

Abstract
Peptides that self-assemble, at low concentration, into bilayer-spanning pores which allow the passage of macromolecules would be beneficial in multiple areas of biotechnology. However, there are few, if any, natural or designed peptides that have this property. Here we show that the 26-residue peptide “MelP5”, a synthetically evolved gain-of-function variant of the bee venom lytic peptide melittin identified in a high-throughput screen for small molecule leakage, enables the passage of macromolecules across bilayers under conditions where melittin and other pore-forming peptides do not. In surface-supported bilayers, MelP5 forms unusually high conductance, equilibrium pores at peptide:lipid ratios as low as 1:25000. The increase in bilayer conductance due to MelP5 is dramatically higher, per peptide, than the increase due to the parent sequence of melittin or other peptide pore formers. Here we also develop two novel assays for macromolecule leakage from vesicles, and we use them to characterize MelP5 pores in bilayers. We show that MelP5 allows the passage of macromolecules across vesicle membranes at peptide:lipid ratios as low as 1:500, and under conditions where neither osmotic lysis nor gross vesicle destabilization occur. The macromolecule-sized, equilibrium pores formed by MelP5 are unique as neither melittin nor other pore-forming peptides release macromolecules significantly under the same conditions. MelP5 thus appears to belong to a novel functional class of peptide that could form the foundation of multiple potential biotechnological applications.
Introduction
Peptides that form pores in lipid bilayer membranes can be useful in many capacities, including biosensor design,1 targeted cancer therapy,2 channel replacement therapy,3 HIV therapy,4 drug delivery,5,6 and others. In particular, peptides that create macromolecule-sized pores at low peptide concentrations could be especially useful, for example as biosensors, or for promoting efficient release of macromolecular cargoes from endosomes in drug delivery applications.7 However, true equilibrium pore-forming peptides are rare, and there are few, if any, peptides that are known to form macromolecule-sized pores in membranes at low concentrations. Just as importantly, we do not have a molecular understanding of the sequence/structure requirements for pore-forming peptides of any pore size, and thus we cannot rationally design or optimize peptides that cause efficient poration of lipid bilayers.
To circumvent the roadblock caused by the lack of knowledge about the mechanism of pore formation, we recently performed a synthetic molecular evolution-based high-throughput screen of a rational combinatorial peptide library that used the sequence of the membrane lytic bee venom peptide melittin as a template.8 Melittin, at high concentration (P:L ≥ 1:50), releases the small molecule contents of lipid vesicles and has been reported to self-assemble into toroidal pores.9,10 At intermediate concentrations melittin releases small probe molecules from lipid vesicles, but the leakage is partial and occurs through transient, not equilibrium, structures.8,11−13 At low concentration (P:L ≤ 1:1000) in the absence of osmotic stress and anionic lipids, melittin does not efficiently permeabilize all lipid vesicles.8 We used an iterative high-throughput approach to identify gain-of-function variants of melittin that have vesicle permeabilizing activity at very low concentration. In the melittin-based library, we varied 10 critical residues in the 26-residue sequence of melittin. To identify gain-of-function variants we used an orthogonal high-throughput assay for peptides that release probe molecules from lipid vesicles at very low peptide:lipid ratios (P:L ≤ 1:1000) where native melittin is not active. We also assayed independently for the continued presence of detectable pores hours after peptide addition. This step selects for equilibrium pore formers and selects against transient pore-formers, such as melittin.11,14 Only about a dozen peptides from the 7800-member library were highly active under the conditions of these stringent, orthogonal assays. The most active gain-of-function analog identified, “MelP5”, differs from melittin in only 5 amino acids out of 26. Yet, the initial characterization revealed distinct differences between melittin and MelP5: (i) The concentration of MelP5 required to permeabilize synthetic lipid vesicles is up to 20-fold lower than that for melittin in leakage assays based on dye molecule release;8,15 (ii) the pores formed by MelP5 are present in membranes indefinitely whereas melittin’s pores are transient and detectable only for a short time after peptide addition; (iii) oriented circular dichroism shows MelP5 forms a membrane-spanning helix at equilibrium, unlike the helix of melittin which lies parallel to the membrane surface8 under most conditions.10,16−18 These initial results suggested that the mechanism of action of MelP5 may be fundamentally different from that of melittin.
Here we examine the uniqueness and potential utility of the pores formed by MelP5. Furthermore, we examine the ability of MelP5 to form equilibrium pores that pass macromolecules through membranes under conditions where common experimental artifacts such as osmotic lysis, vesicle aggregation and fusion, and global vesicle disruption caused by detergent-like bilayer solubilization do not occur. We compare the behavior of MelP5 to its parent sequence, melittin, and to two other well characterized α-helical peptide pore formers, alamethicin19 and GALA.20 In all experiments, a fundamentally different bilayer response is observed for MelP5 compared to the other peptides, suggesting the formation of large equilibrium pores in membranes. Specifically, we demonstrate the formation of macromolecule-sized pores in membranes by MelP5 at low concentration. Thus, MelP5 belongs to a novel class of pore-forming peptide that may be a useful starting point for multiple biotechnology applications.
Materials and Methods
Reagents
MelP5 and GALA were synthesized and purified by Bio-Synthesis Inc. Peptide purity and identity were verified by HPLC and MALDI mass spectrometry. Melittin and alamethicin were purchased from Sigma-Aldrich. Melittin and MelP5 were synthesized as N-terminal amines and C-terminal amides. 1-Palmitoyl-2-oleoly-sn-3-glycero-phosphocholine (POPC) and cholesterol were purchased as lyophilized powders from Avanti Polar Lipids and dissolved in chloroform. All other solvents and reagents were all purchased from Sigma-Aldrich.
Vesicle Preparation
Vesicles were prepared using previously established techniques.21,22 Briefly, lipids were dried from chloroform under nitrogen at an initial concentration of 25 mg/mL and under vacuum for an additional 30 min. Lipids were resuspended in buffer containing 100 mM potassium chloride and 10 mM sodium phosphate at either pH 7 or 5. These vesicles were then extruded 10 times through a 0.1 μm Nucleopore polycarbonate filter to give unilamellar vesicles of 0.1 μm diameter. Gel filtration chromatography was used to remove external probes from vesicles with entrapped contents.
Supported Bilayer Preparation
The process for supported bilayer formation was the same as described elsewhere.11,23,24 The top leaflet of the bilayers in these experiments consisted of 75% POPC and 25% cholesterol. The bottom leaflet contained an additional 5.9% PEG2k-DSPC along with 69.1% POPC and 25% cholesterol. Silicon of orientation (111) was used for this study. The substrate was washed using a standard technique of isopropanol, acetone, and then isopropanol, followed by a 1 h long piranha etch. Lipid solutions of 69.1% POPC, 25% cholesterol, and 5.9% PEG2k-DSPE were deposed onto a Langmuir–Blodgett (LB) trough from chloroform at a concentration of 1 mg/mL. The washed silicon wafers were immersed in the LB trough, and the surface was compressed to a final surface pressure of 32 mN/m (±0.5mN). The silicon substrate was then withdrawn at a rate of 15 mm/s while a constant surface pressure was maintained. Once removed from the LB trough, the wafers were clamped in place onto a custom electrochemical cell and 450 μL of the vesicle solutions were added to the electrochemical cell. The vesicles in the electrochemical cell were allowed to fuse with the monolayer-coated silicon surface for 1 h, after which 10 mL of phosphate buffer solution were added. Electrical contacts were made as described,11 and the electrochemical cells were then allowed to equilibrate for up to 24 h in buffer before EIS measurements.
Electrochemical Impedance Spectroscopy
Impedance measurements were made using a three-electrode setup with a silver/silver chloride reference electrode and a platinum counter electrode. Experimental details can be found elsewhere.11,25−27 The impedance was measured over the frequency range from 105 to 1 Hz using a 20 mV rms AC perturbation and at a potential of 0 V with respect to the reference electrode. Spectra were recorded at 2 min intervals and fit to an equivalent circuit model to determine the values of resistance and capacitance of the semiconductor–liquid interface (Rct and Cp) and the bilayer membrane (Rm and Cm).26,27 These values were then used to determine the normalized membrane resistance over time (Rm/Rm(t = 0)) and thus to describe the kinetics of peptide-induced bilayer permeabilization.
The kinetics of the normalized resistance drop were described by fitting to the model equation
| 1 |
In this equation there is both a fast component (τfast) describing the initial resistance decrease and a slow component (τslow) which was only invoked when a single time constant failed to fit the data.28 The constant y is a baseline resistance, and A and B are the contributions of the fast and slow processes to the total resistance decrease, respectively.
Calculation of Conductance
The change in bilayer conductance was calculated from the difference of the inverse bilayer resistances at time 0 and 60 min after addition of the peptide. The conductance per peptide was calculated from the experimental P:L value, and assuming a footprint for each lipid of 70 Å2.29
Macromolecule Release from Bilayers
For the dextran release experiment, a 10 kD dextran labeled with both TAMRA and biotin was entrapped in lipid vesicles made from 75% POPC and 25% cholesterol, followed by incubation with immobilized streptavidin to remove external dextran. In a leakage experiment, streptavidin labeled with AlexaFluor 488 was added outside the vesicles. Released dextran binds strongly to external streptavidin, and as a result FRET occurs between AF488 and TAMRA. Peptide-induced macromolecule release occurred within 5–10 min of peptide addition. Following incubation with peptides for 1 h, the fluorescence of the AlexaFluor 488 was measured and compared to controls without peptide (0% leakage) and controls with the detergent Triton X-100 (100% leakage).
For a chymotrypsin release assay, large unilamellar vesicles made from 75% POPC and 25% cholesterol were made in the presence of 1 mg/mL chymotrypsin, followed by gel filtration chromatography to remove external chymotrypsin. Incubation of vesicles with peptides for 1 h was done in 96 well plates, followed by the addition of Texas Red labeled casein (EnzChek reagent) to each well. The Texas Red fluorescence was measured for each well simultaneously as a function of time to reveal the rate of cleavage, which is proportional to the concentration of released chymotrypsin. The addition of excess peptide was used to determine the rate for 100% released chymotrypsin.
Results and Discussion
Assessment of Pore-Forming Peptides
There are many peptides that interact with bilayers and cause the permeation of ions and small probe molecules; e.g., see refs (8 and 30−43.) There are also many mechanisms by which permeabilization can occur,44−48 most of which do not involve the formation of explicit pores. Some membrane permeabilizing peptides cause the release of macromolecules from lipid vesicles.43,49−53 However, macromolecule release usually occurs only at a very high peptide concentration: 2–10 mol % peptide or a peptide:lipid ratio (P:L) of 1:50 to 1:10. Under these conditions, the release of macromolecules may be the result of cooperative, detergent-like global destabilization of bilayer integrity rather than the formation of explicit transmembrane pores. In any case the two are difficult to distinguish at high peptide concentration. Furthermore, in vesicle release experiments, factors such as osmotic stress (due to concentrated entrapped probe macromolecules) and large-scale vesicle aggregation/fusion (caused by the addition of cationic peptides to anionic vesicles) often drive global vesicle destabilization at high peptide concentration, which can allow for macromolecule release without true pores.
Here, we characterize the pores formed by the gain-of-function peptide MelP5 to assess the possibility that it forms true macromolecule-sized pores in membranes. We compare the behavior of MelP5 to the parent peptide, melittin, and other pore formers first by examining permeation of small ions through bilayers, followed by a characterization of macromolecular leakage from lipid vesicles. For all of these experiments we use a lipid composition of 75% phosphatidylcholine and 25% cholesterol. PC/cholesterol bilayers are physically robust and are resistant to nonspecific, detergent-like destabilization. Furthermore, because PC/cholesterol bilayers are zwitterionic, they do not readily aggregate or fuse in the presence of charged peptides. Finally, PC/cholesterol bilayers also roughly mimic the external surface of mammalian cells, thereby increasing the relevance of our results to potential biotech applications.
Melittin and MelP5
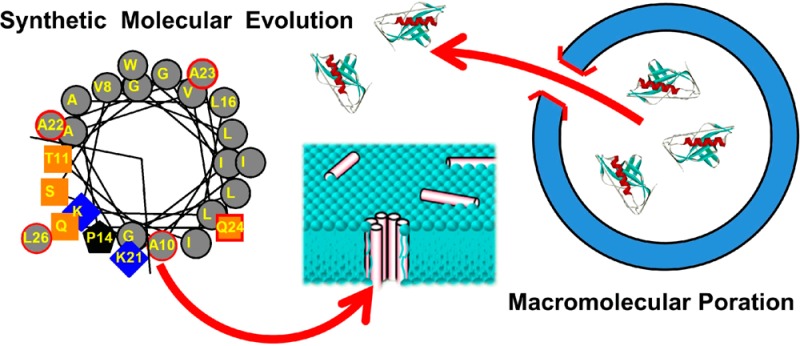
In Figure 1 we show sequences and helical wheel projections for the parent peptide melittin and the gain of function variant, MelP5, that we discovered by synthetic molecular evolution.8 Both peptides bind equally well to membranes and form amphipathic α-helices containing a polar face and a nonpolar face.8 The five differences in amino acid sequence between the two sequences, shown in red, were selected in a high-throughput screen.8 Four of the changes occur in the C-terminal tail, where the uniformly polar/cationic KRKRQQ sequence of melittin is replaced with KAAQQL in MelP5. These changes, in particular lysine 23 to alanine, significantly improve the potential helicity and amphipathicity of the C-terminal sequence such that MelP5, unlike melittin, can form a continuous amphipathic helix along its entire length. The only other change in MelP5 is the substitution of alanine for threonine in position 10. This change, which was found in multiple gain-of-function sequences,8 also improves the ideality of the amphipathic helix of MelP5 compared to melittin. We have hypothesized that the length and ideality of the amphipathic helix is the main structural basis for the novel activity of MelP5.8
Figure 1.

Structural comparison of melittin and MelP5. The sequences of melittin and MelP5 are shown at the top. Residues with numbers are those that were varied in the library from which MelP5 was selected. The helical wheel diagrams show the spatial arrangement of the side chains under the assumption of an ideal helix formed by all the residues. Gray symbols are hydrophobic residues, blue symbols are basic residues, and orange symbols are polar residues. Symbols outlined in red (right) are those for which a variation was selected during screening.
Structural Basis for the Activity of MelP5
MelP5 and melittin bind to bilayers with almost identical free energies (−8.2 kcal/mol in mole fraction units8). We have shown that MelP5 is more helical than melittin in PC bilayers and PC bilayers with phosphatidylglycerol.8 To determine if MelP5 is also more helical than melittin in the PC/cholesterol bilayers used in this work, we performed solution circular dichroism spectroscopy using essentially identical samples of MelP5 and melittin in the presence of lipid vesicles made from POPC with 25% cholesterol (Figure 2). Under these conditions both peptides are ≥95% membrane bound and thus the mean residue ellipticities reflect the fractional helix content of the bound peptide. Consistent with the expectation that the C-terminal tail of MelP5 is helical, while the C-terminal tail of melittin is not, MelP5 has about 20% more α-helix than melittin in cholesterol-containing lipid bilayers, as assessed by the ellipticity at 222 nm.54
Figure 2.

Circular dichroism spectra of melittin and MelP5 in bilayers composed of POPC and 25% cholesterol. CD spectra were taken for solution of 25 μM peptide in the presence of 1 mM POPC/cholesterol vesicles. Vesicle-only background has been subtracted.
Bilayer Response to MelP5
Electrochemical impedance spectroscopy (EIS) was used here because it is highly sensitive to small changes in membrane properties and gives direct information about a peptide’s effect on bilayer resistance and capacitance as a function of time. We have recently used EIS to provide new insights into the mechanism of action of antimicrobial peptides,25 other membrane permeabilizing peptides (including melittin),11 and membrane translocating peptides.24 For these studies, we use PEG-cushioned, surface supported lipid bilayers made from POPC with 25% cholesterol. POPC/cholesterol bilayers are robust, fluid phase bilayers that mimic the zwitterionic plasma membranes of eukaryotic cells. In a published study of dye leakage from vesicles8 we showed that MelP5 is more potent than melittin by a factor of about 20 in PC/cholesterol bilayers. Importantly, we note that the activity of MelP5, unlike that of melittin, is not strongly dependent on lipid composition.8
We prepared the supported bilayers as described above. The impedance response of the pure bilayer was then measured to establish the baseline characteristics. The average starting resistance was (2.29 ± 0.23) × 103 Ω cm2. At this point, peptide was added without removing free vesicles, and the impedance response was measured every 2 min for 1 h. The bilayer resistance and capacitance at each time point were determined using curve fitting as described elsewhere.11,23,24 The addition of MelP5 caused an initial rapid decrease in bilayer resistance followed by a slow decrease toward a steady-state value. This behavior was observed for all peptide concentrations, even down to peptide:lipid ratios as low as 1:25000 (Figure 3A). The steady-state resistance decreased with increasing peptide-to-lipid ratio. The time dependence of the bilayer response could not be fit with a single exponential, but was fit very well by two exponentials, suggesting that there are two rate processes with halftimes of about 2 min and 20–40 min, respectively. The bilayer response to MelP5 is dramatically different from the bilayer’s response to the parent peptide melittin, which we have previously characterized.11 As shown in Figure 3B, melittin causes a similar initial decrease in bilayer resistance, but is followed by a subsequent recovery. Given the similarities between the two sequences, and the similarity in the early EIS time course, we speculate that the initial membrane-bound state is similar for the two peptides, but that MelP5 then self-assembles into an equilibrium transmembrane pore while melittin equilibrates into a nonpore, nonmembrane-permeabilizing state discussed elsewhere.11,14 Consistent with these conclusions, the helical axis of melittin lies mostly parallel to the bilayer surface8,10,16−18 while MelP5 has a membrane spanning orientation,8 consistent with true pores.
Figure 3.

The bilayer resistance response to MelP5 by electrochemical impedance spectroscopy. (A) Aliquots of MelP5 were added to the 60 μM lipid vesicles in the sample chamber to give peptide:lipid ratios of 1:1000, 1:2500, 1:5000, 1:10000, and 1:25000. The resistance of the POPC/cholesterol bilayers was measured before peptide addition, and every 2 min after. Each trace represents an average of at least three independent experiments. The standard deviation of the three measurements is indicated by an error bar at the end. (B) Comparison of the resistance changes caused by melittin and MelP5 at P:L = 1:1000.
Impedance spectroscopy also allows the determination of bilayer capacitance, which reports on the thickness and overall integrity of the bilayer. In these experiments, the average starting capacitance of the bilayers was 0.90 ± 0.11 μF/cm2, corresponding to a bilayer thickness of about 5 nm.29 In Figure 4 we show normalized capacitance over the 60 min experiment. For all peptide concentrations, except P:L = 1:1000, the addition of MelP5 did not change the bilayer capacitance, indicating that the overall bilayer structure remains intact and is not globally perturbed by the peptide, despite the fact that the bilayers have very low resistance. The increase in capacitance at the highest concentration is due to the low resistance which results in a decrease in the frequency range of the capacitive region and hence a decrease in the phase angle, which can increase fitting uncertainties in capacitance. The addition of melittin also resulted in no measurable changes in bilayer capacitance. In summary, the capacitance measurements indicate that MelP5 and melittin result in local perturbations of the membrane but do not destabilize the bilayers globally. As described above, the resistance measurements show that the mechanisms of these local perturbations are significantly different for the two peptides.
Figure 4.

Bilayer capacitance response to MelP5. Aliquots of MelP5 were added at various concentrations relative to the 60 μM lipid vesicles in the sample chamber to give peptide:lipid ratios of 1:1000, 1:2500, 1:5000, 1:10000, and 1:25000. The capacitance of the POPC/cholesterol bilayers was measured every 2 min by electrochemical impedance spectroscopy before and after the addition of MelP5. Each trace represents an average of at least three independent experiments. The standard deviation of the three measurements is indicated by an error bar at the end. The inset shows the capacitance of PC/cholesterol bilayers after addition of melittin at P:L = 1:1000.
Comparison of MelP5 to Other Pore-Forming Peptides
To gain further insight into the bilayer response to MelP5, we also studied the well-characterized, pH-dependent pore-forming peptide, GALA.20,55−57 At pH 7, GALA does not bind to bilayers and does not cause membrane permeabilization55 while, at pH 5, it partitions strongly into membranes as an α-helix and self-assembles into membrane spanning, equilibrium pores.20,57 Thus, at pH 7 GALA serves as a negative control, while at pH 5 it serves as a positive control for equilibrium, transmembrane pores. The EIS results for GALA are unequivocal (Figure 5). At pH 7, GALA has little effect on bilayer electrical properties, with only a small, time-independent decrease in bilayer resistance at the highest concentration studied, P:L = 1:200. This result shows that the EIS signatures we observe are specific for membrane-permeabilizing peptides. At pH 5, the effect of GALA is consistent with the formation of equilibrium transmembrane pores;20,57 it causes a large and immediate decrease in bilayer resistance that is measurable at concentrations as low as P:L = 1:5000. The time course of the resistance change for GALA does not have a slow phase like MelP5, but instead is a single exponential with a time constant of <2 min suggesting a one-step insertion of GALA into the membrane.
Figure 5.

Bilayer response to GALA at pH 7 and pH 5. The resistance (top row) and capacitance (bottom row) of POPC/cholesterol bilayers were measured by electrochemical impedance spectroscopy after addition of the peptide GALA at pH 7 (left column) or pH 5 (right column). Aliquots of GALA were added to the 60 μM lipid vesicles in the sample chamber to give peptide:lipid ratios of 1:200, 1:1000, and 1:5000, and the resistance and capacitance values were determined from EIS spectra taken every 2 min over a period of 1 h. Each trace represents an average of at least three independent experiments. The standard deviation of the three measurements is indicated by an error bar at the end.
We have previously described the EIS signature of the membrane response to the fungal antibiotic peptide alamethicin,11 which is also an amphipathic α-helix that forms small, equilibrium, transmembrane pores in bilayers.15,19,58−60 Like GALA at pH 5, alamethicin decreases bilayer resistance rapidly with a single exponential time dependence, and like GALA it causes a measurable decrease in bilayer resistance even at low peptide concentration. Both GALA at pH 5 and alamethicin have the EIS signature expected of a transmembrane equilibrium pore, supporting our mechanistic interpretation of the EIS data for MelP5. In Figure 6A we compare the peptide concentration dependence of the equilibrium resistance values for all of the peptides studied, measured 1 h after peptide addition. MelP5, GALA at pH 5, and alamethicin all behave like equilibrium pore formers where the concentration of the pores in the membrane is driven by the law of mass action. Melittin and GALA at pH 7 are dramatically different. Their equilibrium values of bilayer resistance are not dependent on peptide concentration and are about 80–90% at all concentrations studied11 indicating an absence of true pores. MelP5 is, by far, the most active peptide and causes a resistance drop that is an exponential function of peptide concentration with an asymptote of zero bilayer resistance.
Figure 6.

Comparison of bilayer response to peptides. (A) The equilibrium reduction in normalized bilayer resistance is shown for each of the peptides studied here. Each point is the bilayer resistance measured at the 60 min time point of EIS experiments, relative to the resistance before peptide addition. (B) The peptide-induced change in membrane conductance between 0 and 60 min, calculated and then normalized to the number of peptides in the 1 cm2 area of the supported bilayer.
In Figure 6B, we compare the peptide-induced increase in bilayer conductance, at 60 min, calculated on a per-peptide basis, showing that MelP5 belongs to a uniquely active peptide class, with much higher specific activity than even the archetypal equilibrium pore forming peptides GALA at pH 5 and alamethicin.
Macromolecular Poration
The extraordinary properties of MelP5 in electrochemical impedance spectroscopy experiments prompted us to investigate whether the MelP5 pores are large enough to pass macromolecules. For this, we developed two macromolecule release assays specifically designed to avoid the artifacts discussed earlier: osmotic stress, peptide induced vesicle aggregation/fusion and global bilayer destabilization. First, we use zwitterionic, fluid phase PC/cholesterol bilayers which are robust and inert bilayers that do not readily aggregate or fuse in the presence of cationic peptides. Second, we entrap a low concentration (≤1 mg/mL) of probe macromolecules to essentially eliminate osmotic stress on the vesicle. Third, we are interested only in macromolecule release at P:L ratios that do not promote global vesicle disruption, i.e. P:L < 1:50. In the first assay, we entrapped a 10 kD dextran (labeled with both TAMRA and biotin) inside vesicles. To the outside solution we added streptavidin (∼53 kD) labeled with AlexaFluor488. Peptide-induced release of dextran leads to dextran–streptavidin complex formation, which is measured by resonance energy transfer between the fluorophores that leads to quenching of AlexaFluor488. In the second macromolecule leakage assay, we entrapped chymotrypsin, 24 kD, at low concentration in lipid vesicles61 and used the 23 kD milk protein, casein, labeled with multiple Texas Red fluorophores as a probe of released protease. The Texas Red fluorescence, which is strongly self-quenched in the intact casein molecule, increases upon proteolysis. Released chymotrypsin is quantitated by measuring the initial rate of Texas Red fluorescence change compared with that of intact and lysed vesicles.
Using these two novel assays, we examined the ability of MelP5 to release macromolecules from lipid vesicles and compared MelP5 to melittin, alamethicin, and GALA at pH 5. In both assays MelP5 released vesicle-entrapped macromolecules with a midpoint concentration around P:L ≈ 1:500 (Figure 7). Macromolecule release by MelP5 is essentially complete within about 5 min, a time scale that is consistent with the time scale of resistance changes observed in impedance measurements. Here we measured the release after 60 min. In contrast, neither melittin nor alamethicin release macromolecules significantly until much higher concentrations; the concentrations producing 50% release for these peptides are above P:L = 1:50, conditions under which there are thousands of peptide molecules bound to each vesicle45 and under which amphipathic helices can exhibit detergent-like global vesicle destabilization. GALA at pH 5 does not release macromolecules significantly at these conditions. We conclude that only MelP5 forms macromolecule-sized pores in bilayers at low concentration.
Figure 7.

Peptide induced release of macromolecules from POPC/cholesterol vesicles. (A) Release of a 10 kD dextran from PC/cholesterol vesicles by membrane permeabilizing peptides measured 60 min after addition of peptide. (B) Comparison of MelP5-induded release of small molecule probes ANTS/DPX8 and two macromolecules: the 24 kD protein chymotrypsin and a 10 kD dextran. Fractional leakage was measured 1 h after peptides were added to 1 mM PC/cholesterol vesicles. Each experiment was repeated at least three times. The average ± SD is shown.
GALA at pH 5 has been reported, in one paper, to release a 4 kD dextran from PC vesicles at P:L ratios as low as 1:500.62 However, we observed little release of a 10 kD dextran up to P:L = 1:50. We do not have an explanation for the discrepancy. MelP5 behaved identically at pH 5 as at pH 7 (not shown); thus, we conclude that the assay is unaffected by pH. Our experiments were conducted in parallel and show unequivocally that MelP5 releases macromolecules at low concentration, under conditions where GALA at pH 5 does not. This observation is entirely consistent with the fact that the conductance of MelP5 pores in impedance measurements is much higher than the conductance of GALA pores at pH 5.
In Figure 7B we compare MelP5-induced release of small molecule probes to the release of macromolecular probes from PC/cholesterol vesicles. There is an obvious distinction between the two classes of probe, suggesting that the MelP5 pores increase in size with increasing peptide concentration.
Because melittin has been so broadly studied, it is informative to compare our macromolecular leakage results to the literature. One of the earliest reports on macromolecule release by melittin was a description of hemoglobin release from osmotically balanced erythrocytes by DeGrado and colleagues.13 In that report, melittin was shown to form transient leakage pathways in cell membranes (with a half-life of ∼15 s) that rapidly release a fraction of the cellular hemoglobin (64 kD). Hemoglobin release occurred at very high bound P:L, when about 45% of the outer membrane surface area was occupied by peptide,13 leading the authors to postulate that the “large increase in the area caused by the melittin inserted into erythrocyte membranes should result in local expansion of the outer leaflet of the membrane...leading to further penetration of the melittin and ultimate destruction of the continuity of the membrane.” Explicit pores were not postulated and are not needed to explain the data. Melittin has also been shown to release macromolecules from synthetic lipid vesicles, yet only at high P:L, just as we have reported here. For example, Ladokhin et al.49 showed that melittin (at P:L = 1:50) caused ∼90% release of a 4 kD dextran from PC vesicles and ∼20% release of a 70 kD dextran under the same conditions. Poolman and colleagues53 showed that 50% release of a 10 kD dextran occurs at P:L = 1:50 from PC vesicles and occurs at P:L = 1:20 from PC/cholesterol vesicles. Release of 50% of the 14 kD protein α-lactalbumin occurs at P:L = 1:10.53 Similarly, we previously reported that melittin caused ∼40% release of a 3 kD dextran and 12% release of a 40 kD dextran at P:L = 1:50 in PC vesicles.37 At P:L = 1:500 we previously showed that dextran release by melittin was only marginally greater than background, ≤10%. These published results are consistent with the observation reported here (using the two novel assays) that melittin releases only a small amount of entrapped macromolecules even at P:L = 1:50.
Other peptides, such as cationic antimicrobial peptides, also have been reported to cause macromolecule release from vesicles. These include human defensins,50 rabbit defensins,51,52 small β-sheet peptides,38 and magainin.63 As in the case of melittin, most of these reports describe partial release of macromolecules only at high peptide concentration (P:L ≥ 1:50). Furthermore, cationic AMPs are often active only in anionic bilayers, and not in PC bilayers, suggesting that vesicle fusion and aggregation may be contributing to, and perhaps dominating, macromolecule leakage, a possibility that has been shown to be true in at least one case.50 Macromolecular-sized pores are not likely to account for the observed macromolecule release by any cationic antimicrobial peptide, which probably destabilize bilayers globally due to their interfacial activity.45 Similarly, reports of transient macromolecule-sized leakage in giant unilamellar vesicles63,64 correspond to experiments with high P:L ratios where transient, global destabilization is likely.
MelP5 is a unique pore-forming peptide
Our data show that MelP5, the synthetically evolved, gain-of-function variant of the natural membrane permeabilizing peptide melittin, is unique. It forms very high conductance, equilibrium pores that release macromolecules from lipid vesicles at concentrations as low as P:L = 1:500. The pores form within a few minutes of peptide addition, and leakage is essentially complete within 5–10 min. If we assume a circular “barrel-stave” pore (by no means the only possible pore structure) a minimum of six to eight MelP5 molecules would be required to form a pore large enough to release chymotrypsin. In contrast, the parent peptide melittin forms only transient pores in membranes and only releases macromolecules at very high concentration, P:L ≥ 1:50. Thus, one generation of synthetic molecular evolution, in which just five amino acids were changed, has fundamentally altered the mechanism of membrane permeabilization and has changed both the structure and function of the pores formed. These results highlight the power of iterative combinatorial libraries and orthogonal high-throughput screening (i.e., synthetic molecular evolution) to discover and engineer pore-forming peptides. Furthermore, the novel function and structure of MelP5 could form the foundation for the next generation of engineered or synthetically evolved, self-assembling, macromolecule-sized peptides pores.
Acknowledgments
Funded by NIH (GM095930 to K.H.), NSF DMR (1003441 to K.H. and P.S.), NSF (DMR 1003411) to W.C.W., and by the NSF REU program DBI-1062975 to P.S..
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Cornell B. A.; Braach-Maksvytis V. L. B.; King L. G.; Osman P. D. J.; Raguse B.; Wieczorek L.; Pace R. J. Nature 1997, 387, 580–583. [DOI] [PubMed] [Google Scholar]
- Soman N. R.; Baldwin S. L.; Hu G.; Marsh J. N.; Lanza G. M.; Heuser J. E.; Arbeit J. M.; Wickline S. A.; Schlesinger P. H. J. Clin. Invest. 2009, 119, 2830–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukovnik U.; Gao J.; Cook G. A.; Shank L. P.; Seabra M. B.; Schultz B. D.; Iwamoto T.; Chen J.; Tomich J. M. Biochim. Biophys. Acta 2011, 1818, 1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood J. L.; Jallouk A. P.; Campbell N.; Ratner L.; Wickline S. A. Antivir. Ther. 2013, 18, 95–103. [DOI] [PubMed] [Google Scholar]
- Gerlach S. L.; Rathinakumar R.; Chakravarty G.; Goransson U.; Wimley W. C.; Darwin S. P.; Mondal D. Biopolymers 2010, 94, 617–625. [DOI] [PubMed] [Google Scholar]
- Salomone F.; Cardarelli F.; Di Luca M.; Boccardi C.; Nifosi R.; Bardi G.; Di Bari L.; Serresi M.; Beltram F. J. Controlled Release 2012, 163, 293–303. [DOI] [PubMed] [Google Scholar]
- Duncan R. Nat. Rev. Drug Discovery 2003, 2, 347–360. [DOI] [PubMed] [Google Scholar]
- Krauson A. J.; He J.; Wimley W. C. J. Am. Chem. Soc. 2012, 134, 12732–12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Harroun T. A.; Weiss T. M.; Ding L.; Huang H. W. Biophys. J. 2001, 81, 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. T.; Sun T. L.; Hung W. C.; Huang H. W. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 14243–14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedman G.; Herman K.; Searson P.; Wimley W. C.; Hristova K. Biochim. Biophys. Acta 2013, 1828, 1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladokhin A. S.; Wimley W. C.; White S. H. Biophys. J. 1995, 69, 1964–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGrado W. F.; Musso G. F.; Lieber M.; Kaiser E. T.; Kézdy F. J. Biophys. J. 1982, 37, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon-Grossman M.; Zimmermann H.; Wolf S. G.; Shai Y.; Goldfarb D. J. Phys. Chem. B 2012, 116, 179–188. [DOI] [PubMed] [Google Scholar]
- Krauson A. J.; He J.; Wimley W. C. Biochim. Biophys. Acta 2012, 1818, 1625–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladokhin A. S.; White S. H. J. Mol. Biol. 1999, 285, 1363–1369. [DOI] [PubMed] [Google Scholar]
- Frey S.; Tamm L. K. Biophys. J. 1991, 60, 922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristova K.; Dempsey C. E.; White S. H. Biophys. J. 2001, 80, 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafiso D. S. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 141–165. [DOI] [PubMed] [Google Scholar]
- Parente R. A.; Nir S.; Szoka F. Biochemistry 1990, 29, 8720–8728. [DOI] [PubMed] [Google Scholar]
- Hope M. J.; Bally M. B.; Mayer L. D.; Janoff A. S.; Cullis P. R. Chem. Phys. Lipids 1986, 40, 89–109. [DOI] [PubMed] [Google Scholar]
- Mayer L. D.; Hope M. J.; Cullis P. R. Biochim. Biophys. Acta 1986, 858, 161–168. [DOI] [PubMed] [Google Scholar]
- Lin J.; Szymanski J.; Searson P. C.; Hristova K. Langmuir 2010, 26, 3544–3548. [DOI] [PubMed] [Google Scholar]
- Cruz J.; Mihailescu M.; Wiedman G.; Herman K.; Searson P. C.; Wimley W. C.; Hristova K. Biophys. J. 2013, 104, 2419–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.; Motylinski J.; Krauson A. J.; Wimley W. C.; Searson P. C.; Hristova K. Langmuir 2012, 28, 6088–6096. [DOI] [PubMed] [Google Scholar]
- Lin J.; Merzlyakov M.; Hristova K.; Searson P. C. Biointerphases 2008, 3, FA33. [DOI] [PubMed] [Google Scholar]
- Nikolov V.; Lin J.; Merzlyakov M.; Hristova K.; Searson P. C. Langmuir 2007, 23, 13040–13045. [DOI] [PubMed] [Google Scholar]
- Bevington P. R.Data reduction and error analysis for the physical sciences; McGraw-Hill Book Company: New York, 1969. [Google Scholar]
- Wiener M. C.; White S. H. Biophys. J. 1992, 61, 434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamba Y.; Yamazaki M. J. Phys. Chem. B 2009, 113, 4846–4852. [DOI] [PubMed] [Google Scholar]
- Matsuzaki K. Biochim. Biophys. Acta 1998, 1376, 391–400. [DOI] [PubMed] [Google Scholar]
- Nguyen L. T.; Schibli D. J.; Vogel H. J. J. Pept. Sci. 2005, 11, 379–389. [DOI] [PubMed] [Google Scholar]
- Matsuzaki K.; Yoneyama S.; Fujii N.; Miyajima K.; Yamada K.-I.; Kirino Y.; Anzai K. Biochemistry 1997, 36, 9799–9806. [DOI] [PubMed] [Google Scholar]
- Falla T. J.; Karunaratne D. N.; Hancock R. E. W. J. Biol. Chem. 1996, 271, 19298–19303. [DOI] [PubMed] [Google Scholar]
- Girshman J.; Greathouse D. V.; Koeppe R. E. II; Andersen O. S. Biophys. J. 1997, 73, 1310–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maget-Dana R.; Ptak M. Biophys. J. 1997, 73, 2527–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch J. M.; Marks J. R.; Rathinakumar R.; Wimley W. C. Biochemistry 2007, 46, 12124–12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinakumar R.; Wimley W. C. J. Am. Chem. Soc. 2008, 130, 9849–9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J. P.; Wu C.; Yang J. L. Eur. J. Biochem. 2000, 267, 3289–3300. [DOI] [PubMed] [Google Scholar]
- Krauson A. J.; He J.; Hoffmann A. R.; Wimley A. W.; Wimley W. C. ACS Chem. Biol. 2013, 8, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. T.; Chen F. Y.; Huang H. W. Biochemistry 2004, 43, 3590–3599. [DOI] [PubMed] [Google Scholar]
- Raghuraman H.; Chattopadhyay A. Biosci. Rep. 2007, 27, 189–223. [DOI] [PubMed] [Google Scholar]
- Ladokhin A. S.; White S. H. Biochim. Biophys. Acta 2001, 1514, 253–260. [DOI] [PubMed] [Google Scholar]
- Bechinger B.; Lohner K. Biochim. Biophys. Acta 2006, 1758, 1529–1539. [DOI] [PubMed] [Google Scholar]
- Wimley W. C. ACS Chem. Biol. 2010, 5, 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai Y. Biopolymers 2002, 66, 236–248. [DOI] [PubMed] [Google Scholar]
- Almeida P. F.; Pokorny A. Biochemistry 2009, 48, 8083–8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epand R. F.; Maloy W. L.; Ramamoorthy A.; Epand R. M. Biochemistry 2010, 49, 4076–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladokhin A. S.; Selsted M. E.; White S. H. Biophys. J. 1997, 72, 1762–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley W. C.; Selsted M. E.; White S. H. Protein Sci. 1994, 3, 1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristova K.; Selsted M. E.; White S. H. J. Biol. Chem. 1997, 272, 24224–24233. [DOI] [PubMed] [Google Scholar]
- Hristova K.; Selsted M. E.; White S. H. Biochemistry 1996, 35, 11888–11894. [DOI] [PubMed] [Google Scholar]
- van den B. G.; Kusters I.; Velasquez J.; Mika J. T.; Krasnikov V.; Driessen A. J.; Poolman B. Methods 2008, 46, 123–130. [DOI] [PubMed] [Google Scholar]
- Scholtz J. M.; Hong Q.; York E. J.; Stewart J. M.; Baldwin R. L. Biopolymers 1991, 31, 1463–1470. [DOI] [PubMed] [Google Scholar]
- Subbarao N. K.; Parente R. A.; Szoka F. C.; Nadasdi L.; Pongracz K. Biochemistry 1987, 26, 2964–2972. [DOI] [PubMed] [Google Scholar]
- Parente R. A.; Nadasdi L.; Subbarao N. K.; Szoka F. C. Biochemistry 1990, 29, 8713–8719. [DOI] [PubMed] [Google Scholar]
- Goormaghtigh E.; Demeutter J.; Szoka F.; Cabiaux V.; Parente R. A.; Ruysschaert J. M. Eur. J. Biochem. 1991, 195, 421–429. [DOI] [PubMed] [Google Scholar]
- Leitgeb B.; Szekeres A.; Manczinger L.; Vagvolgyi C.; Kredics L. Chem. Biodiversity 2007, 4, 1027–1051. [DOI] [PubMed] [Google Scholar]
- Qian S.; Wang W.; Yang L.; Huang H. W. Biophys. J. 2008, 94, 3512–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K.; Ludtke S. J.; Worcester D. L.; Huang H. W. Biophys. J. 1996, 70, 2659–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks J. R.; Placone J.; Hristova K.; Wimley W. C. J. Am. Chem. Soc. 2011, 133, 8995–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol F.; Nir S.; Szoka F. C. Jr. Biophys. J. 2000, 78, 818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamba Y.; Ariyama H.; Levadny V.; Yamazaki M. J. Phys. Chem. B 2010, 114, 12018–12026. [DOI] [PubMed] [Google Scholar]
- Fuertes G.; Garcia-Saez A. J.; Esteban-Martin S.; Gimenez D.; Sanchez-Munoz O. L.; Schwille P.; Salgado J. Biophys. J. 2010, 99, 2917–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]