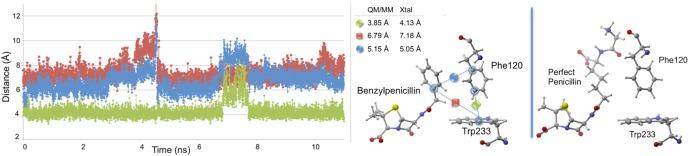
Left: distance analysis of π–π network stability is monitored and compared to the QM/MM minimized and crystal structures. The PENG–Phe120 (blue), Phe120–Trp233 (green), and PENG–Trp233 (red) distances are measured for the PENG–R61 11 ns MD simulation. Right: the PPEN–Phe120–Trp233 π–π network is absent due to PPEN’s butyl moiety versus the benzyl moiety of PENG.
The second aim of this investigation is to use peptidase and lactamase active site stabilization information to shed light on the structural basis of common antibacterial resistance mechanisms. To this end, a ProBiS binding site search was performed on the PPEN–R61 complex. 280 similar structures were found of which 24 had a Z-score over 2.00. Multiple sequence alignment of these 24 was performed to examine conservation in these evolutionarily competitive enzymes. Results showed no significant conservation of aromatic residues in this active site pocket (see the Supporting Information). Hence, structural analysis is used to gain more insight into these enzymes. The 24 top scoring proteins were structurally aligned50 with PPEN–R61 and inspected. Results indicate that both peptidases and lactamases have aromatic residue(s) that could form similar π–π networks as seen in R61. Phenylalanine and tryptophan are more commonly found in peptidases, whereas tyrosine is more prevalent in lactamases (see the Supporting Information).
Overall, this provides significant new insights into the role of Phe120 and Trp233 in peptidases; the importance of which has been well documented,48,49,51,52 although the underlying causes, until now, have been unknown. Trp233 is particularly interesting as its mutation to Ser leads to an unstable and poorly active enzyme whereas W233L greatly increases the deacylation rate of β-lactams (300-fold) hence conferring β-lactamase-like activity.52 In fact, sequence alignment results show that Trp233 is largely conserved as a Leu in β-lactamases (see the Supporting Information). This highlights its importance not just for peptidases, but also for understanding the structural basis of resistance mechanisms.
The other half of the π–π network, Phe120, plays a possibly more important biological role. For example, it is known that R61’s Arg285 is critical for stabilizing both native substrates and inhibitors (e.g., PENG); however, the lack of this residue in β-lactamases does not prevent hydrolysis.11,12,49 Current results indicate that the structurally conserved aromatic moiety in β-lactamases is critical to overcoming the lack of this Arg285 stabilization. This has wide-ranging implications as conservation of this protein–ligand π–π intermolecular interaction could be a major reason why β-lactamases are so efficient.
Several residues are significantly more stabilizing in PPEN–R61 compared to PENG–R61: Asp217, Tyr306, Ser326, and Asn327. All of these protein–ligand interactions are located in the peptidomimetic binding region of the active site. ΔΔE is broken into two components, ΔEPPEN and ΔEPENG. For nearly all of the residues located in PPEN’s peptidomimetic region, ΔEPENG is negligible, indicating minimal electrostatic effects on the ligand. This differs for Asp217, which has a ΔEPPEN and ΔEPENG contribution of 14.3 and −18.5 kcal mol–1, respectively (ΔEPENG is destabilizing, Table 1—Supporting Information). This destabilization results from an extended unfavorable dipole–dipole network, which consists of a water buffer sandwiched between Asp217 and benzylpenicillin’s aromatic moiety. The ΔEPPEN stabilization is a result of a water mediated salt bridge (Figure 6a) formed between PPEN’s terminal N4H3+, water, and the carboxylate on Asp217. The N4···Asp217 OD1 distance was monitored over the course of a PPEN–R61 11 ns MD simulation. This interaction is not expected to be as ubiquitous as the PENG–Phe120–Trp233 π–π network due to R61’s secondary structure. Asp217 resides in a surface exposed loop, whereas Phe120 exists in an α-helix. Despite differences in secondary structure, PPEN’s interaction with Asp217 remains largely intact, and similar to that of the QM/MM structure (rN4–OD1 = 4.94 Å), throughout the simulation (Figure 6b).
Figure 6.
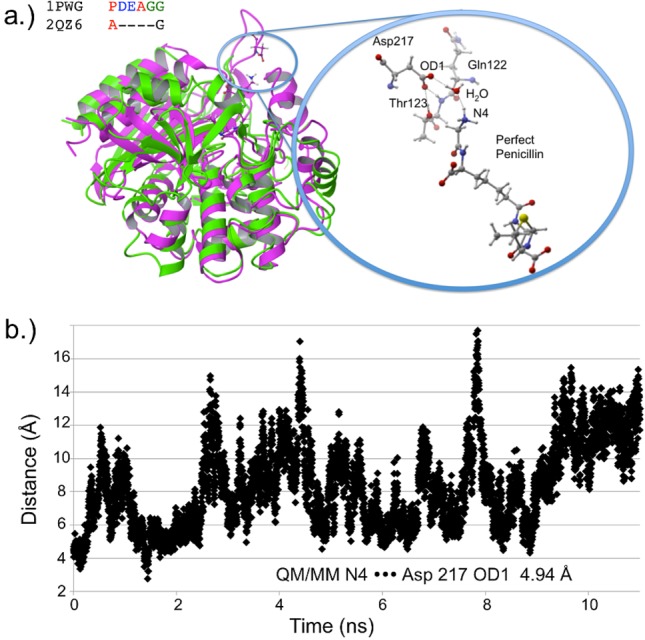
(a) PDB 1PWG (purple) and β-lactamase PDB 2QZ6 (green, Z-score = 2.60) are structurally aligned. The DD-peptidase has the domain housing Asp217, whereas the β-lactamase does not. The closeup structure shows active site detail including neighboring residues Thr123 and Gln122. (b) The distance is measured between N4 and Asp217’s OD1 for the 11 ns PPEN–R61 MD simulation.
As with the π–π network, Asp217 was examined to gain insight into possible structure–resistance relationships. Once again, alignment results showed no sequence conservation in either DD-peptidases or β-lactamases. However, the loop (domain containing Asp217 in R61) is somewhat conserved in peptidases, whereas being completely absent in all β-lactamases (see the Supporting Information). Structural alignment was again performed to examine possible molecular level interactions. In most cases, peptidases possess both the relevant domain and either an Asp or Glu capable of forming the interaction with N4. Lactamases, in contrast, do not contain this loop, thus PPEN’s (or a peptidase native substrate) terminal N4 would be completely solvent exposed, making significant protein–ligand stabilization highly unlikely. This suggests that β-lactams that form an interaction with Asp217, such as a peptidomimetic, could increase both binding and specificity to DD-peptidases rather than class C β-lactamases. Previous experimental studies of R61 with both PENG/PPEN11,12 and native substrates48,49 provide evidence that supports this hypothesis.
CPA pinpoints Tyr306, Ser326, and Asn327 (Figure 4) that all act as hydrogen bond donors to PPEN’s carboxylate 2 group, which contributes significant electrostatic stabilization to the PPEN–R61 system. The Tyr306 side chain (PPEN LP O5 → Tyr 306 σ* O–H), Ser326 side chain (PPEN LP O5 → Ser 326 σ* O–H), and Asn327 backbone amide (PPEN LP O5 → Asn 327 σ* N–H) interact with O5 of PPEN and constitute 13% of all orbital stabilization gained from direct protein–ligand interactions (Figure 7). Carboxylate 2 is further stabilized by water molecules that also serve as hydrogen bond donors (PPEN LP O5/O6 → H2O σ* O–H). O5 and O6 accept one and four hydrogen bonds from water molecules, respectively, which accounts for an additional 17% of the PPEN–R61 orbital stabilization. Of particular interest is the Tyr306–O5 hydrogen bond, which accounts for 8% of the total NBO stabilization.
Figure 7.
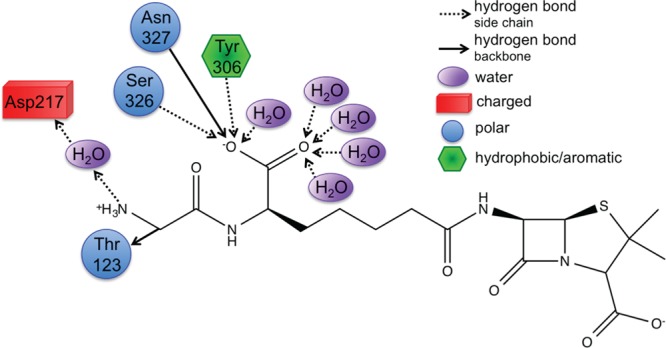
Tyr306, Ser326, Asn327, and water form a hydrophilic pocket that strongly interact with carboyxlate 2 of perfect penicillin. Asp217 forms a water mediated salt bridge with N4.
Sequence and structure alignment were again employed to gain further insight into resistance mechanisms. In contrast to previous interactions, the hydrophilic pocket defined by Tyr306–Ser326–Asn327 did show some type conservation in both β-lactamases and DD-peptidases (i.e., potential hydrogen bond donors). Results from MD simulations were analyzed to confirm the stability of these protein–ligand interactions. Unlike the π–π and Asp217 interactions, the PPEN carboxylate 2–hydrophilic pocket hydrogen bonds were not maintained throughout the simulation. At approximately 2 ns, this hydrogen bonding pattern breaks and is not reformed. This is attributed to flexibility in the aliphatic portion of PPEN’s tail. This moiety undergoes a rotational conformation change, which we confirm by examining two dihedral angles of the ligand (Figure 8). Following rotation, water molecules enter the active site and ultimately stabilize both carboxylate 2 and the three hydrophilic residues. It remains unclear if these interactions would reform during extended simulations; however, it seems likely that a more rigid framework would ensure they are maintained.
Figure 8.
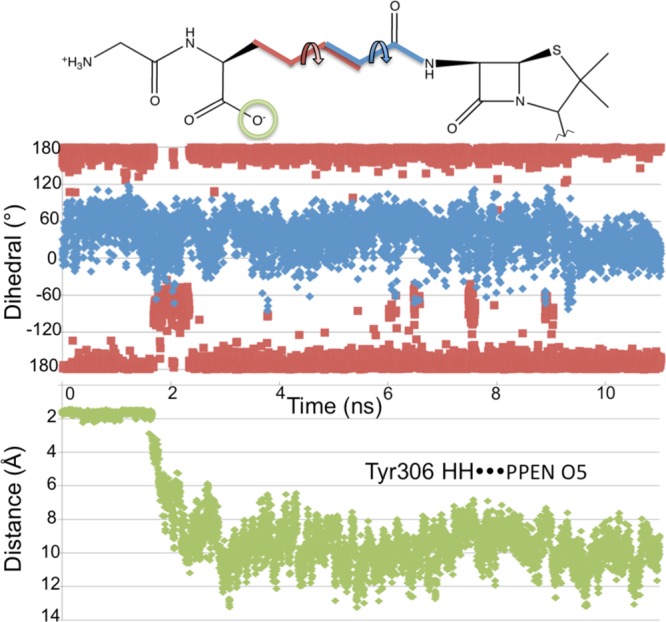
Top: Dihedral angles are measured for the 11 ns PPEN–R61 MD simulation, which are denoted by D1 (black) and D2 (gray). Bottom: the hydrogen bond distance between Tyr306HH···PPEN O5 is measured for the 11 ns PPEN–R61 MD simulation.
3.2. Fragment Scoring, Virtual Screening, and Physiochemical Analysis
Fragment scoring was performed to gain additional insight into receptor (QM/MM optimized)–ligand substructure (Figure 9) interactions. PENG was divided into three fragments: a β-lactam unit, an amide group, and the aromatic moiety. These were compared to PPEN, which was divided into six fragments: a β-lactam unit, two amide groups (β-lactam neighbor and peptidomimetic moiety), a butyl chain, carboxylate group, and the ammonio group. Hydrogen atoms were added to each fragment to satisfy valency requirements and G-scores were computed in place using the GlideXP scoring function. XP descriptors were generated by decomposing the composite scoring function to gain additional insight into interaction energies (Tables 1 and 2).
Figure 9.
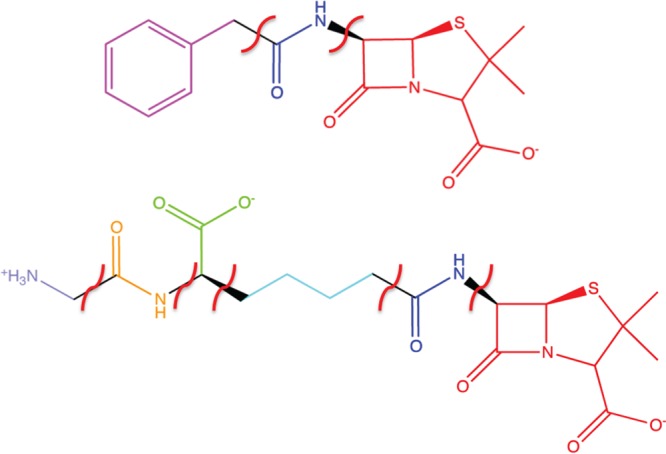
PENG and PPEN fragments used in the XP descriptor analysis.
Table 1. XP Descriptor Analysis from the Glide XP Fragment Decomposition Scoring Function for Perfect Penicillina.
| fragment | G- score | LipoEvdW | PhobEnPairHB | H bond | electro | sitemap | low MW | penalties | expos penal | rot penal |
|---|---|---|---|---|---|---|---|---|---|---|
| lactam unit | –14.8 | –1.3 | –2.0 | –4.6 | –6.3 | –0.2 | –0.5 | 0.0 | 0.0 | 0.0 |
| carboxylate 2 | –5.3 | 0.0 | 0.0 | –2.8 | –2.0 | 0.0 | –0.5 | 0.0 | 0.0 | 0.0 |
| N4H3+ | –3.4 | 0.3 | 0.0 | 0.0 | –3.2 | 0.0 | –0.5 | 0.0 | 0.0 | 0.0 |
| carbonyl 3/N3 amide | –2.6 | 0.0 | 0.0 | –1.3 | –0.7 | 0.0 | –0.5 | 0.0 | 0.0 | 0.0 |
| PPEN butyl chain | –0.2 | –1.1 | 0.0 | 0.0 | 0.0 | 0.0 | –0.5 | 0.0 | 0.2 | 1.1 |
| carbonyl 2/N2 amide | –3.1 | –0.2 | 0.0 | –1.7 | –0.7 | –0.2 | –0.5 | 0.0 | 0.2 | 0.0 |
All results are in kcal mol–1.
Table 2. XP Descriptor Analysis from the Glide XP Fragment Decomposition Scoring Function for Benzylpenicillina.
| fragment | G-score | LipoEvdW | PhobEnPairHB | H bond | electro | sitemap | low MW | penalties | expos penalites | rot penalties |
|---|---|---|---|---|---|---|---|---|---|---|
| lactam unit | –13.0 | –1.8 | –3.9 | –3.8 | –2.8 | –0.2 | –0.5 | 0.0 | 0.0 | 0.0 |
| amide | –2.7 | 0.0 | 0.0 | –1.9 | –0.8 | 0.0 | –0.5 | 0.5 | 0.0 | 0.0 |
| phenyl ring | –2.7 | –2.0 | 0.0 | 0.0 | –0.1 | 0.0 | –0.5 | 0.0 | 0.0 | 0.0 |
All results are in kcal mol–1.
Fragment scoring complements CPA results by giving valuable insight into the physiochemical properties and affinities of the binding site. As expected, the β-lactam units are important for effective binding due to strong hydrogen bonding and electrostatic contributions from its carboxylate group. A comparison can be made between PENG’s phenyl group and PPEN’s butyl chain due to their similar location in the R61 active site. The fragment decomposition further illustrates the advantage of PENG’s aromatic moiety (−2.7 kcal mol–1) over PPEN’s hydrophobic butyl fragment (−0.2 kcal mol–1). The importance of PPEN’s unique fragments are also evaluated. Favorable contributions are observed from carboxylate 2 and the amide group. Carboxylate 2’s score is dominated by electrostatics and hydrogen bonding due to R61’s hydrophilic pocket interacting with this functional group. PPEN’s terminal CH3–N4H3+ unit has significant electrostatic stabilization attributed to its water mediated salt bridge with Asp217(−3.4 kcal mol–1).
Detailed protein–ligand interactions characterized via atomistic modeling were used to direct virtual screening, a technique commonly employed to examine large libraries of biologically relevant compounds. Although virtual screening has proved to be invaluable to medicinal chemists,53,54 some key limitations exist; e.g., uncertainty of water mediated interactions, unknown binding/allosteric sites, description of ligand/receptor flexibility, imperfect force fields and/or scoring functions.55 Therefore, the coupling of virtual screening with atomistic modeling (e.g., MD simulations, QM/MM) provides an improved connection to reality.
This virtual screening effort began via construction of a virtual library focused on penam and cepham pharmacophores, which was screened against QM/MM minimized structures, both PPEN–R61 and PENG–R61, using rigid GlideXP docking (see the Supporting Information for details). Eight compounds (Table 3) were found to have a better docking score than PPEN in PPEN–R61. The top five ligands in this virtual screening set have a seven membered amphiphilic ring fused to a phenyl group (Figure 10). The fused ring structure induces hydrophobic interactions, π–π stacking, and hydrogen bonding with multiple active site residues. Bonsignore et al.56 have previously synthesized the series of top scoring compounds and performed cellular assays against Staphylococcus aureus; moderate antimicrobial activity was observed. To date, these β-lactam compounds have not been cocrystallized or characterized by direct enzyme assay; therefore, their true effectiveness remains unclear.
Table 3. XP Descriptor Analysis for the Top Scoring Compound (Listed by CID number) from the Glide XP study for the PPEN–R61 Structurea.
| ligand | G-score | LipoEvdW | PhobEnPairHB | H bond | electro | sitemap | low MW | rot penalties |
|---|---|---|---|---|---|---|---|---|
| 10070689 | –23.7 | –3.6 | –3.9 | –9.6 | –6.3 | –0.4 | –0.1 | 0.1 |
| 10454697 | –23.7 | –3.8 | –3.9 | –9.3 | –6.3 | –0.4 | –0.1 | 0.1 |
| 11742866 | –23.3 | –3.8 | –3.9 | –9.0 | –6.3 | –0.5 | –0.1 | 0.1 |
| 10003026 | –21.8 | –3.3 | –3.9 | –8.1 | –6.3 | –0.3 | –0.1 | 0.1 |
| 10026664 | –21.7 | –2.7 | –3.9 | –8.7 | –6.3 | –0.4 | 0.0 | 0.1 |
| 54250959 | –21.7 | –2.1 | –5.9 | –6.9 | –6.3 | –0.6 | –0.1 | 0.1 |
| 53464041 | –20.4 | –3.3 | –3.9 | –6.3 | –6.3 | –0.5 | –0.3 | 0.1 |
| 4773847 | –20.3 | –2.8 | –2.0 | –10.7 | –7.8 | –0.4 | –0.1 | 0.3 |
All results are in kcal mol–1.
Figure 10.

Final binding poses obtained from the IFD investigations. Top scoring compounds were docked into PPEN–R61 and are listed using their CID number.
The top scoring ligands from GlideXP (G-scores ∼ −23 to −25 kcal mol–1) were redocked via IFD (Table 4) into the PPEN–R61 protein. Five compounds scored better than PPEN. The β-lactam framework for these fused ring structures retained the same hydrogen bonding network observed in the case of PPEN. Further, a carbonyl group of the seven membered ring mimics the function of PPEN’s carboxylate 2. However, the heteroatoms of the seven membered ring do not hydrogen bond with neighboring residues except for CID 11742866. In this case, the oxygen accepts a hydrogen bond from the Gln303 side chain (Figure 10e).
Table 4. IFD and ADME Results for the Top Scoring Compoundsa.
| ligand | #stars | G-score | LipoEvdW | PhobEnPairHB | H bond | electro | sitemap | low MW | rot penalties | penalties |
|---|---|---|---|---|---|---|---|---|---|---|
| 10026664 | 0 | –22.3 | –2.7 | –3.9 | –7.6 | –6.3 | –0.4 | 0.0 | 0.1 | 1.6 |
| 10454697 | 0 | –21.1 | –3.8 | –3.9 | –7.5 | –6.3 | –0.4 | –0.1 | 0.1 | 0.0 |
| 10003026 | 0 | –20.8 | –3.3 | –3.9 | –7.6 | –6.3 | –0.3 | –0.1 | 0.1 | 0.0 |
| MPPEN | 4 | –20.5 | –5.8 | –2.0 | –7.8 | –7.8 | –0.4 | 0.0 | 0.2 | 3.0 |
| 10070689 | 0 | –20.5 | –3.6 | –3.9 | –7.5 | –6.3 | –0.4 | –0.1 | 0.1 | 1.2 |
| 11742866 | 0 | –19.6 | –3.8 | –3.9 | –6.7 | –6.3 | –0.5 | –0.1 | 0.1 | 1.5 |
| PPEN | 6 | –18.9 | –2.8 | –2.0 | –8.1 | –7.8 | –0.4 | –0.1 | 0.3 | 3.0 |
| PENG | 0 | –15.5 | –4.4 | –2.0 | –5.8 | –2.9 | –0.3 | –0.4 | 0.2 | 0.0 |
All results are in kcal mol–1.
Inhibitor design is multifarious in nature. One aspect not yet considered is physiochemical properties. These properties can be grouped into five broad categories: size, shape, flexibility/rigidity, electronic nature, and solubility in both water and organic solvents.57 Nearly all of the top scoring compounds follow Jorgensen’s rule of three58 and Lipinski’s rule of five,59,60 an indication of potential drugability. To gain detailed insight into these properties, the ADME tool (QikProp) was used to investigate all top scoring compounds from IFD studies. QikProp estimates drug-likeness by #stars, where a #star is assigned to a compound if a physiochemical property (see the Supporting Information for detailed information about the 24 physiochemical descriptors) is an outlier compared to known drugs (≥95%). There are 24 possible #stars, one for each physiochemical property computed. If a compound has 6 or more #stars, it is not considered drug-like; however, 5 or less #stars falls within the recommended range. All top scoring IFD compounds and PENG fall in the recommended range of drug-like properties. However, PPEN does not due to 6 violations (Table 5), which are attributed to the hydrophilicity of heteroatoms affecting the solvent accessible surface area, van der Waals surface, aqueous solubility, human serum albumin binding, and the octanol/water and brain/blood barrier partition coefficients.
Table 5. XP Descriptor Analysis for the Top Scoring Compounds (Listed by CID number) from the Glide XP Study for the PENG–R61 Structurea.
| ligand | G-score | LipoEvdW | PhobEnPairHB | H bond | electro | sitemap | low MW | rot penalties |
|---|---|---|---|---|---|---|---|---|
| 107602 | –19.4 | –3.9 | –5.9 | –6.1 | –3.0 | –0.5 | –0.3 | 0.2 |
| 56607984 | –19.3 | –3.7 | –5.9 | –6.1 | –3.0 | –0.5 | –0.3 | 0.2 |
| 103613 | –19.2 | –4.0 | –5.9 | –5.8 | –3.0 | –0.4 | –0.3 | 0.1 |
| 56613099 | –18.8 | –3.4 | –5.9 | –5.9 | –3.0 | –0.4 | –0.3 | 0.1 |
| 56627471 | –17.6 | –3.7 | –5.9 | –4.6 | –3.0 | –0.3 | –0.3 | 0.1 |
| 2349 | –17.3 | –3.4 | –5.9 | –4.6 | –3.0 | –0.2 | –0.4 | 0.2 |
All results are in kcal mol–1.
3.3. Modifying the Perfect Penicillin
Present results indicate that both PPEN and PENG have advantageous structural features that improve binding affinity and specificity. A logical next step is to combine the most favorable moieties from each inhibitor and propose new lead compounds. The first modification would be to replace the butyl chain of PPEN with a phenyl ring to mimic PENG. This not only ensures that π–π stacking occurs in the aromatic pocket, but the compound’s overall flexibility is decreased. A drug’s effectiveness generally increases by decreasing the number of rotatable bonds (ideal range: 0–15) and increasing the number of ring atoms.57
The next suggestion would be to keep the peptidomimetic tail of PPEN, namely the −CO2– and terminal N4H3+ moieties. Upon combining these structural features, a “more perfect, perfect penicillin” (MPPEN) is imagined (Figure 11).
Figure 11.
The more perfect, perfect penicillin is shown using the 2D view of its docking placement in the R61 active site.
MPPEN was docked into the PPEN–R61 enzyme structure using IFD and yielded a better docking score (−20.5 kcal mol–1) than PPEN (−19.6 kcal mol–1) itself. ADME properties were also evaluated and resulted in a decrease (from 6/24 for PPEN to 4/24 for MPPEN) of #star violations. This satisfies the requirement of less than 6 #star violations, specifically the structure improvement eliminated #stars related to aqueous solubility and human serum albumin binding. Each of the remaining violations are also closer to the ideal range, which indicates further improvement (see the Supporting Information for full results).
4. Conclusions
The present work identifies several protein–ligand interactions that play key roles in binding and specificity of β-lactam inhibitors. In particular, we characterize underlying intermolecular interactions that contribute to common antibiotic resistance mechanisms. Benzylpenicillin and a novel β-lactam peptidomimetic (perfect penicillin) complexed to Stremptomyces R61 were examined using an arsenal of computational techniques. Noncovalent interactions were investigated by combining MD simulations, QM/MM calculations, charge perturbation analysis, QM/MM NBO, bioinformatics, virtual screening, flexible docking, and physiochemical property prediction (i.e., ADME). Several molecular level interactions were identified that differentially stabilize the aforementioned model inhibitors. Benzylpenicillin’s phenyl group forms an extended π–π network with Phe120 and Trp233 that contributes significantly to its efficacy in DD-peptidase. Further, structural analysis revealed that this aromatic stabilization is conserved in β-lactamases. This led us to a novel hypothesis that suggests antibacterial resistance has evolved, in part, due to stabilizing aromatic interactions. Additionally, interactions between the protein and the peptidomimetic tail region (i.e., mimic of the native substrate), particularly carboxylate 2 and the terminal N4H3+ unit, form unique hydrogen bonding and strong electrostatic interactions. Of particular interest is the water mediated salt bridge between Asp217 and the N4H3+. Structural alignment revealed that the enzyme domain housing Asp217 does not exist in class C β-lactamases. This highlights a key interaction that should confer specificity to peptidomimetic inhibitors. Finally, interaction information is used to suggest modifications to current β-lactam compounds (i.e., perfect penicillin) that should improve binding and specificity in DD-peptidases and physiochemical properties. The resulting compound, “a more perfect, perfect penicillin”, is posited for future experimental studies and structure-based inhibitor design.
Acknowledgments
The authors thank Professors Y. Chen and W. Guida for helpful discussion. HLW3 acknowledges NIH (1K22HL088341-01A1) and the University of South Florida (start-up) for funding. Computations were performed at the USF Research Computing Center (NSF Grant No. CHE-0722887) and XSEDE (MCB120133), both centers are greatly appreciated.
Supporting Information Available
MD simulation details, link-atom information for NBO QM/MM calculations, full CPA results, PENG parameters, PPEN parameters, virtual screening details (i.e., setup and results), additional ADME results and definitions, and additional informatics results (i.e., ProBiS and Clustal Omega). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Salton M. R. J.The Bacterial Cell Wall; Elsevier: Amsterdam, 1964. [Google Scholar]
- Wise E. M. J.; Park J. T. Penicillin: its basic site of action as an inhibitor of a peptide cross-linking reaction in cell wall mucopeptide synthesis. Proc. Natl. Acad. Sci. U. S. A. 1965, 54, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipper D. J.; Strominger J. L. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-d-alanyl-d-alanine. Proc. Natl. Acad. Sci. U. S. A. 1965, 54, 1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipper D. J.; Strominger J. L. Biosynthesis of the peptidoglycan of bacterial cell walls. J. Biol. Chem. 1968, 243, 3169–3179. [PubMed] [Google Scholar]
- Frere J. M.; Joris B. Penicillin-sensitive enzymes in peptidoglycan biosynthesis. CRC Crit. Rev. Microbiol. 1985, 11, 299–396. [DOI] [PubMed] [Google Scholar]
- Waxman D. J.; Strominger J. L. Penicillin-binding proteins and the mechanism of action of betalactam antibiotics. Annu. Rev. Biochem. 1983, 52, 825–869. [DOI] [PubMed] [Google Scholar]
- Ghuysen J. M. Molecular structures of penicillin-binding proteins and beta-lactamases. Trends Microbiol. 1994, 2, 372–380. [DOI] [PubMed] [Google Scholar]
- Ghuysen J. M. Penicillin-binding proteins. wall peptidoglycan assembly and resistance to penicillin: facts, doubts and hopes. Int. J. Antimicrob. Agents 1997, 8, 45–60. [DOI] [PubMed] [Google Scholar]
- Fisher J. F.; Meroueh S. O.; Mobashery S. Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 2005, 105, 395–424. [DOI] [PubMed] [Google Scholar]
- Holten K. B.; Onusko E. M. Appropriate prescribing of oral beta-lactam antibiotics. Am. Fam. Physician 2000, 62, 611–620. [PubMed] [Google Scholar]
- Silvaggi N. R.; Josephine H. R.; Kuzin A. P.; Nagarajan R.; Pratt R. F.; Kelly J. A. Crystal structures of complexes between the r61 dd-peptidase and peptidoglycan-mimetic beta-lactams: a non-covalent complex with a “perfect penicillin”. J. Mol. Biol. 2005, 345, 521–533. [DOI] [PubMed] [Google Scholar]
- Josephine H. R.; Kumar I.; Pratt R. F. The perfect penicillin? inhibition of a bacterial dd-peptidase by peptidoglycan-mimetic beta-lactams. J. Am. Chem. Soc. 2004, 126, 8122–8123. [DOI] [PubMed] [Google Scholar]
- Miller B. T.; Singh R. P.; Klauda J. B.; Hodoscek M.; Brooks B. R.; Woodcock H. L. Charmming: a new, flexible web portal for charmm. J. Chem. Inf. Model. 2008, 48, 1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; A. D. MacKerell J. Automation of the charmm general force field (cgenff) i: Bond and perception and atom typing. J. Chem. Inf. Mod. 2012, 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Raman E. P.; A. D. MacKerell J. Automation of the charmm general force field (cgenff) ii: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks I. C. L.; Mackerell J. A. D.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. Charmm: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherman B. F.; Goldberg S. D.; Cornish V. W.; Friesner R. A. Mixed quantum mechanical/ molecular mechanical (qm/mm) study of the deacylation reaction in a penicillin binding protein (pbp) versus in a class c beta-lactamase. J. Am. Chem. Soc. 2004, 126, 7652–7664. [DOI] [PubMed] [Google Scholar]
- Li H.; Robertson A. D.; Jansen J. H. Very fast empirical prediction and interpretation of protein pk(a) values. Proteins 2005, 61, 704–721. [DOI] [PubMed] [Google Scholar]
- Bas D. C.; Rogers D. M.; Jensen J. H. Very fast prediction and rationalization of pka values for protein-ligand omplexes. Proteins 2008, 73, 765–783. [DOI] [PubMed] [Google Scholar]
- Olsson M. H. M.; Sondergaard C. R.; Rostkowski M.; Jensen J. H. Propka3: Consistent treatment of internal and surface residues in empirical pka predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D. J.; Bashford D.; Bellott M.; Dunbrack R. L. J.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S. others, All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; MacKerell A. D. J. others, Charmm general force field: A force field for drug-like molecules compatible with the charmm all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock H. L.; Hodošček M.; Gilbert A. T. B.; Gill P. M. W.; Schaefer H. F.; Brooks B. R. Interfacing q-chem and charmm to perform qm/mm reaction path calculations. J. Comput. Chem. 2007, 28, 1485–1502. [DOI] [PubMed] [Google Scholar]
- Becke A. D. A new mixing of hartree–fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- Lonsdale R.; Harvey J. N.; Mulholland A. J. Inclusion of dispersion effects significantly improves accuracy of calculated reaction barriers for cytochrome p450 catalyzed reactions. J. Phys. Chem. Lett. 2010, 1, 3232–3237. [Google Scholar]
- Bash P. A.; Field M. J.; Davenport R. C.; Petsko G. A.; Ringe D.; Karplus M. Computersimulation and analysis of the reaction pathway of triosephosphate isomerase. Biochem. 1991, 30, 5826–5832. [DOI] [PubMed] [Google Scholar]
- Cui Q.; Karplus M. Triosephosphate isomerase: a theoretical comparison of alternative pathways. J. Am. Chem. Soc. 2001, 123, 2284–2290. [DOI] [PubMed] [Google Scholar]
- Lee Y. S.; Worthington S. E.; Krauss M.; Brooks B. R. Reaction mechanism of chorismate mutase studied by the combined cotentials of quantum mechanics and molecular mechanics. J. Phys. Chem. B 2002, 106, 12059–12065. [Google Scholar]
- Reed A. E.; Curtiss L. A.; Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar]
- Yang Y.; Cui Q. Interactions between phosphate and water in solution: a natural bond orbital based analysis in a qm/mm framework. J. Phys. Chem. B 2007, 111, 3999–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Cui Q. The hydrolysis activity of adenosine triphosphate in myosin: a theoretical analysis of anomeric effects and the nature of the transition state. J. Phys. Chem. A 2009, 113, 12439–12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y.; Molnar L. F.; Jung Y.; Kussmann J.; Ochsenfeld C.; Brown S. T.; Gilbert A. T. B.; Slipchenko L. V.; Levchenko S. V.; O’Neill D. P.; DiStasio R. A. J.; Lochan R. C.; Wang T.; Beran G. J. O.; Besley N. A.; Herbert J. M.; Lin C. Y.; Voorhis T. V.; Chien S. H.; Sodt A.; Steele R. P.; Rassolov V. A.; Maslen P. E.; Korambath P. P.; Adamson R. D.; Austin B.; Baker J.; Byrd E. F. C.; Dachsel H.; Doerksen R. J.; Dreuw A.; Dunietz B. D.; Dutoi A. D.; Furlani T. R.; Gwaltney S. R.; Heyden A.; Hirata S.; Hsu C.-P.; Kedziora G.; Khalliulin R. Z.; Klunzinger P.; Lee A. M.; Lee M. S.; Liang W.; Lotan I.; Nair N.; Peters B.; Proynov E. I.; Pieniazek P. A.; Rhee Y. M.; Ritchie J.; Rosta E.; Sherrill C. D.; Simmonett A. C.; Subotnik J. E.; Woodcock H. L.; Zhang W.; Bell A. T.; Chakraborty A. K.; Chipman D. M.; Keil F. J.; Warshel A.; Hehre W. J.; Schaefer H. F.; Kong J.; Krylov A. I.; Gill P. M. W.; Head-Gordon M. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [DOI] [PubMed] [Google Scholar]
- Foster J. P.; Weinhold F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar]
- Konc J.; Cesnik T.; Konc J. T.; Penca M.; Jenezic D. Probis-database: Precalculated binding site similarities and local pairwise alignments of pdb structures. J. Chem. Inf. Mod. 2012, 52, 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konc J.; Janezic D. Probis: a web server for detection of structurally similar protein binding sites. Nucleic Acids Res. 2010, 38, W436–W440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konc J.; Janezic D. Probis algorithm for detection of structurally similar protein binding sites by local structural alignment. Bioinformatics 2010, 26, 1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Soding J.; Thompson J. D.; Higgins D. G. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon M.; McWilliam H.; Li W.; Valentin F.; Squizzato S.; Paern J.; Lopez R. A new bioinformatics analysis tools framework at embl-ebi. Nucleic Acids Res. 2010, 38, W695–W699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Bolton E. E.; Bryant S. H. Pubchem3d: biologically relevant 3-d similarity. J. Cheminf. 2011, 3, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2013-2: LigPrep, v. 2.7; Schrödinger, LLC, New York, NY, 2013.
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: a new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- Small-Molecule Drug Discovery Suite 2013-2: QikProp, v. 3.7; Schrödinger, LLC, New York, NY, 2013.
- Sinnokrot M. O.; Valeev E. F.; Sherrill C. D. Estimates of the ab initio limit for pi-pi interactions: the benzene dimer. J. Am. Chem. Soc. 2002, 124, 10887–10893. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Sanders J. K. M. The nature of pi-pi interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar]
- Wheeler S. E.; Houk K. N. Origin of substituent effects in edge-to-face aryl-aryl interactions. Mol. Phys. 2009, 107, 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. W.; Pratt R. F. Dipeptide binding to the extended active site of the Streptomyces R61 D-alanyl-D-alanine-peptidase: the path to a specific substrate. Biochem. 2000, 39, 12200–12209. [DOI] [PubMed] [Google Scholar]
- McDonough M. A.; Anderson J. W.; Silvaggi N. R.; Pratt R.; Knox J. R.; Kelly J. A. Structures of Two Kinetic Intermediates Reveal Species Specificity of Penicillin-binding Proteins. J. Mol. Biol. 2002, 322, 111–122. [DOI] [PubMed] [Google Scholar]
- Yang A. S.; Honig B. An integrated approach to the analysis and modeling of protein sequences and structures. i. protein structural alignment and a quantitative measure for protein structural distance. J. Mol. Biol. 2000, 301, 665–678. [DOI] [PubMed] [Google Scholar]
- Kelly J. A.; Kuzin A. P. The refined crystallographic structure of a dd-peptidase pencillin-targe enzyme at 1.6 a resolution. J. Mol. Biol. 1995, 254, 223–236. [DOI] [PubMed] [Google Scholar]
- Bourguignon-Bellefroid C.; Wilkin J. M.; Joris B.; Aplin R. T.; Houssier C.; Prendergast F. G.; Van Beeumen J.; Ghuysen J. M.; Frère J. M. Importance of the two tryptophan residues in the Streptomyces R61 exocellular DD-peptidase. Biochem. J. 1992, 282Pt 2361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert M. H. J.; Lang M. Essential factors for successful virtual screening. Mini-Rev. Med. Chem. 2008, 8, 1389–5575. [DOI] [PubMed] [Google Scholar]
- Villoutreix B. O.; Eudes R.; Miteva M. A. Structure-based virtual ligand screening: recent success stories. Comb. Chem. High–Throughput Screening 2009, 12, 1000–1016. [DOI] [PubMed] [Google Scholar]
- Scior T.; Bender A.; Tresadern G.; Medina-Franco J. L.; Martinez-Mayorga K.; Langer T.; Cuanalo-Contreras K.; K.Agrafiotis D. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 63–72. [DOI] [PubMed] [Google Scholar]
- Bonsignore L.; Logu A. D.; Loy G.; Lavagna S. M.; Secci D. Synthesis and antimicrobial activity of coumarin and benzodioxazepine-, diazazepine- and benzoxazepine-substituted penicillins. Eur. J. Med. Chem. 1994, 29, 479–485. [Google Scholar]
- Voutchkova A. M.; Ferris L. A.; Zimmerman J. B.; Anastas P. T. Toward molecular design for hazard reduction–fundamental relationships between chemical properties and toxicity. Tetrahedron 2010, 66, 1031–1039. [Google Scholar]
- Jorgensen W. L. Efficient drug lead discovery and optimization. Acc. Chem. Res. 2009, 42, 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev. 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today 2004, 1, 337–341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.