

Abstract
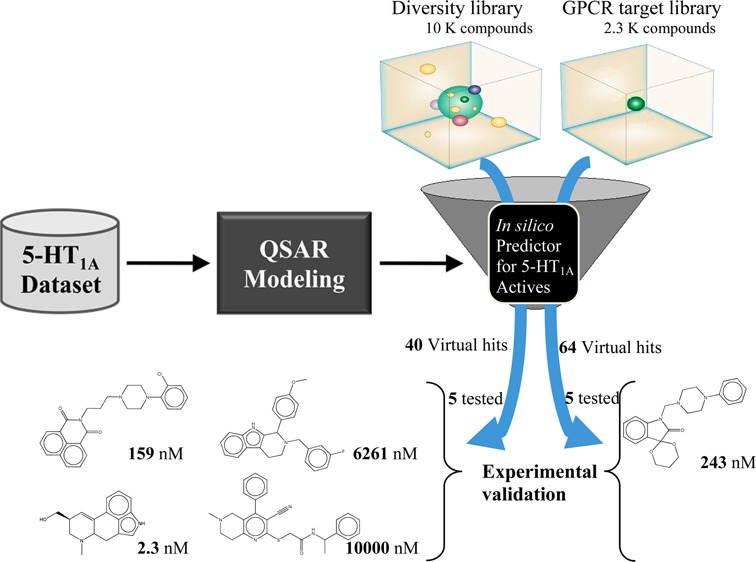
The 5-hydroxytryptamine 1A (5-HT1A) serotonin receptor has been an attractive target for treating mood and anxiety disorders such as schizophrenia. We have developed binary classification quantitative structure–activity relationship (QSAR) models of 5-HT1A receptor binding activity using data retrieved from the PDSP Ki database. The prediction accuracy of these models was estimated by external 5-fold cross-validation as well as using an additional validation set comprising 66 structurally distinct compounds from the World of Molecular Bioactivity database. These validated models were then used to mine three major types of chemical screening libraries, i.e., drug-like libraries, GPCR targeted libraries, and diversity libraries, to identify novel computational hits. The five best hits from each class of libraries were chosen for further experimental testing in radioligand binding assays, and nine of the 15 hits were confirmed to be active experimentally with binding affinity better than 10 μM. The most active compound, Lysergol, from the diversity library showed very high binding affinity (Ki) of 2.3 nM against 5-HT1A receptor. The novel 5-HT1A actives identified with the QSAR-based virtual screening approach could be potentially developed as novel anxiolytics or potential antischizophrenic drugs.
Introduction
The 5-HT1A receptor is one the most abundant subtypes of the 5-HT receptor family; it is highly enriched in the raphe nucleus, cerebral cortex, hippocampus, septum, and amygdale. Because of its presence in the brain regions whose functions are heavily involved in mood and anxiety disorders, the 5-HT1A receptor has been actively explored as a target for antipsychotic, anxiolytic and antidepressant drug discovery. Several 5-HT1A receptor agonists such as buspirone and tandospirone are medications approved to treat anxiety and depression. Some of the atypical antipsychotic drugs, such as aripiprazole, clozapine, and olanzapine, are also partial 5-HT1A receptor agonists and are sometimes used in low doses in combination with standard antidepressants to achieve faster symptom relief and greater overall efficacy.1−3 Furthermore, 5-HT1A receptors have been actively studied as potential drug targets for treating the cognitive deficits in schizophrenia:4 the activation of the 5-HT1A receptor has been linked to the increased dopamine release, which could improve certain symptoms of schizophrenia.5,6 Besides being traditionally explored as targets for psychiatric disorders, 5-HT1A receptors have recently received considerable attention as targets to develop treatments for neurodegenerative diseases.7 Recent discoveries have shown that the modulation of 5-HT1A receptor may present a novel mechanism for treating the Alzheimer’s disease or help relieve the symptoms of Parkinson’s disease.8
Although several drugs are available on market that acting via a 5-HT1A receptor-mediated mechanism, only a few were originally developed to selectively target 5-HT1A receptors. In addition, current 5-HT1A modulators exhibit various side effects or even some severe side effects,9 preventing their widespread clinical use. These side effects could be due to potent off-target actions.10,11 Moreover, some patients have been reported to be nonresponders or poor-responders to current medications.12 Thus, there is still a need for developing novel 5-HT1A receptor modulators.
Virtual screening (VS) is a common and efficient approach for the discovery of new lead compounds.13 Structure-based VS has been the most popular approach to identify putative receptor actives in chemical libraries,14 but in recent years, ligand-based cheminformatics approaches have been used widely in VS applications15 as well. In the case of 5-HT1A receptors, when the experimental structure of the receptor is unknown, ligand-based approaches can be explored for VS. Our group has been investigating the use of quantitative structure activity relationships (QSAR) models as effective VS tools, and several successful applications have been reported.15−17 The QSAR approach explores existing chemical databases with biologically activities to establish statistically significant and externally predictive models that allow one to predict biological activity of untested compounds from their chemical structures. Once a QSAR model has been developed, it can be used to search libraries of chemical structures with the aim of finding new, structurally different hit(s) with the desired biological activity.18 Indeed, the size and diversity of chemical libraries available for both virtual and experimental screening have been growing rapidly in recent years, providing growing opportunities to use VS methods to identify novel hits among available chemicals.
In this paper, we report on the development of rigorously validated QSAR models of the 5-HT1A receptor binders using previously reported bioactivity data for the receptor ligands. We employ these models for virtual screening to arrive at a small number of 15 prioritized computational hits that have been subject to experimental validation. We show that 9 of these 15 hits show appreciable binding affinity ranging between 10 μM and 2.3 nM. This study confirms that QSAR-based virtual screening is an effective tool to discover novel bioactive compounds that can be further pursued as novel antipsychotics.
Methods
Data Sets for Model Building and Validation
Data Sets for QSAR Model Building
The data for 5-HT1A activity were retrieved from the National Institute of Mental Health (NIMH) Psychoactive Drug Screening Program (PDSP) Ki database.19 In this study, we used 10 μM as the cutoff value to define actives versus inactives, and we only retrieved the experimental radioligand binding data with cloned human cell lines using the ligand [3H]-8-OH-DPAT. By submitting such queries, 180 unique compounds were designated as 5-HT1A actives with binding affinities ranging from 0.115 nM to 8.41 μM. We also retrieved 78 inactives, which were shown experimentally to have no binding affinity to the 5-HT1A receptor at 10 μM concentration.
Data Set for Independent External Validation
An additional 66 putative 5-HT1A actives were extracted from the World of Molecular Bioactivity (WOMBAT) database.20 This commercial database is a collection of chemical annotations published in top medicinal chemistry journals; therefore, the binding data therein are considered as highly reliable. Compounds extracted from WOMBAT were regarded as 5-HT1A actives when they satisfied all of the following criteria: (1) compounds were tested on cloned human species cell lines; (2) [3H]-8-OH-DPAT was used as hot ligand; (3) their binding affinities were higher than 10 μM. Notably, all 66 compounds were different structurally from compounds in the modeling set.
Data Set Curation
Prior to QSAR model development, chemical structures were curated following the guidelines we published earlier.21 First, all molecules were cleaned using the “Wash Molecules” module in Molecular Operating Environment (MOE,22 version 2009.10). Second, the routine Standardizer was used for structure canonicalization and transformation, JChem 5.2, 2009, ChemAxon (http://www.chemaxon.com). Finally, duplicates were detected by the analysis of the normalized molecular structures. For chemicals extracted from the PDSP Ki database, 75 duplicate compounds for 5-HT1A actives and 17 for inactives, i.e., different salts or isomeric states, were detected. The functional data for duplicates were verified to be identical, so in each case a single example was removed. The curated subset of the 5-HT1A ligands from PDSP included 166 unique organic compounds (105 actives and 61 inactives). The chemical structures of these compounds are available in the Supporting Information.
Libraries for Virtual Screening
Drug-Like Screening Libraries
Drug-like libraries are collections of currently marketed drugs or drug candidates in the approval process. For our study, we used the World Drug Index (WDI) database and the Prestwick Chemical Library (PCL) (http://www.prestwickchemical.fr/). The WDI library is maintained by Derwent Publications and our version contains 59 000 pharmacologically active compounds, including all marketed drugs and those in the development. The PCL contains 1200 small molecules, all of which are marketed drugs. By design, compounds in the PCL feature a high chemical and pharmacological diversity as well as a high degree of bioavailability and safety in human.
Targeted Screening Libraries
The 5-HT1A receptor belongs to the large family of GPCRs; therefore, GPCR-targeted libraries such as TimTec (http://www.timtec.net/) AntiTarg-G library and ASINEX (http://www.asinex.com/) Synergy GPCRs CNS library were used for virtual screening to identify new putative 5-HT1A ligands. The TimTec AntiTarg-G library is a plated screening set of 2300 molecules that contain chemical scaffolds present in compounds reported in the technical or patent literature to bind GPCRs. Similarly, the ASINEX Synergy GPCRs CNS library is composed of 3233 compounds rich in GPCRs drug-like pharmacophore fragments.
Diversity Screening Libraries
The diversity libraries were also from TimTec and ASINEX, namely TimTec Diversity Set 10K and ASINEX Diverse Set-Platinum 5K. The TimTec diversity screening set contains 10 000 samples selected from the company’s stock of over 180 000 compounds as the most structurally diverse and competitively priced collection. The assorted set stands out as having a diverse selection of singletons identified in the TimTec stock of readily available compounds. In addition, it is also a compound collection that complies with Lipinski’s Rule of Five. ASINEX Diversity Set-Platinum 5K, which contains 5072 compounds, is an assortment of all ASINEX libraries based on the compounds’ structural diversity. This set of compounds is claimed by ASINEX to be a great starting point that requires a diverse chemical collection.
Training, Test, and External Validation Sets Selection
We have followed the rigorous QSAR modeling workflow for model building, validation, and virtual screening (Figure 1) developed in our laboratory.23 This workflow requires that an external predictive power should be established for every QSAR model. Thus, we have employed the external 5-fold cross-validation protocol where the modeling set is randomly split into five subsets of approximately equal size (20% of compounds). Each time, one subset is used as an external validation set, and the union of four other subsets is used as the modeling set, i.e., each modeling set contains 80% of compounds. Modeling sets were further partitioned into multiple pairs of representative training and test sets of different sizes using the Sphere Exclusion algorithm developed in our laboratory,24,25 which ensures the closeness in chemical spaces within the paired data sets.
Figure 1.

The workflow of QSAR model building, validation and virtual screening as applied to the 5-HT1A data set of 105 actives and 61 inactives from PDSP.
Generation of 2D Molecular Descriptors
The SMILES26 strings of each compound in the 5-HT1A data set were converted to 2D chemical structures using the MOE software package. The Dragon27 software (version 5.5) was used to calculate a wide range of topological indices of molecular structures. Dragon descriptors with zero values or zero variance were excluded, whereas the remaining descriptors were range-scaled within the interval of 0–1 prior to distance calculations and model building because the absolute scales for the variety of Dragon descriptors can differ by orders of magnitude.28
QSAR Modeling Methods
k Nearest Neighbors (kNN) Classification Method
The kNN classification QSAR method28,29 is based on the idea that the class that a compound belongs to can be predicted by the class membership of its nearest neighbors (i.e., most similar compounds), taking into account weighted similarities between the compound and its nearest neighbors. Because our implementation of kNN approach includes variable selection, the similarity is evaluated using only a subset of all descriptors, which is optimized by a simulated annealing (SA) approach to achieve the best correct classification rate (CCR):30
| 1 |
where N1total and N2 are the number of actives and inactives in the data set, and N1corr and N2 are the number of known actives correctly predicted as actives (true positives) and the number of inactives correctly predicted as inactives (true negatives), respectively. Unlike total accuracy, CCR inherently took into account the imbalance in class membership of objects in the data set, which was important because the 5HT1A data set was imbalanced containing 105 actives and 61 inactives. The accuracy of the models was characterized by the leave-one-out cross-validation (LOO-CV) CCRtrain for the training sets and predictive CCRtest for the test sets. Additional details of this approach can be found elsewhere.28,31 Models with high CCRtrain and CCRtest were used to predict compounds included neither in the training nor in the test set as a matter of external validation. Theoretically, any compound represented by the corresponding chemical descriptors can be assigned to a class (predicted class) using the classification kNN approach. However, if the distances between the query compound and all of its k nearest neighbors in the training set are higher than some threshold, the query compound is considered as highly dissimilar from all of the training set compounds, and the prediction of its activity using the kNN approach is considered unreasonable. Therefore, a similarity threshold (or model applicability domain, AD) was introduced to avoid making predictions for compounds that differ substantially from the training set molecules.32 The distance threshold is defined as follows:
| 2 |
Here, y̅ is the average Euclidean distance between each compound and its k nearest neighbors within the training set, σ is its standard deviation of these distances, and n is an arbitrary parameter called the n-cutoff to control the significance level. Typically, we set n to 0.5, which places the boundary for deciding whether a compound is within or outside of the AD at one-half of the standard deviation from y̅. It is important to notice that increasing the value of n would increase the number of compounds in the external set that are considered within the AD but could decrease the accuracy of the prediction due to the inclusion of dissimilar nearest neighbors.
Random Forest (RF) Classification Method
Random Forest is a machine learning technique that consists of many decision trees and outputs the consensus prediction from the individual trees.33 In this study, the implementation of an RF34 algorithm available in the R Project35 (Version 2.14.1) was used. In the RF modeling procedure, N samples (modeling set compounds) are randomly drawn with replacement from the original data set. These samples were used to construct n training sets and to build n trees. In these studies, n was equal to 500. Predictions were made by averaging predicted activities over all trees in the final forest.
Support Vector Machines (SVM) Classification Method
The original version of SVM was developed by V. Vapnik36 and the description of the SVM algorithm can be found in many publications.37,38 Briefly, molecular descriptors are first mapped onto a high dimensional feature space using various kernel functions. Then, SVM finds a separating hyperplane with the maximal margin in this high dimensional space to separate compounds with different activities. Models built with SVM allow for the prediction of the target property using a set of descriptors solely calculated from the structure of a given compound.
In this study, we used the WinSVM program developed in our group (freely available for academic laboratories upon request) implementing the open-source LIBSVM package.37 The WinSVM program provides users with a convenient graphical interface to prepare input data, perform SVM modeling, and select models for external evaluation. The program also allows one to visualize molecular structures and produce various plots, making the use of SVM easier and more appropriate for QSAR modeling to obtain robust and predictive models and apply them to virtual libraries.39
Robustness of QSAR Models
A Y-randomization test was used to ensure the model robustness.40 This test includes rebuilding the training set models using randomized activities (Y-vector) of the training set and comparing the resulting model statistics with that of the models built with original data. It is expected that models built with randomized activities should have significantly lower CCR values for both the training and test sets. The one-tail hypothesis testing was applied to confirm the robustness of QSAR models. In this approach, two alternative hypotheses are formulated: (1) for H0, h = μ; (2) for H1, h > μ, where μ is the average value of CCRtrain for Y-randomization models and h is that for the actual models. The null hypothesis (H0) states that the QSAR models for the actual data set are not significantly better than random models, whereas the H1 hypothesis assumes the opposite, suggesting that the actual models are significantly better than the random models. Hypothesis rejection is based on a standard one-tail test, which involves the following three steps: (1) determine the average value of CCRtrain (μ) and its standard deviation (σ) for random models; (2) calculate the Z score that corresponds to the average value of CCRtrain (h) for the actual models using the following equation:
| 3 |
(3) Compare this Z score with the tabular critical values of Zc at different levels of significance (α)60 to determine the level at which H0 should be rejected. If the Z score is higher than tabular values of Zc, one concludes that at the level of significance that corresponds to that Zc, H0 should be rejected while H1 should be accepted. The Y-randomization test was applied to all data sets considered in this study, and the test was repeated twice in each case.
Virtual Screening using Consensus Models
As illustrated in the workflow of Figure 1, QSAR models that passed both internal and external validation were employed for virtual screening. A global applicability domain (calculated using all descriptors) was applied first to filter out compounds that were structurally highly different (beyond the AD threshold calculated with n-cutoff = 0.5, cf. eq 2) from compounds in the modeling set. All 105 known 5-HT1A actives extracted from PDSP were used as probes in the chemical similarity calculations. Then, all acceptable models obtained with various machine learning techniques, kNN, RF, and SVM, were applied in consensus to predict the class of compounds in the external library that were found within the global applicability domain. Furthermore, the results were accepted only when the compound was found within the applicability domains of more than 50% of all models used in consensus prediction and the standard deviation of estimated means across all models was small. During the consensus prediction of kNN, we restricted ourselves to the most conservative AD for each model using the n-cutoff = 0.5 in eq 2.
All the modeling and virtual screening calculations were carried out at a 352-processor Beowulf Linux cluster of the ITS Research Computing Division of the University of North Carolina at Chapel Hill (UNC—CH). The CPU nodes are Intel Xeon IBM BladeCenter of Dual Intel Xeon 2.8 GHz, with 2.5GB RAM on each node. The cluster runs the Red Hat Enterprise Linux 4.0 (32-bit) and the nodes communicate via a Gigabit Ethernet network. The processing speed of QSAR-based screening is ca. 100K compounds per minute, fairly high compared to other methods. In addition, the data processing speed was found to be able to scale linearly with the size of the screening library.
Fingerprint Based Similarity Search
The chemical similarity search was conducted with the MOE 2006.08 package using the standard protocol. The MACCS structural keys were utilized with the Tanimoto Coefficient (Tc) as the similarity metric. In the case that the hits from individual searches were in duplicate, a special Scientific Vector Language (SVL) script was employed to remove them by considering both chemical topology and chirality.
Radioligand Binding Assays
The experimental tests were performed by the National Institute of Mental Health PDSP program (http://pdsp.med.unc.edu/indexR.html). The five computational hits from Prestwick library were purchased from Sigma-Aldrich, and the ten additional compounds were purchased from TimTec LLC (cf. Certificate of Analysis in the Supporting Information). Radioligands were purchased by PDSP from Perkin-Elmer or GE Healthcare. Competition binding assays were performed using transfected or stably expressing cell membrane preparations as previously described41,42 and all experimental details are available online (http://pdsp.med.unc.edu/pdspw/binding.php).
Results
QSAR Model Development to Classify 5-HT1A Actives versus Inactives
Calculation of Descriptors
Dragon27 software (version 5.5) was used; initially, 880 chemically relevant 0D-2D descriptors were calculated. A total of 672 descriptors were eventually used for 5-HT1A data set after removing descriptors with zero value or zero variance. Furthermore, all descriptors were range-scaled to fall between the values of zero and one.
kNN Classification
The kNN QSAR method with variable selection afforded multiple models with optimal accuracy characterized by CCR for both training and test sets. For the five internal modeling sets (each one is comprised of approximately 80% of the entire 5-HT1A data set) generated after applying the external 5-fold cross-validation protocol (cf. Methods), there were a total of 838 models with both CCRtrain and CCRtest equal to or higher than 0.80. Most models with CCRtest ≥ 0.80 also had corresponding CCRtrain ≥ 0.80, but the opposite was not always true. The high accuracy of the models implied that these models could correctly identify the majority of actives and inactives in both the training and test sets.
RF Classification
The RF QSAR classification method was applied for the same five modeling sets used for kNN modeling. For each modeling set, the decision trees were tuned and selected under the RF algorithm and only the final model (a set of classification trees) was reported (cf. Table 1). The external 5-fold cross-validation procedure (same as when using kNN) was employed using the same division of the data set into five folds; the resulting external accuracy ranged between 0.68 and 0.84 (cf. Table 1).
Table 1. QSAR Model Validations on the External 5-Fold CV Sets As Well As the Additional Independent External Set from WOMBAT.
| confusion matrix |
statistics |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| machine learning methods | external sets | prediction CCR | N(1)a | N(2)a | TP | TN | FP | FN | SE | SP | EN(1) | EN(2) | |
| 1 | 0.86 | 19b | 14 | 17 | 11 | 3 | 2 | 0.89 | 0.79 | 1.61 | 1.76 | ||
| 2 | 0.61 | 20 | 13 | 15 | 6 | 7 | 5 | 0.75 | 0.46 | 1.16 | 1.30 | ||
| k-nearest neighbor | 3 | 0.77 | 22 | 11 | 20 | 7 | 4 | 2 | 0.91 | 0.64 | 1.43 | 1.75 | |
| 4 | 0.86 | 20 | 13 | 19 | 10 | 3 | 1 | 0.95 | 0.77 | 1.61 | 1.88 | ||
| 5 | 0.68 | 23 | 10 | 22 | 4 | 6 | 1 | 0.96 | 0.40 | 1.23 | 1.80 | ||
| Cumulative | 0.76 | 104 | 61 | 93 | 38 | 23 | 11 | 0.89 | 0.62 | 1.41 | 1.71 | ||
| WOMBAT | N/A | 66 | 0 | 62 | N/A | N/A | 4 | 0.94 | N/A | N/A | N/A | ||
| 1 | 0.80 | 20 | 14 | 16 | 11 | 3 | 4 | 0.80 | 0.79 | 1.58 | 1.59 | ||
| 2 | 0.68 | 20 | 13 | 15 | 8 | 5 | 5 | 0.75 | 0.62 | 1.32 | 1.42 | ||
| random forest | 3 | 0.84 | 22 | 11 | 21 | 8 | 3 | 1 | 0.95 | 0.73 | 1.56 | 1.88 | |
| 4 | 0.74 | 20 | 13 | 19 | 7 | 6 | 1 | 0.95 | 0.54 | 1.35 | 1.83 | ||
| 5 | 0.83 | 23 | 10 | 22 | 7 | 3 | 1 | 0.96 | 0.70 | 1.52 | 1.88 | ||
| Cumulative | 0.78 | 105 | 61 | 93 | 41 | 20 | 12 | 0.89 | 0.67 | 1.46 | 1.71 | ||
| WOMBAT | N/A | 66 | 0 | 62 | N/A | N/A | 4 | 0.94 | N/A | N/A | N/A | ||
| 1 | 0.87 | 20 | 14 | 19 | 11 | 3 | 1 | 0.95 | 0.79 | 1.36 | 1.88 | ||
| 2 | 0.68 | 20 | 13 | 18 | 6 | 7 | 2 | 0.90 | 0.46 | 1.25 | 1.64 | ||
| support vector machines | 3 | 0.95 | 22 | 11 | 22 | 10 | 1 | 0 | 1.00 | 0.91 | 1.83 | 2.00 | |
| 4 | 0.76 | 20 | 13 | 18 | 8 | 5 | 2 | 0.90 | 0.62 | 1.40 | 1.72 | ||
| 5 | 0.76 | 23 | 10 | 21 | 6 | 4 | 2 | 0.91 | 0.60 | 1.39 | 1.75 | ||
| Cumulative | 0.80 | 105 | 61 | 98 | 41 | 20 | 7 | 0.93 | 0.67 | 1.48 | 1.82 | ||
| WOMBAT | N/A | 66 | 0 | 62 | N/A | N/A | 4 | 0.96 | N/A | N/A | N/A | ||
N(1) = number of actives, N(2) = number of inactives, TP = true positive (actives predicted as actives), FP = false positives (inactives predicted as actives), FN = false negatives (actives predicted as inactives), TN = true negative (inactives predicted as inactives), SE = sensitivity = TP/N(1), SP = specificity = TN/N(2), EN = the normalized enrichment, EN(1) = (2TP × N(2))/(TP × N(2) + FP × N(1)), EN(2) = (2TN × N(1))/(TN × N(1) + FN × N(2)), and CCR = correct classification rate.
Some N(1) actives of and N(2) inactives were out of application domain of all consensus models, thus having no prediction. Only data for compounds found within the AD were used for statistical summaries.
SVM Classification
The same five sets of modeling compounds were also used to build SVM QSAR classification models. Due to the limited number of models selected by using 0.80 as the cutoff for both CCRtrain and CCRtest, and the potential unreliable predictions on external compounds by using only few models, the models with both CCRtrain and CCRtest ≥ 0.65 were considered acceptable and were selected for consensus prediction; a total of 207 of such models were retained.
QSAR Model Validations
In addition to the internal validation of kNN, RF, and SVM models using test sets, Y-randomization and external validation are the critical steps of our QSAR workflow (Figure 1). Only models that have been validated by these two steps can be employed for external prediction and virtual screening.32 Furthermore, a data set of 66 5-HT1A actives from WOMBAT was used as the independent external validation set.
Y-Randomization Test
The binary annotations of 5-HT1A as actives or inactives in the training set were randomly shuffled, and kNN, RF, and SVM classification models were built with the same parameter settings as used in the original data modeling. The Y-randomization test was performed once for each training/test set split and all its runs showed that almost all models had both CCRtrain and CCRtest around 0.50, which is equivalent to random guess (cf. Figure 2). Because no classification rules or hyperplanes can be identified by SVM classification methods after the random shuffling of the original 5-HT1A annotations, no prediction could be further made for the test set compounds, thus no statistics were reported in Figure 2. Moreover, the one-tail hypothesis was applied, and a Z score of 2.17 was calculated. After comparing this Z score with the tabular critical values of Zc at different levels of significance (α)60, we concluded that with 98.48% confidence the null hypothesis H0 should be rejected, implying that the difference of CCR for models built with the original data versus those built with the data subjected to Y-randomization was significant.
Figure 2.

Box plots for the external prediction accuracy (CCRevs) of different QSAR classification models for 5-HT1A actives. Lower horizontal line of the box, 20th quantile; middle line, median; upper line, 80th quantile. Lower vertical line, range of data between 20th quantile and the minimum; upper vertical line, range of data between the 80th quantile and the maximum. Square dot, mean.
External Cross-Validation
The external 5-fold cross-validation approach was employed for the external prediction, i.e., for each split, models were built using ∼80% of the 5-HT1A data set to predict the remaining randomly excluded ∼20% of compounds. Consensus predictions were carried out using models with both high CCRtrain and CCRtest. Exactly the same external sets were employed for validation of kNN, RF, and SVM classification models, and the results are compared and summarized in both Figure 2 and Table 2. It is noticed that only 19 out of 20 5-HT1A actives in the first external set were predicted by kNN classification models; the remaining 5-HT1A active compound could not be predicted by our consensus kNN models because it was outside of the models’ applicability domain. The consensus score, in terms of the average class number in classification QSAR, was calculated by the fraction of models that predicted a compound as inactive divided by the total number of models used for prediction plus one; this formula is based on the annotation of actives as having class label of “1” and inactives as those with the class label of “2”. Thus, if a set of classification QSAR models is used to predict a compound’s activity, the mean predicted class could range between 1 (when all models predict this compound as active) and 2 (when all models predict this compound as inactive). Obviously, when models disagree, the mean class label may take any value between 1 and 2. Under n-cutoff = 0.5 (cf. eq 2), most of the external validation set achieved a rather high prediction accuracy. The highest accuracy for the kNN classification models across all external validation folds was observed for the fourth external set split (Table 1), with sensitivity of 95% (for actives) and specificity of 77% (for inactives), leading to CCRevs = 0.86. Increasing n-cutoff (eq 2) raised the model coverage for predicting of both active and inactive compounds because of the extended applicability domain for individual models. However, the prediction with extended applicability domain for consensus models also comes with lower confidence level (data not shown). Generally speaking, to have reliable predictions but also broad model coverage, a reasonable n value should be selected.
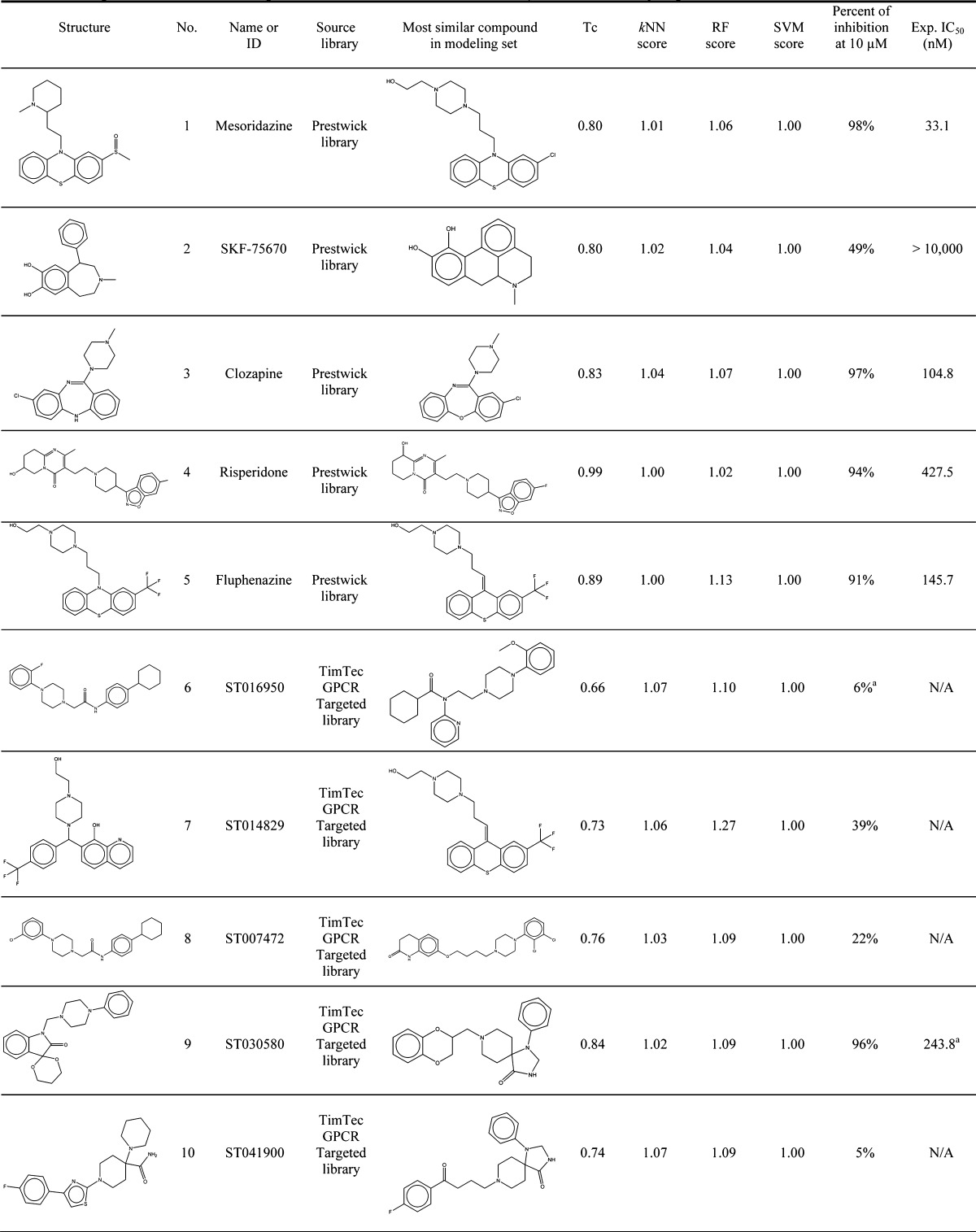
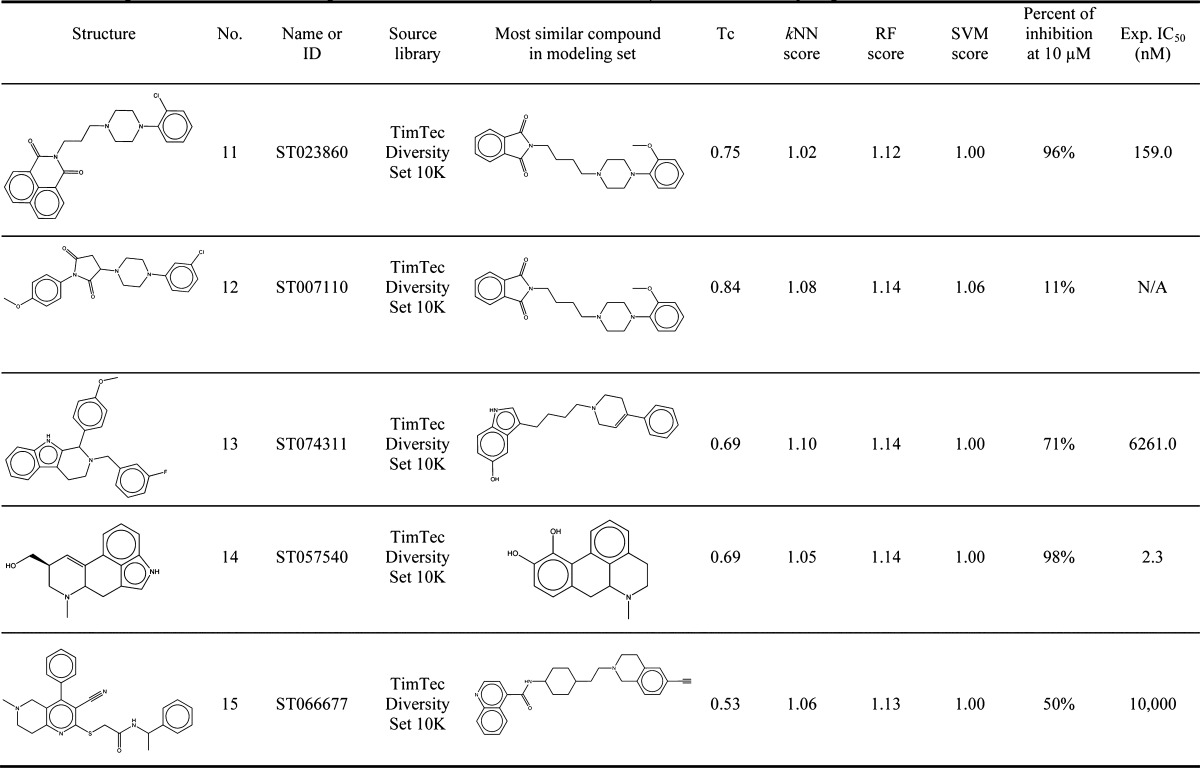
Table 2. Prediction Scores and Experimental Data for 15 Hits Identified by Virtual Screening As Putative 5-HT1A Actives.
The full IC50 curve was generated in further experiments and the Ki value was determined.
The consensus scores for each of the compounds in the external sets predicted by all three (kNN, RF, and SVM) classification models are shown in Table 1. The models with qualifying CCRtrain and CCRtest values in excess of 0.80 and the highest CCRevs resulting for a single split of the data in the 5-fold validation protocol were used in consensus for virtual screening. Notably, the kNN models chosen for the prediction had relatively small ncutoff (0.5) and relatively broad coverage (≥50%) for compounds in external data sets.
Independent External Validation
We used models built for a PDSP data set of 166 5-HT1A active/inactive compounds to predict the class label for 66 known 5-HT1A actives from WOMBAT. We should emphasize that these latter compounds had unique structures that were different from the existing PDSP actives. Among the 66 actives (all were within the applicability domain), 62 were accurately annotated by kNN consensus prediction (SE = 0.94, Table 1). Thus, the majority of ligands were predicted correctly by our consensus models. The only four incorrectly predicted compounds had the consensus prediction scores of 1.51, 1.54, 1.55, and 1.67, respectively. As illustrated above, consensus prediction is based on the results obtained by all validated predictive models. The closer the value is to 1.0, the greater the confidence in the prediction of a compound being 5-HT1A active, whereas the value closer to 2.0 implies greater confidence in predicting a compound to be inactive. Because the predicted class labels for the four false negative 5-HT1A actives did not exceed 1.67, and compounds were within the applicability domain of only 70 models (i.e., approximately 30% of all models), the kNN prediction is considered as of low confidence. When RF and SVM were applied, the prediction accuracy for the additional 66 actives from WOMBAT was also high, ranging from CCRevs = 0.94 to 0.96 (Table 1).
The success of this independent external validation highlights the power of our QSAR models in predicting the possible 5-HT1A binding classifications, so that these models can be reliably applied for virtual screening to identify novel 5-HT1A receptor actives.
Model-Based Virtual Screening
Models with the highest predicted CCRevs for each machine learning method, i.e., 217 kNN models with both internal and external CCRtrain and CCRtest equal to or greater than 0.80 and CCRevs equal to 0.86, one RF model with CCRevs equal to 0.84, and 47 SVM models with CCRevs equal to 0.95, were used in consensus for virtual screening. Initially, 55 ,384 compounds from the Prestwick and WDI libraries were screened to identify putative 5-HT1A actives; the number of compounds falling within the AD when the n-cutoff (cf. eq 2) was varied are shown in Figure 4. It should be noted that there is a big overlap between compounds screened by Prestwick and WDI libraries, and hits from WDI also share the same or highly similar structures with compounds in the modeling set. Therefore, only the screening statistics from the Prestwick library are shown in Figure 4. The compounds within the AD defined by n-cutoff = 0.5 were further predicted by kNN consensus models. 234 compounds from the Prestwick library were predicted as actives by at least one of the kNN consensus models. To narrow the hit list and obtain the higher confidence level for each prediction, we took both the consensus score (average class number) and model coverage into consideration; model coverage was defined as a fraction of models for which a compound was found to fall within the respective applicability domain. In particular, only hits with an average class number between 1.0 and 1.1 and the model coverage over 50% were selected. In total, 125 compounds from PCL and 181 from WDI were identified that satisfied both criteria.
Figure 4.

The hit rates of putative 5-HT1A actives identified in five different types of screening libraries by the global similarity search at three different values of the applicability domain n-cutoff (−0.5; 0; +0.5).
The majority of these virtual hits were found to be highly similar to the compounds already known (compounds in the QSAR modeling set), so it would be less interesting to test these hits experimentally. To verify the diversity of those virtual hits, pairwise similarity calculations were performed. Each compound was represented by a fingerprint of 166 substructure keys (MACCS structural keys43), indicating the presence or absence of a particular chemical substructure. The pairwise similarity was assessed by Tanimoto coefficients44 (Tc) between PCL hits, between PCL hits and each hit’s nearest neighbor from the actives in the modeling set (identified by the lowest Euclidean distances calculated with the Dragon descriptors), and between PCL hits and all actives in the modeling set. The majority of compound pairs formed by Prestwick’s virtual hits with each hit’s nearest neighbor within the modeling set had Tc over 0.90, whereas other pairwise similarity scores show a normal distribution, suggesting that the virtual hits are structurally dissimilar from each other but highly similar with known 5-HT1A actives (Figure 5). Furthermore, the principal component analysis (PCA) was also conducted and it could be easily seen that there is a big overlap between the distribution of chemical space occupied by the Prestwick library and modeling set compounds (Figure 3).
Figure 5.

The distribution of the pairwise structural similarity within the sets of screening hits from three types of libraries in comparison to modeling sets using the kernel density plot for Tc distribution: Aa, between hits from the Prestwick library; Ab, between the Prestwick hits and all modeling set compounds; Ac, between the Prestwick hits and their nearest neighbors in the modeling set. Ba, Between TimTec GPCR-targeted library hits; Bb, between the TimTec GPCR-targeted library hits and all modeling set compounds; Bc, between the GPCR-targeted library hits and their nearest neighbors in the modeling set. Ca, Between TimTec diversity library hits; Cb, between the TimTec diversity library hits and all modeling set compounds; Cc, between the TimTec diversity library hits and their nearest neighbors in the modeling set.
Figure 3.

PCA plot of three types of virtual screening libraries along with the modeling set; the plots were generated from calculating the top three principal components by using multiple chemical descriptors (Dragon 5.5, 0D-2D descriptors) of compound in respective libraries. Compounds from the modeling set are colored in red; compounds from the Prestwick library are colored in green; compounds from the TimTec GPCR-targeted library are colored in blue and compounds from the TimTec diversity library are colored in orange.
To explore more structurally diverse 5-HT1A compounds, we further screened GPCR-targeted libraries and diversity libraries from the commercial chemical sources of TimTec and ASINEX. Thus, the additional collection of 24 000 compounds was screened, which included the TimTec ActiTarg-G (GPCR-targeted) library of about 2300 compounds, the ASINEX Synergy GPCRs CNS (GPCR-targeted) library of about 7000 compounds, the TimTec Diversity Set 10K (diversity library) of 10 000 compounds and the ASINEX Diversity Set-Platinum 5K (diversity library) of about 5100 compounds. The putative hit rates for different screening libraries are shown in Figure 4 with various n-cutoff values (representing different applicability domain), and the exact numbers of compounds chosen from these libraries are also available in the Supporting Information (Table S1). It is obvious that many more chemicals were selected from the GPCR-targeted library than from the diversity library by applying the same n value, verifying that the diversity library has much more structural-varied compounds than the GPCR-targeted library.
The binding potential for compounds within the AD of n = 0.5 was further predicted by kNN consensus models. 445 compounds from the TimTec ActiTarg-G library, 487 from the TimTec Diversity Set 10K, 2177 from the ASINEX Synergy GPCRs CNS library and 782 from the ASINEX Diversity Set-Platinum 5K were predicted as actives by at least one of the kNN consensus models. To narrow the hit list and obtain the higher confidence level for each prediction, both the consensus score and model coverage were taken into consideration. In particular, only the hits with average class numbers between 1.0 and 1.1 and the model coverage over 50% were selected. We found that there were 64 compounds from the TimTec AntiTarg-G library and 40 from the TimTec Diversity Set 10K that satisfied both criteria. As for ASINEX libraries, there were still hundreds of compounds that met those strict criteria, but we decided not to pursue these compounds at this time.
Several structural classes were observed by screening different libraries according to the Tc values. Notably, many of the 64 virtual hits from the TimTec AntiTarg-G library were found to be structurally similar to actives used in model building, whereas the 40 virtual hits from the TimTec Diversity Set 10K had highly different structures. The pairwise similarity measured by Tc values was also compared between virtual hits, hits versus their nearest neighbor within the modeling set compounds, virtual hits versus modeling set compounds, and between modeling set compounds (Figure 5). It is clearly seen that the virtual hits from the TimTec AntiTarg-G library had chemical structures with a much lower similarity (Figure 5Ac) to the known 5-HT1A actives than Prestwick virtual hits (Figure 5Bc). The average Tc value between TimTec Anti-Targ-G library hits and their nearest neighbors in the modeling set was 0.60 compared to 0.90 for the hits screened from Prestwick. It should be noted that the cutoff value for Tc to be defined as the hits by major commercial packages for virtual screening is 0.80, when combined with the 166 MACCS structural keys. For our virtual hits screened from the TimTec Diversity Set 10K, the Tc value between hits and their nearest neighbors in the modeling set was as low as around 0.45, suggesting that they are highly structurally different. Although these hits are also predicted to be 5-HT1A actives with a high confidence by our consensus models as well as RF and SVM, it would be interesting and exciting to test them experimentally, in a hope of revealing 5-HT1A actives with a new scaffold. Moreover, the visualization of the PCA plot also confirmed the broader distribution in chemical space for compounds from the TimTec diversity library compared with TimTec GPCR-targeted library (cf. Figure 3).
From all virtual screening hits chosen by kNN, 15 chemicals were further selected for the experimental testing, including five compounds from PCL, five from TimTec AntiTarg-G library, and five from TimTec Diversity Set 10K. The following selection criteria were used: (1) high confidence of consensus prediction by RF and SVM; (2) low structural similarity between hits and the 5-HT1A actives we already known; (3) commercial availability.
Experimental Validation
The validations of our in silico hits by the NIMH PDSP yielded many actives that were subsequently confirmed to have appreciable 5-HT1A binding activity. We should stress that only binary QSAR models were used for virtual screening so no prediction of exact binding affinities (Ki values) could be made. Nine out of 15 in silico hits were found to have the percent of inhibition equal to or higher than 50% (i.e., Mesoridazine, Clozapine, Risperidone and Fluphenazine from PCL; ST030580 from GPCR targeted library; ST023860, ST074311, ST057540 and ST066677 from the diversity library) and five of them displayed >95% inhibition at 10 μM. For these more potent compounds, Ki values were obtained (see Methods). The five in silico hits from PCL showed the highest success rate (80%), though most of them were similar to the modeling set compounds (Tc ranged from 0.80 to 0.99, with an average Tc value of 0.86) and no novel core scaffolds were found. They were also found to be less interesting from the drug repurposing prospective. Mesoridazine (Ki = 33.1 nM) and fluphenazine (Ki = 145.7 nM) belong to the typical antipsychotic class whereas clozapine (Ki = 104.8 nM) and risperidone (Ki = 427.5 nM) are atypical antipsychotics; all four drugs are commonly used in the treatment of schizophrenia and bipolar disorder. The known mechanism of action for mesoridazine as a phenothiazine antipsychotic is through its potent binding with 5-HT2A and dopamine receptors,45 whereas the binding with human 5-HT1A receptors have not been reported before. The Ki of fluphenazine,47 clozapine48 and risperidone49 for 5-HT1A have been reported elsewhere; however, this data was not included in PDSP and was unknown to us at the time of our computational studies. Thus, these observations may be considered as (re)discovery of the known mechanism of action for known antipsychotics.
To our surprise, only one in silico hit from the GPCRs targeted library was found to be active (Ki = 243.8 nM). This compound, ST030580, showed a quite different ring arrangement from its nearest neighbor compound in the modeling set, while maintaining the azaspiro-bicyclic structural element. Among the three confirmed hits from the TimTec Diversity Set 10K, compound ST057540 (also known as Lysergol ([(8α)-6-methyl-9,10-didehydroergolin-8-yl]methanol)) yielded 98.20% binding inhibition against 5-HT1A receptor at 10 μM and its Ki value was found to be 2.3 nM afterward (Figure 6). Furthermore, the Tc between this compound and its nearest neighbor in the modeling set (ID: 27405, with dibenzo[de,g]quinolone structure) is only 0.69, suggesting low structural similarity between this molecule and any compound in the modeling set. Lysergol is an alkaloid of the ergoline family that occurs as a minor constituent in some species of fungi. Lysergol is sometimes also utilized as an intermediate in the manufacturing of some ergoloid medicines (e.g., nicergoline). This compound fully satisfies the “Lipinski’s Rule of Five”,50 with a LogP value of 1.76, which is considered to be ideal for both oral absorption and CNS penetration. It was also predicted to have a very low probability of rapid biodegradation by EPI-Suite.51 Literature search indicates that Lysergol binds mainly to the GPCR targets and shows more selectivity toward 5-HT1 versus 5-HT2 receptors. Thus, our discovery that Lysergol is a potent low nanomolar 5HT1A binder confounded by its known high-affinity binding to 5-HT1B and 5-HT1D52 suggests that Lysergol may find application in treating schizophrenia as was suggested independently (e.g., by Groo and Palosi).53 Two other active hits, compounds ST023860 and ST074311, also feature relatively different scaffolds in comparison to modeling set compounds with Tc of 0.75 and 0.69, respectively. In addition, one confirmed hit from the Diversity Set, i.e. ST066677, showed the Tc as low as 0.53 to its most similar compound. These compounds represent unique scaffolds opening opportunities for the further investigation of them as well as their close chemical analogs as novel antipsychotic agents. In summary, the above results once again prove the predictive power of our binary kNN, RF and SVM classification QSAR models built from 5-HT1A actives/inactives. These studies illustrate that QSAR models generated by following the validated QSAR workflow, as employed in this paper, could be used as a general tool for identifying promising hits by the means of virtual screening of various types of chemical libraries.
Figure 6.

The full dose response curve for hit compounds ST057540 (arrow-up triangles, Ki = 2.3 nM), ST074311 (arrow-down triangles, Ki = 8194 nM) and the positive control, Methysergide (solid squares, Ki = 26 nM) measured by human 5-HT1A receptor radioligand binding assay. The radioligand is [3H]-8-OH-DPAT at the concentration of 0.5 nM with the standard binding buffer.
Discussion
We should emphasize that rigorous model validation is an inherent feature of our QSAR modeling workflow. This issue of model validation has been given a lot of attention by the QSAR research community.54 In the past, most practitioners merely presumed that internally cross-validated models built from available training set data should be externally predictive. We and others have demonstrated that internal validation techniques such as leave-one-out (LOO) or even leave-many-out (LMO) cross-validation applied to the training set are insufficient to ensure the external predictive power of QSAR models.32,55 Thus, we used external 5-fold cross-validation approach as well as the Y-randomization test in this study to ensure the robustness and predictive power of our QSAR models. Needless to say, the use of externally validated models and applicability domains is especially critical when the models are employed in virtual screening.
A set of unique 66 5-HT1A actives from different resources, i.e., WOMBAT library, were further validated by our consensus models. All three QSAR methods (kNN, RF and SVM) could accurately annotate the majority of compounds, with CCRevs ranged from 0.94 to 0.96. The success of this independent external validation reassured us that our QSAR models were indeed externally predictive, robust, and applicable to virtual screening.
We have employed our QSAR models for virtual screening of several chemical libraries, including two drug-like libraries, two GPCR-targeted libraries and two diversity libraries. Both the global similarity search (using AD) and the subsequent QSAR model predictions confirmed our expectations that drug-like libraries and GPCR-targeted libraries had a much higher computational hit rate than diversity libraries when the same n-cutoff values were applied. The screening hits from drug-like libraries had much higher structural similarity to our modeling set compounds than hits from GPCR-targeted or diversity libraries. As described in Methods, 15 top hits (five best from each of the diversity library, GPCR-targeted library, and drug-like library) were chosen for the experimental validation and nine out of these 15 compounds suggested by our QSAR models were confirmed to be 5-HT1A actives. Interestingly, overall the number of screening hits was higher for the GPCR-targeted library than for the diversity library (cf. Figure 4) as could be expected. However, the number of experimentally confirmed hits was much higher for the diversity library (four out five screening hits were validated experimentally; cf. Table 2) than that for the GPCR-targeted library (only one of five screening hits was confirmed; cf. Table 2). Interestingly, the most potent nanomolar 5-HT1A active compound (compound No. 14 in Table 2; Ki = 2.3 nM) was identified from the TimTec diversity library, sharing very low structural similarity (Tc = 0.69) with its nearest neighbor compound in the modeling set. These findings verified that model-based virtual screening yielded hits that would not be detected using simple similarity search because of their structural novelty as compared to the training set compounds.
Conclusions
Our studies demonstrate that binary classification QSAR models built with Dragon descriptors can accurately differentiate true 5-HT1A actives from inactives. A state-of-the-art QSAR modeling scheme was applied, and the models were rigorously validated using both internal (multiple training/test set divisions and Y-randomization) as well as external (5-fold CV sets) validation approaches. We have demonstrated that this strategy afforded multiple QSAR models with high internal as well as external predictive power. The predictors were further validated on the WOMBAT hits (66 literature extracted compounds tested for 5-HT1A binding). We found that our predictions agreed highly with the experimental annotation of 66 compounds as 5-HT1A actives as reported in various publications. Furthermore, our models used in the most conservative way, i.e., in consensus fashion and with the strictest applicability domain criteria, identified 43 putative 5-HT1A actives by mining three major types of screening libraries including drug-like libraries (WDI and PCL), GPCR-targeted libraries, and diversity libraries. Fifteen of them were tested experimentally in the NIMH PDSP at UNC-Chapel Hill and nine showed significant inhibition activities (≥50% inhibition) in a single concentration. Interestingly, the five virtual hits identified from the TimTec diversity library showed higher success ratio (60% versus 20%) than the other five from the TimTec GPCR-targeted library; slightly better results were also reported for the PCL drug-like library (80%). One compound (ST057540) was found to have the highest Ki of 2.3 nM among all hits, whereas the Tc values between this compound and its nearest neighbor in the modeling set (ID: 27405) was only 0.69. It was of great interest to find out that this compound, Lysergol, though used as an intermediate in manufacturing of some ergoloid medicines, had unclear pharmacological indication, but many drug-like chemical properties suggesting that we may have identified a yet unknown antischizophrenic drug candidate. In summary, we have demonstrated that QSAR models can be successfully used for finding promising and structurally diverse hits by the means of virtual screening of chemical libraries. All QSAR models developed and validated in this study as virtual screening tools to identify 5HT1A ligands are available from the Chembench portal (chembench.mml.unc.edu).
Acknowledgments
We acknowledge access to the computing facilities at the ITS Research Computing Division of the UNC—CH. The studies reported in this paper were supported in part by the NIH research grants GM066940 and GM096967 (A.T.), the NIMH Psychoactive Drug Screening Program and RO1MH61887 to B.L.R. and the UNC—CH University Research Council Research Grant A3-12988 to X.S.W.
Glossary
Abbreviations
- 5-HT1A
5-hydroxytryptamine receptor subtype 1A
- AD
applicability domain
- CCR
correct classification rate
- CCRtrain
correct classification rate for training set
- CCRtest
correct classification rate for test set
- CCRevs
correct classification rate for external validation set
- CCRrand
correct classification rate of the random models using the external validation set
- CV
cross-validation
- FN
false negative
- FP
false positive
- GPCRs
G-protein coupled receptors
- HTS
high throughput screening
- kNN
k nearest neighbor
- LOO-CV
leave-one-out cross-validation
- MLSCN
Molecular Library Screening Center Network
- MOE
molecular operating environment
- PCL
Prestwick Chemical Library
- PDSP
Psychoactive Drug Screening Program
- QSAR
quantitative structure–activity relationship
- RF
random forest
- SA
simulated annealing
- SAR
structure–activity relationship
- SE
sensitivity
- SP
specificity
- SSRIs
selective serotonin reuptake inhibitors
- SVM
support vector machines
- Tc
Tanimoto coefficients
- TN
true negative
- TP
true positive
- WDI
World Drug Index
- WOMBAT
World of Molecular Bioactivity
Supporting Information Available
The number of compounds chosen from all six screening libraries by different ncutoff values, model predicted and experiment tested data for 5-HT1A screening hits, chemical structures for 5-HT1A modeling data set (actives and inactives) and independent validation set, the Certificate of Analysis for commercial compounds, as well as other supplementary data indicated in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ Exploratory Clinical and Translational Research, Bristol-Myers Squibb, Princeton, NJ 08543.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Simon J. S.; Nemeroff C. B. Aripiprazole augmentation of antidepressants for the treatment of partially responding and nonresponding patients with major depressive disorder. J. Clin. Psychiatry 2005, 66, 1216–1220. [DOI] [PubMed] [Google Scholar]
- Jeyapaul P.; Vieweg R. A case study evaluating the use of clozapine in depression with psychotic features. Ann. Gen. Psychiatry 2006, 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corya S. A.; Andersen S. W.; Detke H. C.; Kelly L. S.; Van Campen L. E.; Sanger T. M.; Williamson D. J.; Dube S. Long-term antidepressant efficacy and safety of olanzapine/fluoxetine combination: a 76-week open-label study. J. Clin. Psychiatry 2003, 64, 1349–1356. [DOI] [PubMed] [Google Scholar]
- Bantick R. A.; Deakin J. F.; Grasby P. M. The 5-HT1A receptor in schizophrenia: a promising target for novel atypical neuroleptics?. J. Psychopharmacol. (London, U. K.) 2001, 15, 37–46. [DOI] [PubMed] [Google Scholar]
- Li Z.; Ichikawa J.; Dai J.; Meltzer H. Y. Aripiprazole, a novel antipsychotic drug, preferentially increases dopamine release in the prefrontal cortex and hippocampus in rat brain. Eur. J. Pharmacol. 2004, 493, 75–83. [DOI] [PubMed] [Google Scholar]
- Bantick R. A.; De Vries M. H.; Grasby P. M. The effect of a 5-HT1A receptor agonist on striatal dopamine release. Synapse 2005, 57, 67–75. [DOI] [PubMed] [Google Scholar]
- Schechter L. E.; Dawson L. A.; Harder J. A. The potential utility of 5-HT1A receptor antagonists in the treatment of cognitive dysfunction associated with Alzheimer’s disease. Curr. Pharm. Des. 2002, 8, 139–145. [DOI] [PubMed] [Google Scholar]
- Mizukami K.; Ishikawa M.; Akatsu H.; Abrahamson E. E.; Ikonomovic M. D.; Asada T. An immunohistochemical study of the serotonin 1A receptor in the hippocampus of subjects with Alzheimer’s disease. Neuropathology 2011, 31, 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton R. E.; Marunycz J. D.; Alderdice M. T.; Napoliello M. J. Review of the side-effect profile of buspirone. Am. J. Med. 1986, 80, 17–21. [DOI] [PubMed] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X. P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser M. J.; Setola V.; Irwin J. J.; Laggner C.; Abbas A. I.; Hufeisen S. J.; Jensen N. H.; Kuijer M. B.; Matos R. C.; Tran T. B.; Whaley R.; Glennon R. A.; Hert J.; Thomas K. L.; Edwards D. D.; Shoichet B. K.; Roth B. L. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerner R. J. [Generalized anxiety disorder (GAD)—diagnosis and therapy]. Med. Monatsschr. Pharm. 2007, 30, 401–408. [PubMed] [Google Scholar]
- Tropsha A.; Golbraikh A. Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr. Pharm. Des. 2007, 13, 3494–3504. [DOI] [PubMed] [Google Scholar]
- Lyne P. D. Structure-based virtual screening: an overview. Drug Discov. Today 2002, 7, 1047–1055. [DOI] [PubMed] [Google Scholar]
- Varnek A.; Tropsha A.. Chemoinformatics Approaches to Virtual Screening: An Approach to Virtual Screening; RSC Publishing: Cambridge, U.K., 2008. [Google Scholar]
- Hoffman B.; Cho S. J.; Zheng W.; Wyrick S.; Nichols D. E.; Mailman R. B.; Tropsha A. Quantitative structure-activity relationship modeling of dopamine D(1) antagonists using comparative molecular field analysis, genetic algorithms-partial least-squares, and K nearest neighbor methods. J. Med. Chem. 1999, 42, 3217–3226. [DOI] [PubMed] [Google Scholar]
- Tropsha A.; Zheng W. Identification of the descriptor pharmacophores using variable selection QSAR: applications to database mining. Curr. Pharm. Des. 2001, 7, 599–612. [DOI] [PubMed] [Google Scholar]
- Wang J. X.; Dipasquale A. J.; Bray A. M.; Maeji N. J.; Spellmeyer D. C.; Geysen H. M. Systematic study of substance P analogs. II. Rapid screening of 512 substance P stereoisomers for binding to NK1 receptor. Int. J. Pept. Protein Res. 1993, 42, 392–399. [PubMed] [Google Scholar]
- National Institute of Mental Health’s Psychoactive Drug Screening Program. 2009. Contract # HHSN-271-2008-00025-C (NIMH PDSP).
- Olah M e. al.WOMBAT and WOMBAT-PK: bioactivity databases for lead and drug discovery. In Chemical Biology: From Small Moleculars to Systems Biology and Drug Design; Schreiber S. L., Ed.; Wiley-VCH: New York, 2007, 2013; pp 760–786. [Google Scholar]
- Fourches D.; Muratov E.; Tropsha A. Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc., 1010 Sherbooke St.West, Suite #910, Montreal, QC H3A 2R7, Canada, 2012.
- Tropsha A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inf. 2010, 29, 476–488. [DOI] [PubMed] [Google Scholar]
- Golbraikh A.; Shen M.; Xiao Z.; Xiao Y. D.; Lee K. H.; Tropsha A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 2003, 17, 241–253. [DOI] [PubMed] [Google Scholar]
- Golbraikh A.; Tropsha A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. Mol. Diversity 2002, 5, 231–243. [DOI] [PubMed] [Google Scholar]
- Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar]
- DRAGON for Windows and Linux; Talete: Milano, Italy, 2007. (Available at http://www.talete.mi.it/help/dragon_help).
- Zheng W.; Tropsha A. Novel variable selection quantitative structure--property relationship approach based on the k-nearest-neighbor principle. J. Chem. Inf. Comput. Sci. 2000, 40, 185–194. [DOI] [PubMed] [Google Scholar]
- Tropsha A.Recent Trends in Quantitative Structure-Activity Relationships. In Burger’s Medicinal Chemistry and Drug Discovery; Abraham D., Ed.; John Wiley & Sons, Inc.: New York, 2003; pp 49–77. [Google Scholar]
- de Cerqueira L. P.; Golbraikh A.; Oloff S.; Xiao Y.; Tropsha A. Combinatorial QSAR modeling of P-glycoprotein substrates. J. Chem. Inf. Model. 2006, 46, 1245–1254. [DOI] [PubMed] [Google Scholar]
- Itskowitz P.; Tropsha A. kappa Nearest neighbors QSAR modeling as a variational problem: theory and applications. J. Chem. Inf. Model. 2005, 45, 777–785. [DOI] [PubMed] [Google Scholar]
- Tropsha A.; Gramatica P.; Gombar V. K. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar]
- Breiman L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar]
- Liaw A.; Wiener M. Classification and Regression by random Forest. R News 2002, 2318–22. [Google Scholar]
- Dalgaard P.Introductory Statistics with R; Springer: New York, 2008. [Google Scholar]
- Vapnik V.The Nature of Statistical Learning Theory; Springer-Verlag: Heidelberg, 1995. [Google Scholar]
- Chang C.; Lin C. LIBSVM: A Library for Support Vector Machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 27:1–27:27. [Google Scholar]
- Fine S.; Scheinberg K.; Effcient S. V. M. Training Using Low-rank Kernel Representations. J. Mach. Learn. Res. 2001, 2, 243–264. [Google Scholar]
- Fourches D.; Barnes J. C.; Day N. C.; Bradley P.; Reed J. Z.; Tropsha A. Cheminformatics analysis of assertions mined from literature that describe drug-induced liver injury in different species. Chem. Res. Toxicol. 2010, 23, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold S.; Eriksson L.. Statistical Validation of QSAR Results. In Chemometrics Methods in Molecular Design (Methods and Principles in Medicinal Chemistry), Vol 2; Waterbeemd H. v. d., Ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1995; pp 309–318. [Google Scholar]
- Shapiro D. A.; Renock S.; Arrington E.; Chiodo L. A.; Liu L. X.; Sibley D. R.; Roth B. L.; Mailman R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003, 28, 1400–1411. [DOI] [PubMed] [Google Scholar]
- Roth B. L.; Baner K.; Westkaemper R.; Siebert D.; Rice K. C.; Steinberg S.; Ernsberger P.; Rothman R. B. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 11934–11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACCS Structural Keys; MDL Information Systems Inc: San Ramon, CA, 2005.
- Tanimoto T.IBM Internal Report; IBM Corp: Armonk, November 17, 1957.
- Roth B. L.; Tandra S.; Burgess L. H.; Sibley D. R.; Meltzer H. Y. D4 dopamine receptor binding affinity does not distinguish between typical and atypical antipsychotic drugs. Psychopharmacology (Berlin) 1995, 120, 365–368. [DOI] [PubMed] [Google Scholar]
- Kroeze W. K.; Hufeisen S. J.; Popadak B. A.; Renock S. M.; Steinberg S.; Ernsberger P.; Jayathilake K.; Meltzer H. Y.; Roth B. L. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 2003, 28, 519–526. [DOI] [PubMed] [Google Scholar]
- Bymaster F. P.; Calligaro D. O.; Falcone J. F.; Marsh R. D.; Moore N. A.; Tye N. C.; Seeman P.; Wong D. T. Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology 1996, 14, 87–96. [DOI] [PubMed] [Google Scholar]
- Schotte A.; Janssen P. F.; Gommeren W.; Luyten W. H.; Van G. P.; Lesage A. S.; De L. K.; Leysen J. E. Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (Berlin) 1996, 124, 57–73. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- US EPA . Estimation Programs Interface Suite for Microsoft Windows, v 4.11. United States Environmental Protection Agency, Washington, DC, 2011. [Google Scholar]
- Weinshank R. L.; Zgombick J. M.; Macchi M. J.; Branchek T. A.; Hartig P. R. Human serotonin 1D receptor is encoded by a subfamily of two distinct genes: 5-HT1D alpha and 5-HT1D beta. Proc. Natl. Acad. Sci. U. S. A 1992, 89, 3630–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groo D.; Palosi A. Substituted ergolines: potential antipsychotics with unique profile. I. Psychopharmacological characterization. Pol. J. Pharmacol. Pharm. 1988, 40, 593–601. [PubMed] [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. QSAR/QSPR and Proprietary Data. J Chem. Inf. Model. 2006, 46, 937. [Google Scholar]
- Golbraikh A.; Tropsha A. Beware of q(2)!. J. Mol. Graphics Modell. 2002, 20, 269–276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.