

Abstract
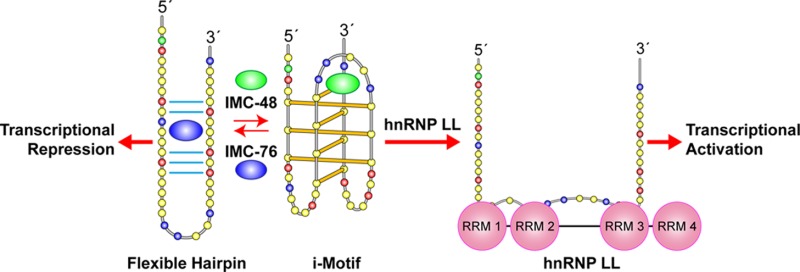
In a companion paper (DOI: 10.021/ja410934b) we demonstrate that the C-rich strand of the cis-regulatory element in the BCL2 promoter element is highly dynamic in nature and can form either an i-motif or a flexible hairpin. Under physiological conditions these two secondary DNA structures are found in an equilibrium mixture, which can be shifted by the addition of small molecules that trap out either the i-motif (IMC-48) or the flexible hairpin (IMC-76). In cellular experiments we demonstrate that the addition of these molecules has opposite effects on BCL2 gene expression and furthermore that these effects are antagonistic. In this contribution we have identified a transcriptional factor that recognizes and binds to the BCL2 i-motif to activate transcription. The molecular basis for the recognition of the i-motif by hnRNP LL is determined, and we demonstrate that the protein unfolds the i-motif structure to form a stable single-stranded complex. In subsequent experiments we show that IMC-48 and IMC-76 have opposite, antagonistic effects on the formation of the hnRNP LL–i-motif complex as well as on the transcription factor occupancy at the BCL2 promoter. For the first time we propose that the i-motif acts as a molecular switch that controls gene expression and that small molecules that target the dynamic equilibrium of the i-motif and the flexible hairpin can differentially modulate gene expression.
Introduction
While the presence of G-quadruplexes in telomeric sequences, promoter elements, and 5′UTRs is well documented, and in some cases with a biological role proposed, similar research is lacking for the complementary DNA secondary structure, the i-motif, although such roles have been suggested.1 In promoter elements where duplex DNA is found, the possibility exists that the G-quadruplex and the i-motif form on opposite strands, but whether they can coexist or are mutually exclusive remains unresolved, except in the case of the insulin promoter where the formation of the two structures is mutually exclusive.2 If the latter were the case more generally, then one might imagine that the G-quadruplex could act as a signal to silence gene expression, as is the case with the MYC promoter,3 and the i-motif as an activator signal. In support of this, the activating transcriptional factor hnRNP K binds to the CT boxes on the C-rich strand in the MYC promoter and induces MYC expression.4
Recent findings in our companion paper (DOI: 10.021/ja410934b) further support the idea of DNA secondary structures serving as switches to turn gene transcription on or off.5 We observed two different small molecules that bound to different topological forms of the C-rich strand of the BCL2 cis-regulatory element and either repressed or activated transcription.5 The compound (IMC-48) that bound to the i-motif species to populate this species relative to the flexible hairpin increased BCL2 gene expression. In contrast, the other compound (IMC-76), which selected for the flexible hairpin species, decreased gene expression. Antagonism between the two molecules was found to occur with the DNA species in solution as well as within a cellular system.5 On the basis of these results, we postulated the presence of transcriptional factors that would similarly bind to the two different DNA structures, thereby mimicking the effect of the two compounds on BCL2 gene expression. Here we identify hnRNP LL as a transcriptional factor that recognizes the BCL2 i-motif and subsequently unfolds it to activate transcription. Furthermore, hnRNP LL belongs to the same protein family as hnRNP K, which previously was shown to activate MYC transcription by binding to the C-rich strand of the MYC promoter.4 Following the identification of hnRNP LL as an activating transcriptional factor for BCL2, we then demonstrate that the two small molecules that bind exclusively to one or the other of the two equilibrating species of the BCL2 C-rich strand exert their activity by modulating the amount of the i-motif available for binding to hnRNP LL. Importantly, this principle was shown at both the level of the DNA species bound to hnRNP LL in solution and the cellular level. These results suggest that the BCL2 i-motif can be considered as a molecular switch similar in principle to a riboswitch found in RNA.6
Results and Discussion
Directly upstream (∼25 bases) from the BCL2 P1 promoter is a GC-rich element known to form G-quadruplex and i-motif structures (Figure 1A). Under negative superhelicity induced by transcriptional activity it can be expected that either the i-motif or the G-quadruplex will exist in the promoter element. Previous in vitro studies using synthetic oligomers demonstrated that the BCL2 G-rich promoter element forms three different G-quadruplexes; the major one exhibits a mixed parallel/antiparallel structure.7,8 The opposite strand is highly dynamic, existing as a mixed population of two molecules at a pH of 6.6, an i-motif and a flexible hairpin (Figure 1B,C). The relationship between these two DNA secondary structures, the interaction of IMC-48 and IMC-76, and the subsequent effect on BCL2 gene expression are also shown in Figure 1C.
Figure 1.

Diagram of the BCL2 gene promoter region with the GC-rich element located directly upstream of the P1 promoter and targeting with IMC-48 and IMC-76. The C-rich i-motif-forming sequence is shown. Three and one-half sets of two intercalated hemiprotonated cytosine+–cytosine base pairs form the i-motif structure. The bases in bold correspond to the bases involved in base pairing within each of the structures. Here and in subsequent figures, the yellow, green, red, and blue circles represent the deoxynucleotides cytosine, adenine, guanine, and thymine, respectively. In the lower portion of the figure we also show the proposed partial hairpin that is in equilibrium with the i-motif and the proposed binding of IMC-48 and IMC-76 to the i-motif and partial hairpin, respectively, along with the proposed transcriptional consequences of targeting with IMC-48 and IMC-76.
Identification of hnRNP LL as a BCL2 i-Motif–Binding Protein That Activates BCL2 Transcription
Specific proteins such as nucleolin9 and NM23-H210 recognize and bind to G-quadruplexes in promoter elements. G-quadruplex-binding agents can interfere with protein–DNA complex formation, potentially modulating gene expression. We sought to identify nuclear proteins that could bind to the i-motif or an unfolded form and might also be involved in BCL2 transcriptional modulation. Since the i-motif is highly dynamic, any identified i-motif-binding protein may take advantage of this property and form a stable DNA complex by i-motif remodeling. The C-rich strand that gives rise to the folded i-motif has features more commonly associated with secondary RNA structures than DNA; therefore, RNA-binding proteins were considered. Candidates included RNA recognition proteins belonging to the hnRNP class normally associated with RNA splicing. Although not as yet reported to bind to an i-motif structure, an example is hnRNP K, which binds to the CT element of the MYC promoter to activate transcription.11
Nuclear proteins from standard and commercially available HeLa nuclear extract that putatively bind to the BCL2 i-motif were purified using a biotinylated oligomer–streptavidin bead complex pull-down assay and identified by liquid LC/MS/MS sequencing. Two biotinylated oligomer–bead complexes were used consisting of either the wild-type BCL2 i-motif–forming sequence or a mutant oligomer (which cannot form a stable i-motif) for nonspecific protein binding. Ninety-five proteins were identified that bound either to the wild-type BCL2 i-motif–forming sequence (35 proteins, Supplemental Table 1), a mutant (20 proteins, Supplemental Table 2), or both sequences (40 proteins, Supplemental Table 3). Proteins that bound uniquely to the BCL2 i-motif–forming promoter element were classified into functional groups: (1) transcription, (2) translation or protein-folding, (3) energy metabolism or other enzymatic processes, and (4) cell adhesion or migration functions, mostly related to the cytoskeleton (Supplemental Tables 1–3). Of interest were proteins having documented function related to transcription (Table 1), particularly hnRNP LL. While hnRNP LL has not been extensively studied, the related protein hnRNP L is a pre-mRNA splicing factor that binds to and stabilizes BCL2 mRNA.12 The hnRNP LL protein is a paralog of hnRNP L, shows tissue-specific distribution,13 and activates T-cells by shifting transcriptomes for cellular proliferation and inhibition of cell death.14
Table 1. Proteins Purified Using Py39WT BCL2 i-Motif Biotinylated Oligomer–Streptavidin Bead Complex and Identified by LC/MS/MS That Were Related to Transcription.
| protein symbol | protein identification | accession number |
|---|---|---|
| HMG-I | isoform HMG-I of high-mobility group protein HMG-I/HMG-Y | IPI00179700 |
| hnRNP UL2 | heterogeneous nuclear ribonucleoprotein U-like protein 2 | IPI00456887 |
| hnRNP LL | isoform 1 of heterogeneous nuclear ribonucleoprotein L-like | IPI00103247 |
| GEMIN5 | Gem-associated protein 5 | IPI00291783 |
| HDGF | hepatoma-derived growth factor | IPI00020956 |
| HMGN1 | nucleosome-binding protein 1 | IPI00006157 |
| DDX21 | Isoform 1 of nucleolar RNA helicase 2 | IPI00015953 |
| RBBP4 | histone-binding protein RBBP4 | IPI00328319 |
| RBBP7 | histone-binding protein RBBP7 | IPI00395865 |
To investigate hnRNP LL for its possible involvement in BCL2 transcriptional modulation, we studied the effects of siRNA knockdown in MCF-7 breast cancer cells. Relative mRNA levels of hnRNP LL and BCL2 were determined by qPCR after treatment with hnRNP LL siRNA. Significantly decreased BCL2 mRNA levels resulted from hnRNP LL knockdown (Figure 2A). To demonstrate that this effect is specific and not caused by an off-target siRNA effect, we examined mRNA levels of two other genes (PDGFR-β and kRAS) following hnRNP LL knockdown. The PDGFR-β and kRAS promoter regions also contain the consensus sequences CCGC for hnRNP LL binding, and preliminary data suggest that hnRNP LL binds to the i-motif-forming oligomers (data not shown) with comparable binding to BCL2. However, the PDGFR-β and kRAS transcripts are not lowered by hnRNP LL siRNA, demonstrating selectivity for the BCL2 i-motif at least among these promoter i-motifs (Figure 2A).
Figure 2.

Confirmation of hnRNP LL as a BCL2 i-motif–binding protein. (A) The effects of siRNA knockdown of hnRNP LL on the BCL2, kRAS, and PDGFR-β mRNA level in MCF-7 cells. 50 nM of hnRNP LL siRNA was added to MCF-7 cells for 72 h. GAPDH was used as an internal control (**P < 0.01). (B) Effect on concentration-dependent binding of hnRNP LL on the BCL2 i-motif–forming oligomer (Py39WT) by EMSA at pH 6.8. (C) Competition EMSA showing BCL2 i-motif–specific binding of hnRNP LL at pH 6.8. Nonlabeled (cold) oligomers were incubated with hnRNP LL on ice for 20 min and end-labeled Py39WT was added for 5 min. (D) Comparative Kd values for hnRNP LL binding to the biotin-Py39WT and biotin-Py39MutT at two different pH levels determined by SPR analysis.
EMSA and SPR Studies Show High Affinity Binding of hnRNP LL to the BCL2 i-Motif
Mobility shift assays (Figure 2B,C) were used to determine whether hnRNP LL bound to the BCL2 i-motif specifically. The hnRNP LL protein bound with high affinity to the BCL2 i-motif at pH 6.8 (Figure 2B). The cold BCL2 i-motif (Py39WT) competed with the 32P-labeled BCL2 i-motif; as expected, the cold mutant i-motif oligomer (Py39MutT) did not compete for hnRNP LL binding (Figure 2C). Also, hnRNP LL did not bind to duplex DNA formed with the cold complementary G-rich strand (Pu39WT) annealed to end-labeled Py39WT.
Using surface plasmon resonance (SPR) analysis (Figure 2D and Supplemental Figure 1), hnRNP LL bound strongly to Py39WT with a Kd value of 19.4 pM at pH 6.5. This disassociation was increased by ∼3.6-fold to 69.8 pM at pH 7.9. For the Py39MutT, the Kd value was 2.5-fold higher at each pH compared to the wild-type.
EMSA and siRNA Knockdown Experiments Show That BCL2 i-Motif Recognition by hnRNP LL Involves the Two i-Motif Lateral Loops
The hnRNP LL protein shares 57% sequence identity to hnRNP L.15 Both proteins have four RNA recognition motifs (RRMs), and at least two are required for stable binding to single-stranded RNA or DNA. Two consensus sequences for binding these RRMs are found in the BCL2 i-motif, and both are located in the lateral loops (CCCGC and CGCCC) (Figure 3A). To determine the importance of these loops in comparison to the central loop (having a TTCCT sequence), cold mutant Py39 sequences were designed having one or more of these loops mutated but still maintaining the basic i-motif core structure. Of these four mutated i-motif sequences, the one having both lateral loops mutated (Mut5′,3′L; 35%) was the least effective competitor, whereas the one having the central loop mutated (MutCL; 73%) was the most effective (Figure 3B). The histograms shown to the right of the EMSAs in Figure 3B depict the quantification of band intensity. While the two individually mutated loop oligomers (Mut5′L and Mut3′L) were of intermediate potency in competing with the wild-type for hnRNP LL binding to the BCL2 i-motif, Mut5′L (48%) appeared less effective than Mut3′L (64%). To complement these experiments, we also determined the biological significance of the lateral loops by investigating whether hnRNP LL knockdown depended on the wild-type loop sequences. Two mutant luciferase constructs were prepared, one in which both lateral loops were mutated (Mut5′,3′L) and a second in which the central loop was mutated (MutCL). As anticipated, while knockdown of the hnRNP LL still had an inhibitory effect on luciferase activity with the wild-type and MutCL promoter constructs, there was no significant effect on reporter activity with the Mut5′,3′L (Figure 3C). The knockdown effect of hnRNP LL siRNA on reporter activity (∼25%) could be considered modest; however, this is probably due to the limited (50–60%) knockdown of hnRNP LL and suggests that activation of BCL2 expression may involve other transcriptional factors. To determine the relative importance of the sequence of each of the lateral loops, a similar EMSA competition experiment was carried out in which either the 5′ or 3′ sequence was swapped out (Mut6) or both lateral loops carried either the 5′ (Mut6-1) or 3′ (Mut6-2) loop sequences (Supplemental Figure 2, left). The histograms of band quantification did not reveal significant differences for any of the sequences (64% for Mut6, 65% for Mut6-1, and 70% for Mut6-2), indicating that the variance observed between Mut6-1 and Mut6-2 may be related to the 5′ or 3′ positions of the lateral loop and the associated RRMs rather than sequences (Supplemental Figure 2, right).
Figure 3.

EMSA and siRNA knockdown experiments demonstrate that hnRNP LL recognizes the i-motif through the lateral loops. (A) Folding pattern of the BCL2 i-motif showing the 5′ and 3′ lateral loops (II and V, respectively) and central loop (CL). (B) Competition EMSA showing selective binding of hnRNP LL to the two lateral loops of Py39WT with 8:5:7 loop folding pattern. Four different mutant sequences were used. Mut5′,3′L has mutations in two lateral loops, and Mut5′L and Mut3′L have mutations in the 5′ and 3′ loops, respectively (binding sequences are color coded to match i-motif folding pattern in A). MutCL has mutations in the central loop. The percents above the sets of histograms for Mut5′,3′L, Mut5′L, Mut3′L, and MutCL indicates the addition of free DNA for each concentration of cold oligomers divided by three. (C) A luciferase assay shows that knockdown with hnRNP LL siDNA was dependent on the wild-type sequence in the lateral loops of the i-motif. Three pGL3 constructs of wild-type, Mut5′,3′L, and MutCL were co-transfected with pRL-TK for normalization and siRNA to hnRNP LL for 72 h. Final relative luciferase activities were obtained by normalization of the ratio of firefly to renilla to siRNA-untreated control of each construct P values (****P < 0.0001, ***P < 0.001, ns: not significant) were determined by t-test analysis.
Binding of hnRNP LL to the BCL2 i-Motif Results in Unfolding of the Structure
There was a significant decrease in the ellipticity of Py39WT at 286 nm in the presence of hnRNP LL in a concentration-dependent manner (Figure 4A, left) and a shift to a lower wavelength by about 2.2 nm at 2 equiv as detected by CD analysis. This suggests a partial unfolding of the i-motif or conversion to another topological form by hnRNP LL, which has been supported by single-molecule experiments in a recently published paper.16 To a lesser extent, the CD signal of Py39MutT also decreased upon binding of hnRNP LL (Figure 4A, right) and shifted to a higher wavelength by about 1–2 nm at 2 equiv. To determine the optimum distance between the two lateral-loop binding sites for competition with Py39WT, a series of oligomers containing the two consensus sequence binding sites connected by variable (2–17 nucleotides) spacers was used. A 13-nucleotide spacer was found to be optimal for competition with the end-labeled Py39WT (Figure 4B), which is the exact nucleotide distance between the two lateral loops in the wild-type sequence. It is important to note that the molar ratio of the unlabeled, unstructured 39-mers to the labeled 39-mer i-motif was 150:1. Pre-organization of the consensus binding sequences into the lateral loops of the folded i-motif provides a significant entropic and kinetic binding advantage. We conclude that a key function of the i-motif folded structure is to provide a rigid chemical scaffold upon which to display the preorganized lateral loops for optimum kinetic advantage for binding of hnRNP LL. In addition, the sequential recognition and binding of not just one but both lateral loops also provide a significant kinetic advantage. On the basis of these EMSA experiments we propose that the RRMs of hnRNP LL recognize the mixed cytosine/guanine sequences in the lateral loops by binding to one or both of the lateral loops (the 5′ lateral loop is the favored one). Then, after subsequent protein-facilitated i-motif unfolding, hnRNP LL binds more stably to an unfolded i-motif species not present initially.
Figure 4.

EMSA, Br2 footprinting, and FRET show that hnRNP LL unfolds the BCL2 i-motif after binding. (A) CD analysis shows that binding of hnRNP LL produces a conformational change in the i-motif. hnRNP LL was preincubated with Py39WT or Py39MutT at pH 6.5 for 5 min at room temperature before measuring the CD. (B) Competition EMSA showing that 13 nt is the optimal length between two hnRNP LL binding sites for the binding of hnRNP LL. All oligomers are 39-mers. Competition EMSA experiments were conducted in a binding buffer (pH 6.8) for 20 min of preincubation of 250 nM of cold oligomers with hnRNP LL and subsequent 5 min incubation of end-labeled Py39WT. This represents about a 150 molar excess of cold DNA to labeled i-motif. The histogram below the gel shows the relative binding intensity from the EMSA gel. (C) Bromine footprinting of the BCL2 i-motif and hnRNP LL complex showing the conformational change of Py39WT induced by hnRNP LL. Py39WT and hnRNP LL were incubated for 5 min at room temperature, and bromine generated in situ was added for 30 min. Black and red plots are 0 and 10 μg of hnRNP LL, respectively. The peaks with the black dots correspond to those where maximum inhibition occurs and include C runs II and IV and the central loop. The right panel shows the folding pattern of the BCL2 i-motif with that region protected from Br2 cleavage shown in the blue shading. Experimental conditions are described in the Methods section. (D) FRET experiments showing i-motif-specific unfolding activity by hnRNP LL. FAM/TAMRA dual-labeled probes were incubated at pH 6.5 or 7.9 with hnRNP LL at room temperature for 5 min, and then fluorescence intensity was measured at 495 nm (Ex.)/528 (Em.). Right panel shows the unfolding of the i-motif consistent with the fluorescence enhancement seen in the left panel (WT at pH 6.5). P values (**P < 0.01, ***P < 0.001) were determined by t-test analysis.
The structure of the Py39WT oligomer after hnRNP LL binding was examined by bromine footprinting (Figure 4C). Binding of hnRNP LL to the BCL2 i-motif changed the cleavage pattern of Py39WT following bromination/piperidine treatment (compare lanes 2 and 4 in Figure 4C). Cytosines in run I were more cleaved, while other cytosines in runs III to IV were less cleaved with increasing concentrations of hnRNP LL. Bromine footprinting of the same sequence in the presence of IMC-76 indicated the opposite effect on runs III and IV.5 This suggests that the unfolded form induced by hnRNP LL is not the partial hairpin. Since runs III to IV are positioned between the 5′ and 3′ lateral loops, which the spacer experiment demonstrated must be unfolded in the hnRNP LL–bound species, bromine footprinting inhibition suggests that they are more protected by close association with hnRNP LL than even the lateral loops (Figure 4C).
The i-motif unfolding activity of hnRNP LL was further confirmed by FRET assay (Figure 4D). In this experiment hnRNP LL increased the fluorescence intensity by 1.8-fold at pH 6.5, where the i-motif is expected to be initially present but had little effect at pH 7.9 where the i-motif is absent (Figure 4D, left). In addition, hnRNP LL selectively increased the fluorescence signal of the wild-type sequence compared to the lateral loop mutant (Mut5′,3′L) at pH 6.5 (Figure 4D, right). This result strongly suggests that the binding and associated unfolding activity of hnRNP LL is restricted to the i-motif structure with the wild-type sequence in the lateral loop.
The binding of hnRNP LL to two similar consensus sequences in the C-rich strand of the BCL2 promoter that results in transcriptional activation is quite analogous to hnRNP K binding to the CT elements in MYC NHE III1. The hnRNP K protein contains three KH domains that are spaced apart in a similar manner to hnRNP LL but recognize TCCC sequences.17 Significantly, TCCC elements are found in the lateral loops of the MYC i-motif and are spaced the same distance apart in the unfolded structure as those found in the BCL2 i-motif18 (unpublished results). Thus, hnRNP K and hnRNP LL may have similar roles in transcriptional activation of MYC and BCL2: they recognize similar single-stranded elements in the lateral loops of their respective i-motifs, and both presumably remodel the i-motif to form a thermodynamically stable species prior to transcriptional activation.
The Mutually Exclusive Binding of IMC-76 to the Flexible Hairpin and IMC-48 and hnRNP LL to the i-Motif Results in a Redistribution of These Species in Solution and in Cells
The partitioning of biological molecules between two equilibrating species in which only one is biologically active is well-known in the RNA world.6 These can act as switches if the chemical equilibrium can be changed by the preferential sequestration of one of the forms by a small molecule. To determine whether a similar mechanism might operate in a system consisting of two equilibrating DNA species, we explored the biological outcome (transcriptional silencing or activation) of using a small molecule that bound preferentially to each of the DNA forms. Characterization of the ternary interactions between the DNA, protein, and each small molecule in a cell-free system permitted extension into a cellular system.
We have demonstrated that IMC-76 can change the dynamic chemical populations of equilibrating C-rich strand species in solution by sequestering the flexible hairpin.5 We have also shown that the RRMs of hnRNP LL require the presence of the CGCCC and CCCGC sequences in the lateral loops of the i-motif for optimum binding and subsequent unfolding leading to transcriptional activation. Taken together, this suggests a competition between IMC-76 and hnRNP LL for binding to the equilibrating populations of the flexible hairpin and i-motif. Binding of IMC-76 to the flexible hairpin should increase the population of this species and deplete the population of the hnRNP LL–bound i-motif species. In cells, IMC-76 is expected to decrease the i-motif population in the promoter element and thus reduce hnRNP LL promoter occupancy. In contrast, IMC-48, like hnRNP LL, binds exclusively to the BCL2 i-motif; thus IMC-48 should increase the i-motif population and thereby increase the amount of hnRNP LL–bound i-motif species—assuming that hnRNP LL binds to the i-motif tightly enough to displace IMC-48—and increase the promoter occupancy in cells. Experiments were designed using EMSA, hnRNP LL knockdown, and ChIP analysis to test these hypotheses.
The results from the EMSA experiment in which different amounts of IMC-76 were incubated at pH 6.5 with the BCL2 i-motif and its equilibrating species for 3 h prior to the addition of hnRNP LL are shown in Figure 5A. In the absence of IMC-76 and hnRNP LL, there are two conformationally different oligomer species separated in the gel. We propose that species 1, the predominant form, is most likely the i-motif, which leaves species 2 as the flexible hairpin. Upon addition of hnRNP LL, species 1 is depleted preferentially to form the hnRNP LL high-mobility-shifted complex. However, as IMC-76 concentration is increased, the amount of the hnRNP LL–BCL2 i-motif complex is decreased, and species 2, putatively the flexible hairpin, is increased at the expense of species 1, the presumed i-motif. This is in accord with the idea that IMC-76 and hnRNP LL compete for the pool of equilibrating species to trap (IMC-76) or remodel (hnRNP LL) the i-motif to an unfolded species. In a parallel experiment, the effect of IMC-48 on the distribution of the three species was determined (Figure 5B). As the concentration of IMC-48 increased, there was a depletion of species 1 and 2 and an increased band intensity of the hnRNP LL–BCL2 i-motif complex. This supports the hypothesis that the increase of i-motif population by IMC-48 facilitates the binding of hnRNP LL to the i-motif structure.
Figure 5.

The consequences of sequestration of the flexible hairpin or the BCL2 i-motif by IMC-76 (A) and IMC-48 (B), respectively, on the binding of hnRNP LL to the i-motif. (A) EMSA analysis of the competition between IMC-76 and hnRNP LL for the i-motif (left) and densitometric analysis (right). (B) EMSA analysis of the cooperativity between IMC-48 and hnRNP LL for the i-motif (left) and densitometric analysis (right). IMC-76 or IMC-48 was incubated with Py39WT for 3 h, and hnRNP LL was added for 10 min at pH 6.5 before running the 6% native PAGE. Relative band intensities are plotted against IMC-76 or IMC-48 concentrations (right). Species 1 and 2 are proposed to be the i-motif and flexible hairpin, respectively.
To investigate the IMC-76 and IMC-48 cellular effects, which are known to affect BCL2 transcription, siRNA and ChIP experiments were performed. First, the potentially additive or subtractive inhibitory effects of hnRNP LL siRNA together with IMC-76 or IMC-48 were determined on BCL2 mRNA transcription, following knockdown of hnRNP LL. Second, to determine the effects of IMC-76 or IMC-48 on promoter occupancy by Sp1 and hnRNP LL, ChIP analysis was performed using MCF-7 cells. While the treatment of 50 nM hnRNP LL siRNA alone significantly decreased BCL2 mRNA levels by 24%, addition of either 0.5 or 2 μM of IMC-76 further decreased the mRNA levels to a total of 33% and 47%, respectively (Figure 6A). In contrast, IMC-48 reversed the inhibitory effects of the hnRNP LL siRNA (Figure 6B). This is expected because both knockdown of hnRNP LL and depletion of the i-motif population by IMC-76 should be additive in lowering transcription, although they act on different targets. In contrast, the effect of IMC-48 in cells should antagonize the inhibitory effects of BCL2 mRNA expression knockdown.
Figure 6.
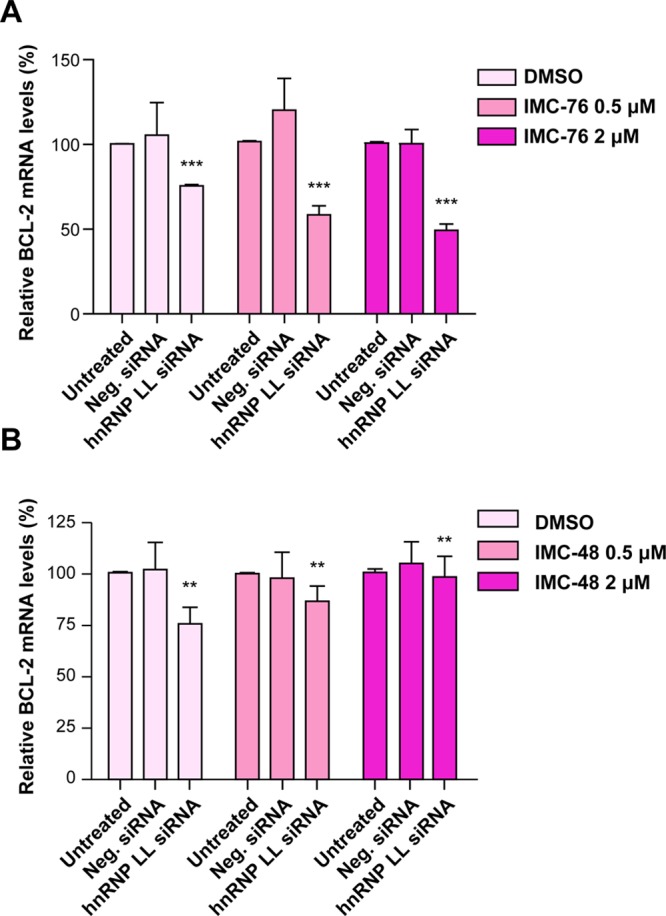
Effect of IMC-76 and IMC-48 combined with knockdown of hnRNP LL on BCL2 mRNA levels. (A) Enhanced effect of IMC-76 treatment on the BCL2 mRNA levels after knockdown of hnRNP LL. (B) Restoration of BCL2 mRNA levels after treatment with IMC-48 following knockdown of hnRNP LL. After transfection of 50 nM of hnRNP LL siRNA into MCF-7 cells for 48 h, IMC-76 or IMC-48 was incubated for a further 24 h (***P < 0.001, **P < 0.01). The P value (**P < 0.01) was determined by one-way ANOVA analysis.
To directly assess the effect of IMC-76 and IMC-48 on recruitment of transcriptional factors to the BCL2 promoter, which is proposed to contain the i-motif-forming element, a ChIP assay was performed on Sp1 and hnRNP LL using MCF-7 and BJAB cells, respectively. The MCF-7 cells, which overexpress BCL2 and have detectable levels of hnRNP LL, were used to determine the inhibitory effect of IMC-76 on BCL2 transcription. Alternatively, the BJAB cells, which only express basal levels of BCL2, were used to evaluate the activating effect of IMC-48 on BCL2 transcription. Sp1 is a ubiquitous transcription factor bound to the GC-rich region in gene promoters. MCF-7 and BJAB cells were treated with IMC-76 and IMC-48 (at 0.5 and 2 μM) for 24 h, respectively. Quantification of immunoprecipitated DNA was performed by SYBR green I qPCR using two specific sets of primers, amplifying either the closest upstream region (−103 to −3 base pairs) or a far upstream region (>3000 base pairs) from the i-motif/G-quadruplex-forming site of the P1 promoter, the latter serving as a negative control for normalization. As shown, IMC-76 decreased the occupancy of both Sp1 and hnRNP LL bound to the BCL2 P1 promoter region in a concentration-dependent manner in MCF-7 cells (Figure 7A). In contrast, IMC-48 increased the promoter occupancy of both Sp1 and hnRNP LL in BJAB cells (Figure 7B). To ensure that the effect of IMC-76 on promoter occupancy by Sp1 and hnRNP LL was not due to inhibition of transcription of these proteins, qPCR was carried out. In a similar way, the effect of IMC-48 on the transcription level of Sp1 and hnRNP LL was tested with BJAB cells (Supplemental Figure 3). In addition, immunoprecipitation (IP) experiments were carried out for both Sp1 and hnRNP LL to verify antibody specificity (Supplemental Figure 4).
Figure 7.

ChIP analysis of the effect of IMC-76 and IMC-48 given either singly (A and B) or in sequential order (IMC-76 followed by IMC-48) (C) on promoter occupancy of Sp1 and hnRNP LL. For single drug treatments (A and B), two concentrations (0.5 and 2 μM) of IMC-76 or IMC-48 with DMSO as a control were incubated with MCF-7 and BJAB cells, respectively, for 24 h. For sequential treatments (C), antagonism between IMC-76 and IMC-48 was shown through restoration of Sp1 and hnRNP LL promoter occupancy levels following administration of IMC-48 after prior knockdown with IMC-76 in MCF-7 cells. MCF-7 cells were treated with DMSO or 2 μM of IMC-76 for 24 h. In a similar way, other MCF-7 cells were treated first with 2 μM of IMC-76 and then with 2 or 4 μM of IMC-48 for a further 24 h. IP was performed with antibodies to Sp1 and hnRNP LL and IgG as a negative control and acetyl-histone H3 (AcH3) as a positive control. The P values (***P < 0.001, **P < 0.01) were determined by one-way ANOVA analysis.
In the companion paper (DOI: 10.021/ja410934b) we have demonstrated that IMC-48 and IMC-76 are antagonistic in redistribution of the two populations of DNA species in solution using 1D NMR studies as well as cellular studies by following the chemosensitization to cyclophosphamide.5 To extend these studies to examine what happens at the promoter level to the transcriptional factors that bind to the BCL2 regulatory element, we carried out an experiment in MCF-7 cells in which we first depleted Sp1 and hnRNP LL from the promoter element by treatment with IMC-76. Then we treated 24 h later with IMC-48, which should reverse these effects relative to the control in which only IMC-76 has been previously added. The results (Figure 7C) showed that the decreased promoter occupancy by both Sp1 and hnRNP LL in MCF-7 cells produced by IMC-76 was reversed by IMC-48 in a concentration-dependent manner, illustrating an antagonistic relationship between IMC-76 and IMC-48 at the promoter level. This result, together with the results from the previous complementary antagonism experiments carried out at the solution level using NMR and at the cellular level using chemosensitization to cyclophosphamide,5 provide very strong evidence for direct competition between IMC-76 and IMC-48 for the two equilibrating populations of the BCL2 i-motif and flexible hairpin, resulting in the cellular consequences mediated via hnRNP LL.
Development of a Molecular Switch Model for the BCL2 i-Motif That Employs DNA Dynamics to Define the Roles of IMC-48 and IMC-76 That Work in Concert with hnRNP LL for BCL2 Transcriptional Activation and Inhibition
Of the noncanonical DNA structures, the i-motif is perhaps the most dynamic at pH levels that are either slightly acidic or even close to neutral. Because the i-motif is formed from hemiprotonated C–CH+ base pairs that have a pKa of 4.58 for the N3 of cytosine,19 their existence in cells has not been generally anticipated. However, an important contributor to their increased stability is favorable van der Waals energies, due to close contacts between deoxyribose sugars in the narrow groove of the tetrad, and this is dependent upon the precise topology of the phosphodiester backbone with intercalation of C–CH+ pairs.20,21 Significantly, i-motifs in RNA cannot be formed, even at low pH, because of the steric hindrance of the 2′-hydroxyl group.22,23 Since the topology of the phosphodiester backbone appears to be critical in stabilization of the i-motif through sugar–sugar interaction, conditions such as molecular crowding, negative superhelicity, and loop constraints may play important roles if they influence these parameters. What is critical for the proposed role of i-motifs as molecular switches in transcriptional regulation is that their dynamic nature is such that they can easily move between folded (i-motif) and unfolded (hairpin) populations of molecules under physiological conditions. Thus, factors such as those described above that influence the topology of the deoxyribose backbone of the intercalated C–CH+ base pairs likely play critical roles in determining the dynamic nature of promoter i-motifs. Recently, the function of i-motifs as reversibly conformational switches for nanobiotechnology has been reviewed.24
In cellular systems, where both transcriptionally induced negative superhelicity and molecular crowding can occur, this molecular plasticity of i-motifs in promoter elements is likely to be evident under physiological conditions, even without considering the presence of transcriptional factors such as hnRNP LL, which can recognize and then unfold the i-motif to form a thermodynamically stable species. Thus we propose that promoter i-motifs may have dynamic properties more like RNA secondary structures than what are typically associated with DNA.1,6 In the studies described here, we show that both small molecules (IMC-76 and IMC-48) and a transcriptional factor (hnRNP LL) can either change the relative population states of the i-motif and its equilibrating conformers or, in the case of hnRNP LL, drive changes in the interhelical conformation of the i-motif to bind most stably to an alternative conformation not originally present. We now show that the competition between these ligand- or protein-associated dynamic states has functional consequences, leading to gene expression modulation. This is analogous to metabolite-sensing riboswitches that regulate gene expression in response to small molecules by causing a redistribution of the conformational states with functional consequences.6 The underlying common feature of the BCL2 i-motif and the riboswitch is the ability of ligands and proteins to take advantage of the intrinsic dynamic chemical behavior of DNA or RNA. This i-motif structure is found in DNA but not in RNA22 and thus is present in the genome at a very low number of copies, implying that there are different drug-receptor characteristics present than those found commonly in protein and RNA or even duplex DNA. Beyond the ability of the molecules such as IMC-48 and IMC-76 to recognize and bind to the i-motif and its equilibrium partners, they must also transport into the cell nucleus and then bind to these structures in open chromatin regions for them to be biologically active. Therefore the chromatin state must also be favorable for transcriptional regulation and involve chromatin-modifying proteins as well as other epigenetic changes. These factors, taken together with the critical need for transcriptionally induced negative superhelicity, imply a complex but inherently attractive new drug receptor class for exploitation in drug combinations, such as with topoisomerase and HDAC inhibitors.
In the dynamic transitional system shown in Figure 8A, we propose two predominant conformational states, of which the flexible hairpin can coexist with either a single-stranded form or the fully folded species (i-motif). For i-motif-forming sequences at neutral pH, both the partially folded and single-stranded states coexist,25 and under molecular crowding conditions, the i-motif conformation can exist even at neutral conditions.26 Under negative superhelicity, the i-motif has been observed in the MYC promoter under physiological conditions.18 For the BCL2 promoter sequence, at pH 6.6 the flexible hairpin and i-motif forms can be observed both by 1H NMR5 and in an EMSA gel (Figure 5). Incremental IMC-76 addition sequesters the flexible hairpin form, which contains five GC base pairs. By analogy with the binding of a steroidal diamine to a poly(dA·dT) duplex,27 IMC-76 most likely binds in the non-Watson–Crick base pair regions where unstacked base pairs exist capped at either side by GC base pairs (Figure 8B). IMC-76 sequestration of the flexible hairpin species will deplete the i-motif. This redistribution of the conformation species in Box A results in a reduction in the amount of the hnRNP LL–shifted band in Figure 5A, which in a cellular context would result in reduction of BCL2 transcriptional activation and chemosensitization. IMC-48 does exactly the opposite and increases the amount of the hnRNP LL–shifted band in Figure 5B. It does this by most likely binding to the central loop of the BCL2 i-motif and further constrains the lateral loops through which hnRNP LL recognizes and binds before unfolding the structure. This would further accelerate the kinetic step and lead to enhanced transcriptional activation of BCL2. Furthermore, it is likely that under varying extents of negative superhelicity produced during transcriptional firing, the intrinsic dynamic behavior of the i-motif and its equilibrating conformational forms will be even more accessible. This may be important when larger energetic barriers are present, such as in the disruption of the C–C+ base pairs.28 The i-motif folding and unfolding kinetics, the latter requiring disruption of base pairing, is slow in comparison to RNA elements like riboswitches.28 However, in cellular promoter elements, where i-motif-binding proteins are present together with dynamic forces that result from negative superhelicity, the kinetics may be much faster.
Figure 8.

Conformational transitions and biological consequences that occur following mutually exclusive binding of IMC-76, IMC-48, and hnRNP LL to the different equilibrating forms of the C-rich strand in the BCL2 promoter. Box A shows the two different major conformational states of the C-rich strand in the BCL2 promoter under different pH conditions. Acidic conditions drive formation of the i-motif, and at pH 6.6 there is a conformational mixture of the flexible hairpin and i-motif. Upon addition of IMC-76, the flexible hairpin form is sequestered (A to B), resulting in depletion of the populations of the i-motif species. Conversely, IMC-48 binds to the central loop of the BCL2 i-motif to sequester this species, and then the RRMs 1 and 2 of the hnRNP LL, which are closely spaced apart, are initially proposed to recognize and bind to both of the lateral loops (II and V) containing the CCCG and CGCC sequences, which are constrained in a single-stranded form (A to C). Following this recognition event, there are hnRNP LL–driven changes in the interhelical conformations such that the two lateral loops are forced apart so that the further spaced apart RRMs 2 and 3 or 4 are able to bind to the 5′ and 3′ CCCG and CGCC recognition sequences to form a stable complex (C to D). Last, hnRNP LL bound to the alternative conformation of the C-rich strand causes transcriptional activation of BCL2 (D to E). The consequence of competition between IMC-76 and hnRNP LL for the different conformational states of the C-rich strand depletes the population undergoing the transition A to C to D to E and repression of BCL2 gene expression. Alternatively, binding of IMC-48 to the BCL2 i-motif leads to an increased amount of i-motif that is bound by hnRNP LL and transcriptional activation (A to C to D).
The recognition and subsequent stable binding of hnRNP LL to the BCL2 i-motif was more complex than we first anticipated. The hnRNP LL protein and its paralog hnRNP L share a 58% overall amino acid identity and contain four classical RRMs that are highly conserved. The overall arrangement of the RRMs in hnRNP L and hnRNP LL is similar, such that in both cases they are separated by linkers of different lengths so they can recognize either adjacent domains or ones spaced further apart. A combination of at least two RRMs (1/2 or 2/3) is required for the high-affinity binding of hnRNP L to RNA. The competition experiments in Figure 4A demonstrate that pre-organization of the binding sequences in the i-motif lateral loops confers entropic and kinetic advantages for hnRNP LL binding. A comparison of the role of hnRNP L in the RNA switch that regulates VEGF expression with the role of hnRNP LL in the regulation of BCL2 expression provides two insightful analogies.29 First, the hnRNP L binding site consists of 21 nucleotides in mRNA 3′UTR approximately equivalent to the 23 combined nucleotides contained in the two lateral loops and the linker region recognized by hnRNP LL. Second, the conformational change in the VEGF 3′UTR is directed by two different signals, hnRNP L and an INF-γ-activated inhibitor of the translational complex, which bind to two different RNA conformers in a mutually exclusive manner, just as hnRNP LL and IMC-76 bind to the i-motif and flexible hairpin in the BCL2 promoter. Whether there is a transcriptional factor equivalent to IMC-76 remains to be determined.
The hnRNP LL protein binds with high affinity to the BCL2 i-motif (20–70 pM), and siRNA knockdown significantly decreased BCL2 expression (Figure 2A). Recognition of the i-motif is through the 5′ and 3′ lateral loops, but subsequent unfolding of the i-motif is presumably required before a stable complex is formed (Figures 3 and 4). It is likely that both lateral loops are initially recognized by adjacent RRMs (1 and 2) before subsequent hnRNP LL–driven changes in the interhelical conformation, so that the 5′ and 3′ lateral loops are driven apart to bind to the RRMs spaced further apart (e.g., 1 or 2 with 3 or 4) (Figure 8A–C and A–D). In cells the competition for the BCL2 i-motif species by IMC-76, which depletes this population, reduces the amount of hnRNP LL bound to the BCL2 promoter, as shown by ChIP analysis (Figure 7A), whereas IMC-48 produces the opposite effect (Figure 7B) by constraining the i-motif structure. As further proof of our overall proposal, IMC-48 and IMC-76 are antagonistic in their effects at three different levels: in solution, as shown by NMR; at the promoter level (Figure 7C), as shown by ChIP analysis; and in cellular consequences, as shown by chemosensitization to cyclophosphamide.5
The mechanism by which hnRNP LL acts as a transcriptional factor is as yet unknown. As noted, hnRNP K activates MYC transcription and binds to the CT elements in the promoter, probably by mechanism similar to that for hnRNP LL. At least two other factors may be important in the mechanism for transcriptional activation by hnRNP LL. First, there is a CA element in the upstream region that is a potential hnRNP L binding site; the binding of the more ubiquitous hnRNP L to this element may result in a looping structure with formation of a heterodimer with hnRNP LL to activate transcription.30 Second, DDX21, an RNA helicase, also binds to the i-motif (Table 1) or to an associated protein, and this may be important in facilitating i-motif unfolding to activate transcription.
Conclusion
Our contribution shows that the intrinsic dynamic state of the i-motif, similar in many respects to the dynamic nature of RNA, makes the dynamic equilibrium of the noncanonical DNA structure an attractive target for small molecule control of gene expression. It is important to note that the biological effects we observe are correlations consistent with the proposed mechanism rather than direct proof for i-motifs in cells. We propose that the correlated evidence between the solution and the cellular effects of IMC-48 and IMC-76 in combination with hnRNP LL are at this point as compelling as that provided for the presence of G-quadruplexes in cells, with the exception of experiments of the type recently published by the Cambridge group using a fluorescent antibody against G-quadruplexes.31 For the BCL2 i-motif, the mutual exclusivity of IMC-76 and hnRNP LL for targeting different conformational forms of the equilibrating i-motif allows the repression of BCL2 gene expression and chemosensitization of drug-resistant lymphoma and breast cancer cells using a steroid molecule. This mutual exclusivity has been supported by single-molecule experiments.16 Conversely, enhanced expression of BCL2 mediated by compounds related to IMC-48 provides a means to protect against neurodegenerative diseases, such as those found in CNS disorders. This brings the i-motif into focus as an alternative structure to the G-quadruplex in promoter elements as a therapeutic target. It is anticipated that the tools of the medicinal chemist can be harnessed to identify additional molecules that function at this locus to control gene expression with important therapeutic consequences.
Methods
i-Motif Protein Binding Purification Assay
All of the following incubations, washes, and centrifugations (1 min at 500 g) were performed at 4 °C. The biotinylated BCL2 i-motif wild-type and mutant oligomers (5′-Biotin-TTTTCTTTTCCCCCACGCCCTCTGCTTTGGGAACCCGGGAGGGGCGCTTACAGCCCCGCTCCCGCCCCCTTCCTCCCGCGCCCGCCCCT-3′) and mutant oligomers (5′-Biotin-TTTTCTTTTCCCCCACGCCCTCTGCTTTGGGAACCCGGGAGGGGCGCTTACAGTTTTGCTCCCGCTTTCTTCCTTTTGCGCCCGCCCCT-3′) (4 μg each) were conjugated to washed streptavidin beads in separate 1.5 mL Eppendorf tubes in binding Buffer B (1 mM DTT, 25 mM Tris-HCl [pH 7.6], 50 mM NaCl, 0.5 mM MgCl2, 1 mM EDTA, 10% glycerol) plus 1× protease inhibitor cocktail overnight, rotating. Following overnight incubation, the beads were washed in Buffer B. The mutant oligomer-conjugated beads were incubated with 500 μg HeLa extract for 3 h, rotating. The beads were centrifuged, and supernatant was transferred to the wild-type oligomer-conjugated beads and incubated for 3 h, rotating. The mutant oligomer/bead/HeLa extract complex was washed in Buffer B, and supernatant from each wash was transferred to the wild-type oligomer. Proteins were eluted off the mutant oligomer-conjugated beads with successive washes of an NaCl gradient (0.1–2 M) in Buffer B, and each supernatant was collected and combined. The wild-type oligomer/bead/HeLa nuclear extract complex was subjected to the same procedure of washing and elution as that described for the mutant oligomer complex. The eluted proteins were processed by the BIO5 Proteomics Core Facility (University of Arizona, BIO5 Institute, Tucson, AZ). The two protein samples were subjected to SDS PAGE and visualized by Coomassie and silver staining. Prominent bands were excised from the gel and analyzed for protein identification by LC/MS/MS.
Purification of Recombinant hnRNP LL
The cDNA of hnRNP LL was purchased from Open Biosystems (Thermo Scientific) and subsequently cloned into the pET28a protein expression vector (Novagen). After sequencing analysis to confirm the pET28a–hnRNP LL, this expression construct was transformed into Rosetta-gami B (DE3) pLysS cells (Novagen). The expression of hnRNP LL was induced by 0.1 mM IPTG (isopropyl β-d-1-thiogalactopyranoside) overnight at room temperature. Harvested cells were resuspended in a lysis buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 1% Triton X-100, 1 mg/mL lysozyme, and 1× protease inhibitor cocktail [Sigma, #8465]) and underwent 10 cycles of the following: incubation on ice for 5 min, vortexing for 30 s, and sonication for 10 s. Cell debris was removed by centrifugation at 14 000 rpm for 30 min at 4 °C, and the supernatant was removed and incubated with HisPur Cobalt resin (Thermo Scientific) while rotating for 30 min at 4 °C to allow for the selective binding of histidine-tagged hnRNP LL. The resin was washed by washing Buffer A (50 mM NaH2PO4 with 0.4× protease inhibitor cocktail) and B (50 mM NaH2PO4 [pH 8.0] and 100 m NaCl with 0.1× protease inhibitor cocktail) sequentially, and elution buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl with 1× protease inhibitor cocktail) was used to separate hnRNP LL from resin. Purified hnRNP LL was subjected to buffer exchange into a protein stock buffer with 20 mM HEPES-NaOH (pH 7.4), 100 mM KCl, 10% glycerol, 2 mM DTT, and 0.1% NP-40 using a centricon (Millipore). Purity of hnRNP LL was confirmed by SYPRO Ruby staining. A Bradford assay was performed to determine the protein concentration.
EMSA
Sequences used for EMSA experiments are shown in Figures 5 and 6. All oligomers for these experiments were purchased from Eurofins MWG Operon and PAGE-purified. Concentrations of purified oligomers were determined using the Lambert–Beer equation with molecular extinction coefficients (M–1 cm–1) as follows: Py39WT, 292 338; Py39MutT, 319 216; Pu39WT, 398 551. The wild-type BCL-2 i-motif (Py39WT) oligomer was end-labeled with [γ-32P]-ATP. The detailed procedure for labeling is described in the literature.32 For competition EMSA, PAGE-purified cold (nonlabeled) oligomers and 160 μM hnRNP LL as a final concentration were preincubated with 20 mM HEPES (pH 6.8), 100 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1 μg/μL BSA, 0.1% Tween 20, 10% glycerol, and 0.01 μg/μL of poly(dI·dC) for 20 min on ice, and end-labeled Py39WT was added for 5 min.
To determine the effect of IMC-76 and IMC-48 on binding of hnRNP LL to the i-motif, end-labeled Py39WT was incubated with several concentrations of IMC-76 in a buffer (20 mM MES [pH 6.5], 100 mM KCl, 4 mM MgCl2, 1 mM DTT, 1 μg/μL BSA, 0.1% Tween 20, 10% glycerol, and 0.01 μg/μL of poly[dI·dC]) for 3 h at room temperature. 160 μM hnRNP LL was added and incubated for 10 min at 4 °C. The DNA–protein complex and free DNA were visualized by 6% native PAGE (0.5× TBE and 1.25% glycerol in gel and running buffer) and phosphorimager scanning.
SPR Analysis
SPR analyses were performed on a Biacore T100 optical biosensor with CM5 sensor chips (GE Healthcare, Piscataway NJ). N-hydroxysuccinimide, 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride, and ethanolamine, 1 M (pH 8.5) were purchased from GE Healthcare. Biotinylated oligomers with wild-type and mutant sequences (Py39WT: 5′-biotinGGCACCACAGCCCCGCTCCCGCCCCCTTCCTCCCGCGCCCGCCCCTCCGCGC3′ and Py39MutT: 5′-biotinGGCACCACAGTTTTGCTTTTGCTTTCTTCCTTTTGCGTTTGTTTTTCCGCGC-3′) were purchased from Eurofins MWG Operon.
Streptavidin (Leinco Technologies, Inc., St. Louis, MO) (SA) was immobilized on a CM5 chip using standard amine coupling. Briefly, carboxy groups on the chip surfaces were activated with an injection of 0.05 M N-hydroxysuccinimide/0.2 M 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride at a flow rate of 10 μL/min for 7 min SA, diluted in 10 mM NaOAc (pH 5.5), 20 μg/mL, and pulsed over the surface at a flow rate of 10 μL/min until 1750 RU was achieved. Temperature was 25 °C, and running buffer was 10 mM HEPES (pH 7.4), 150 mM NaCl, 0.05% Tween 20. Any remaining active esters were blocked by injecting 1 M ethanolamine (pH 8.5) for 7 min at 10 μL/min. The SA surfaces were washed six times with 1 M NaCl/50 mM NaOH at a flow rate of 20 μL/min for 60 s. Both active and reference flow cells had SA.
The biotinylated oligomers were resuspended in 10 mM Tris (pH 8.0), 1 mM EDTA at 100 μM, then diluted to 1 μM in 20 mM HEPES (pH 7.9), 100 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1 μg/μL BSA, 0.1% Tween 20, and 10% glycerol. The diluted oligomers were heated at 95 °C for 5 min, cooled to rt, and centrifuged at 16 000× g for 10 min at rt. The supernatant was diluted to 1 nM in the same buffer and injected over the active surface at 10 μL/min until 5 RU was captured.
hnRNP LL was diluted into running buffer (20 mM HEPES for pH 7.9 and pH 6.8 and 20 mM MES for pH 6.5 were used) together with 100 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1 μg/μL BSA, 0.1% Tween 20, and 10% glycerol. These were then injected over the active and reference flow cells. The dilution range was 0.078–10 nM. Analysis temperature was 18 °C, and flow rate was 50 μL/min. Sample compartment was kept at 10 °C. Association time was 300 s (pH 7.9 and pH 6.8) or 420 s (pH 6.5). Dissociation time was 500 s. Surfaces were regenerated with a 10 s pulse of 20 mM NaOH at a flow rate of 30 μL/min, followed by a stabilization time of 350 s.
Raw data were reference subtracted, and buffer blanks were subtracted (double referencing). Data were fit to a 1:1 binding model using a global fit algorithm (Biacore T100 Evaluation Software) to obtain the kinetic parameters ka, kd, and KD.
Circular Dichroism
CD analyses were conducted as previously described.32 The i-motif-forming oligomers were synthesized by Eurofins MWG Operon. Py39WT and Py39MutT were diluted to 5 μM with a buffer (50 mM MES [pH 6.5], 100 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1 μg/μL BSA, 0.1% Tween 20, and 10% glycerol). Recombinant hnRNP LL was diluted by protein stock buffer to desired concentrations to maintain consistent buffer conditions in each sample. Oligomers and hnRNP LL were incubated for 5 min at room temperature. CD spectra were baseline corrected by subtracting a buffer alone or a buffer with protein.
Bromine Footprinting
For the Br2 footprinting of the BCL-2 i-motif and hnRNP LL complex, recombinant hnRNP LL was incubated with end-labeled Py39WT in a buffer (50 mM MES [pH 6.5], 4 mM MgCl2, 100 mM KCl, 1 mM DTT, 1 μg/μL BSA, 0.1% Tween 20, 10% glycerol, and 0.02 μg/μL poly[dI·dC]) for 5 min at room temperature. Bromination was conducted by addition of 0.1 mM bromine for 30 min at room temperature, and subsequently a phenol/chloroform solution was added to interrupt the bromination and remove the protein. Brominated oligomer was subjected to EtOH precipitation. The pellet was washed with 80% EtOH and treated with 10% piperidine at 93 °C for 15 min to induce the bromination-specific DNA cleavage. Cleaved product was washed with water and visualized by a 20% sequencing gel with 7 M urea.
siRNA Knockdown Assay
siRNAs (ID: SASI_Hs01_00171042 and SASI_Hs01_00171043) targeting hnRNP LL (Sigma) were diluted to 50 nM as a final concentration. As a negative control siRNA, ON-TARGETplus Nontargeting Pool (Dharmacon) was used. For the untreated control, transfection reagent with media only was used. MCF-7 cells (1.5 × 104 per well of a 12-well plate) cultured in 10% FBS and 1% penicillin/streptomycin-supplemented RPMI were treated with hnRNP LL siRNA with Fugene HD transfection reagent for 72 h. For determining the knockdown effect of hnRNP LL along with IMC-76 treatment, siRNA of hnRNP LL was transfected for 48 h followed by addition of IMC-76 for 24 h. Total RNA was extracted using an RNeasy purification kit (Qiagen) and quantitated by measuring absorbance at 260 nm. The cDNA was synthesized by a reverse-transcription kit (Qiagen or Takara with gDNA remover) and used as templates for qPCR with TaqMan probes for hnRNP LL (Hs00293181_m1, FAM-labeled), BCL-2 (HS00608023_m1, FAM-labeled), PDGFR-β (Hs01019589_m1, FAM-labeled), kRAS (Hs00364282_m1, FAM-labeled), and GAPDH (Hs02758991_g1, VIC-labeled) (ABI). The Ct values were obtained by Rotor-Gene Q (Qiagen) to analyze the relative quantity of hnRNP LL and BCL-2 mRNA compared to GAPDH as an internal control.
Promoter Assay
The pGL3-BCL-2 wild-type construct was prepared using the BCL-2 P1 promoter region from −35 to +614, which includes the i-motif starting site. The sequence was inserted into the pGL3-basic vector at the KpnI and NheI restriction sites. The pGL3-Mut5′,3′L and pGL3-MutCL constructs were generated by site-directed mutagenesis. The sequences of each construct were confirmed by sequencing analysis. MCF-7 cells (1.5 × 104) were transfected with 500 ng of pGL3 construct, 10 ng of pRL-TK, and 50 nM of negative control or hnRNP LL siRNA by Fugene HD transfection reagent and incubated for 72 h. Cells were lysed by passive lysis buffer (Promega), and then supernatants were subjected to dual-luciferase assays (Promega) using an FB12 luminometer (Berthold detection system). Data were normalized to the ratio of firefly to renilla luciferase of siRNA-treated sample and to siRNA-untreated control.
ChIP Assay
MCF-7 cells (5 × 105) and BJAB cells (1 × 106) were cultured overnight and then treated for an additional 24 h with 0.5 of IMC-76 or 2 μM IMC-48. Treatment with DMSO served as the control. To determine the antagonistic effect of two compounds, MCF-7 cells (∼3–4 × 105) were treated with DMSO or 2 μM of IMC-76 for 24 h. The next day, DMSO-treated cells were administered with DMSO or IMC-76, and IMC-76-treated cells were administered with 2 or 4 μM of IMC-48 with fresh media for 24 h. The composition of the buffers used for this ChIP assay is the same as those of the EZ ChIP kit (Millipore). Cells were treated with formaldehyde (1%) to cross-link proteins to DNA for 13 min at rt. MCF-7 and BJAB cells were lysed with 1% SDS buffer and sonicated to fragment chromosomal DNA into ∼500 base pairs for 15 and 45 cycles, respectively. Sheared chromosomal DNA was diluted with ChIP dilution buffer and precleaned with protein G-coupled Dynabeads (Invitrogen) for 2 h at 4 °C. Overnight IP with 4 μg of IgG (Cell Signaling, #2729S), acetyl-histone H3 (Millipore, #06-599), Sp1 (Cell Signaling, #5931S), or hnRNP LL (Cell Signaling, #4783S) antibodies at 4 °C was followed by addition of protein G-coupled Dynabeads for 90 min 4 °C. Immunoprecipitants were washed with low salt, high salt, and LiCl immune complex wash buffer. Elution with vortexing for 30 min at rt and reverse cross-linking with 200 mM NaCl at 65 °C overnight were performed sequentially. The DNA was purified using a PCR purification kit (Qiagen), and SYBR Green I qPCR analysis was performed with Rotor-Gene Q (Qiagen) to determine relative quantity of DNA using primers to specifically amplify the −3 to −103 base pairs from the BCL-2 i-motif–forming region within the promoter (BCL-2 P1 promoter region, 5′-AGGAGGGCTCTTTCTTTCTTCTT-3′ [forward] and 5′-GTGCCTGTCCTCTTACTTCATTCT-3′ [reverse]). An upstream region (∼ –3456 base pairs) from this i-motif-forming region33 was also amplified to serve as a negative control for normalization using the primer pair 5′-AGGTTGGGGCCATGGTTTACT-3′ (forward) and 5′-CAGCCTGGGTGACAGACTGATAC-3′ (reverse). Melting analysis of PCR product showed only one detectable Tm (data now shown), and double normalizations were performed to obtain data (2–ΔΔCt). ΔCt values were calculated by subtracting Ct values of negative region (Ct – Ct neg) and then ΔΔCt values were obtained by normalizing to ΔCt of input (ΔCt – ΔCt input).
Quantitative PCR
To determine if IMC-76 and IMC-48 affect the transcription level of Sp1 and hnRNP LL, qPCR was conducted using Rotor-Gene Q (Qiagen). MCF-7 cells (1.5 × 105) and BJAB cells (3 × 105) were treated with 2 μM IMC-76 and IMC-48 for 24 h. Total RNA extraction, cDNA synthesis, and qPCR were performed using the gene-specific TaqMan probes. The specificity and IP quality of Sp1 and hnRNP LL antibodies are demonstrated by the manufacturer and further verified by IP (Supplemental Figure 4).
Immunoprecipitation (IP)
For further verification of the specificity of these antibodies with MCF-7 cells, IP experiments were conducted. For Sp1 IP, nuclei isolated by kit (Sigma, NUC101) were incubated with RIPA buffer (Cell Signaling, #9806) for 15 min on ice. For hnRNP LL IP, whole-cell lysate was used. After homogenization using QIAshredder (Qiagen), the extract was centrifuged at 14 00 rpm at 4 °C for 15 min. The supernatant was precleaned with 50 μL of protein G-coupled magnetic beads (Invitrogen, 10003D) at 4 °C for 1 h. Protein concentration was determined by Bradford assay, and then the supernatant was diluted to 1 μg/μL for Sp1 and 12.5 μg/μL for hnRNP LL by lysis buffer. For IP, antibodies for Sp1 and hnRNP LL were added to reach a 1:100 and 1:10 dilution, respectively. As a negative control, ∼0.5–1 μg of IgG, optimized to adjust the heavy chain signal in IP samples of IgG and Sp1 by Western blot, was used. Binding of antibodies was conducted at 4 °C overnight for Sp1 and 2 h at rt for hnRNP LL. Protein G-coupled magnetic beads with 1% BSA were added and incubated for 1 h to precipitate the immunocomplex. The beads carrying the immunocomplex were washed by lysis buffer three times. To dissociate the immunocomplex, 25 μL of Laemmli buffer was added, heated at 95 °C for 5 min, and subjected to SDS-PAGE (6% or 8%). For Western blot analysis, proteins were transferred to PVDF membrane in TBS buffer with 20% MeOH. After blocking the membrane with 2% BSA/2% nonfat milk in TBS-T (0.1% Tween 20) for 1 h, Sp1 antibody with 1:1000 dilution and hnRNP LL antibody with 1:300 in 1% BSA/TBS-T were treated overnight at 4 °C. As a secondary antibody, goat antirabbit IgG (H+L) Dylight 680 was diluted into 1:10 000 in 1% BSA/TBS-T and incubated for 1 h at rt. LI-COR was used to detect the bands.
Acknowledgments
This research was supported by grants from the National Institutes of Health (GM085585-01 (LHH), GM085585-02S1 (LHH), CA153821 (LHH), T32CA09213 (SLK), GM083117 (SMH). SPR data were acquired by the Arizona Proteomics Consortium supported by NIEHS grant ES06694 to the Southwestern Environmental Health Sciences Center, NIH/NCI grant CA023074 to the Arizona Cancer Center, and by the BIO5 Institute of the University of Arizona. The Biacore T100 biosensor was provided though generous support of the Prescott Friends of the Sarver Heart Center (University of Arizona) with leadership gifts from Jim and Linda Lee, Ron and Laura James, and Swayze and Kathy McCraine. We also thank Dr. David Bishop for his significant contribution to the preparation and editing of the final version of the text and figures displayed in the article.
Supporting Information Available
This information includes proteins identified from pull-down experiments, SPR sensorgrams, competition EMSA gels, IP results for Sp1 and hnRNP LL, cytotoxic potencies, mRNA levels, and uncut examples of EMSA gels. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† These authors contributed equally.
The authors declare the following competing financial interest(s): The corresponding author has a financial interest in TetraGene, a biotech company involved in development of drugs that target G-quadruplexes and i-motifs.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brown R. V.; Hurley L. H. Biochem. Soc. Trans. 2011, 39, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhakal S.; Yu Z.; Konik R.; Cui Y.; Koirala D.; Mao H. Biophys. J. 2012, 102, 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks T. A.; Hurley L. H. Nat. Rev. Cancer 2009, 9, 849. [DOI] [PubMed] [Google Scholar]
- Tomonaga T.; Levens D. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrick S.; Kang H.-J.; Alam M. P.; Mdathil M. M.; Agrawal P.; Gokhale V.; Yang D.; Hecht S. M.; Hurley L. H.. J. Am. Chem. Soc. 2014. [DOI] [PMC free article] [PubMed]
- Dethoff E. A.; Chugh J.; Mustoe A. M.; Al-Hashimi H. M. Nature 2012, 482, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J.; Dexheimer T. S.; Chen D.; Carver M.; Ambrus A.; Jones R. A.; Yang D. J. Am. Chem. Soc. 2006, 128, 1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexheimer T. S.; Sun D.; Fry M.; Hurley L. H. In Quadruplex Nucleic Acids; Neidle S., Ed.; RSCPublishing: Cambridge, 2006, p 180. [Google Scholar]
- González V.; Guo K.; Hurley L.; Sun D. J. Biol. Chem. 2009, 284, 23622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexheimer T. S.; Carey S. S.; Zuohe S.; Gokhale V. M.; Hu X.; Murata L. B.; Maes E. M.; Weichsel A.; Sun D.; Meuillet E. J.; Montfort W. R.; Hurley L. H. Mol. Cancer Ther. 2009, 8, 1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelotti E. F.; Michelotti G. A.; Aronsohn A. I.; Levens D. Mol. Cell. Biol. 1996, 16, 2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.-H.; Lim M.-H.; Youn D.-Y.; Jung S. E.; Ahn Y. S.; Tsujimoto Y.; Lee J.-H. Biochem. Biophys. Res. Commun. 2009, 382, 583. [DOI] [PubMed] [Google Scholar]
- Hung L.-H.; Heiner M.; Hui J.; Schreiner S.; Benes V.; Bindereif A. RNA 2008, 14, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdoerffer S.; Moita L. F.; Neems D.; Freitas R. P.; Hacohen N.; Rao A. Science 2008, 321, 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishna I.Structure-function analysis of heterogeneous nuclear ribonucleoproteins L and LL. Ph.D. Dissertation, Justus-Liebig-Universität, Gießen, Germany, 2010. [Google Scholar]
- Cui Y.; Koirala D.; Kang H.; Dhakal S.; Yangyuoru P.; Hurley L. H.; Mao H. Nucleic Acids Res. 2014, 10.1093/nar/gku185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backe P. H.; Messias A. C.; Ravelli R. B. G.; Sattler M.; Cusack S. Structure 2005, 13, 1055. [DOI] [PubMed] [Google Scholar]
- Sun D.; Hurley L. H. J. Med. Chem. 2009, 52, 2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J. J.; Rytting J. H.; Izatt R. M. J. Phys. Chem. 1967, 71, 2700. [DOI] [PubMed] [Google Scholar]
- Malliavin T. E.; Gau J.; Snoussi K.; Leroy J.-L. Biophys. J. 2003, 84, 3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi S.; Wani A. H.; Krishnan Y. Nucleic Acids Res. 2006, 34, 4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin D.; Gehring K. J. Am. Chem. Soc. 1998, 120, 4069. [Google Scholar]
- Fenna C. P.; Wilkinson V. J.; Arnold J. R. P.; Cosstick R.; Fisher J. Chem. Commun. (Camb.) 2008, 3567. [DOI] [PubMed] [Google Scholar]
- Choi J.; Majima T. Photochem. Photobiol. 2013, 89, 513. [DOI] [PubMed] [Google Scholar]
- Choi J.; Kim S.; Tachikawa T.; Fujitsuka M.; Majima T. J. Am. Chem. Soc. 2011, 133, 16146. [DOI] [PubMed] [Google Scholar]
- Rajendran A.; Nakano S.; Sugimoto N. Chem. Commun. (Camb.) 2010, 46, 1299. [DOI] [PubMed] [Google Scholar]
- Patel D. J.; Canuel L. L. Proc. Natl. Acad. Sci. U.S.A. 1979, 76, 24.284337 [Google Scholar]
- Lieblein A. L.; Buck J.; Schlepckow K.; Fürtig B.; Schwalbe H. Angew. Chem., Int. Ed. 2012, 51, 250. [DOI] [PubMed] [Google Scholar]
- Ray P. S.; Jia J.; Yao P.; Majumder M.; Hatzoglou M.; Fox P. L. Nature 2009, 457, 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preußner M.; Schreiner S.; Hung L.-H.; Porstner M.; Jack H.-M.; Benes V.; Ratsch G.; Bindereif A. Nucleic Acids Res. 2012, 40, 5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G.; Tannahill D.; McCafferty J.; Balasubramanian S. Nat. Chem 2013, 5, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrick S.; Akiyama Y.; Hecht S. M.; Hurley L. H. J. Am. Chem. Soc. 2009, 131, 17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema S. K.; Mishra S. K.; Rangnekar V. M.; Tari A. M.; Kumar R.; Lopez-Berestein G. J. Biol. Chem. 2003, 278, 19995. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.