

Abstract
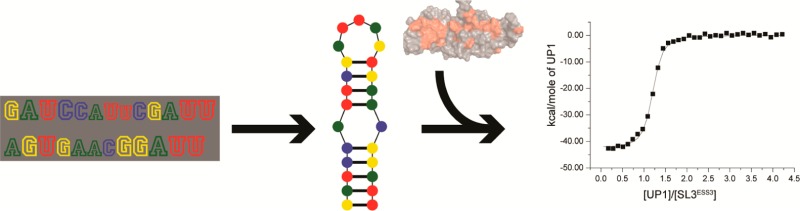
Complete expression of the HIV-1 genome requires balanced usage of suboptimal splice sites. The 3′ acceptor site A7 (ssA7) is negatively regulated in part by an interaction between the host hnRNP A1 protein and a viral splicing silencer (ESS3). Binding of hnRNP A1 to ESS3 and other upstream silencers is sufficient to occlude spliceosome assembly. Efforts to understand the splicing repressive properties of hnRNP A1 on ssA7 have revealed hnRNP A1 binds specific sites within the context of a highly folded RNA structure; however, biochemical models assert hnRNP A1 disrupts RNA structure through cooperative spreading. In an effort to improve our understanding of the ssA7 binding properties of hnRNP A1, herein we have performed a combined phylogenetic and biophysical study of the interaction of its UP1 domain with ESS3. Phylogenetic analyses of group M sequences (x̅ = 2860) taken from the Los Alamos HIV database reveal the ESS3 stem loop (SL3ESS3) structure has been conserved throughout HIV-1 evolution, despite variations in primary sequence. Calorimetric titrations with UP1 clearly show the SL3ESS3 structure is a critical binding determinant because deletion of the base-paired region reduces the affinity by ∼150-fold (Kd values of 27.8 nM and 4.2 μM). Cytosine substitutions of conserved apical loop nucleobases show UP1 preferentially binds purines over pyrimidines, where site-specific interactions were detected via saturation transfer difference nuclear magnetic resonance. Chemical shift mapping of the UP1–SL3ESS3 interface by 1H–15N heteronuclear single-quantum coherence spectroscopy titrations reveals a broad interaction surface on UP1 that encompasses both RRM domains and the inter-RRM linker. Collectively, our results describe a UP1 binding mechanism that is likely different from current models used to explain the alternative splicing properties of hnRNP A1.
Alternative splicing of the human immunodeficiency virus 1 (HIV-1) genome is a highly regulated process that involves site-specific recruitment of host factors to viral RNA control elements.1,2 The splicing pattern is complex, as more than 40 spliced isoforms are produced from a single 9.7 kb genome.3 Recent deep sequencing data using a clinical isolate show the HIV-1 transcript pool may include upward of 100 different spliced products that vary with time, cell type, and infected individual.4 Along with unspliced genomic RNA, the pool encompasses two major classes of spliced transcripts, a 1.8 kb class and a 4 kb class, which derive from the combinatorial usage of five 5′ donor and eight 3′ acceptor sites. The 1.8 kb transcripts are completely spliced and encode viral proteins Tat, Rev, and Nef, whereas the 4 kb class consists of singly spliced transcripts that encode Env, Vpu, Vif, and Vpr. Efforts to understand the splicing mechanism in HIV-1 have revealed all of the 3′ acceptor sites are suboptimal, primarily because of nonconsensus polypyrimidine tracts and branch points.1,2 A proper balance of spliced transcripts is maintained through the use of cis regulatory signals functioning as silencers or enhancers.1,2 The cis signals, collectively termed splicing regulatory elements (SREs), are binding sites for members of the mutually antagonistic SR and hnRNP protein families. Generally, SR proteins bind enhancer elements to activate suboptimal splice sites, whereas hnRNP proteins counteract this activity through interactions with silencer elements.1,2 Despite extensive research that has aimed to understand the HIV-1 splicing mechanism, the series of molecular events that determine whether a 3′ splice site will be activated or repressed remains largely unknown.
Among the 3′ acceptor sites, regulation at splice site A7 (ssA7) has been the most thoroughly studied.5−12 Splicing of site D4 to A7 is required to remove the env intron containing the Rev responsive element (RRE) and produce the 1.8 kb multiply spliced (Rev-independent) transcripts. An intronic splicing silencer (ISS), a bipartite exonic splicing silencer (ESS3a/b), and two exonic splicing enhancers (ESE2 and ESE3) constitute the core SREs that control the activity of ssA7.1,2 RNA secondary structure probing revealed the ISS, ESE, and ESS3b elements are located in the apical portion of three stem loop domains:5,9 SL1ISS, SL2ESE, and SL3ESS3. These sites were additionally shown to bind the host hnRNP A1 (SL1ISS, SL3ESS3, and SL2ESE3) and ASF/SF2 (SL2ESE3) proteins to repress or activate ssA7 usage.5−7,9 Two mutually incompatible models have been proposed to explain the regulation of ssA7.5,9,11,13 The model suggested by Krainer et al. asserts hnRNP A1 initially binds a high-affinity UAG element on ESS3b, followed by cooperative assembly in a 3′–5′ direction along weaker sites.11 Cooperative binding of multiple hnRNP A1 molecules is sufficient to unwind the RNA secondary structure and displace an SR protein bound to its cognate ESE element upstream from ESS3b.11,13 The Kjems and Branlant groups independently proposed an alternative model for ssA7 regulation.5,9 Their model posits hnRNP A1 cooperatively assembles on the ssA7 SRE locus within the context of a conserved RNA secondary structure to effectively occlude ASF/SF2 recognition of the ESE elements. Footprinting studies of multiple hnRNP A1 proteins bound to the ssA7 locus showed discrete protection patterns, inconsistent with binding-induced unwinding of the RNA secondary structure. Given the discrepancies between the two models, it is obvious that a better understanding of the RNA binding properties of hnRNP A1 is needed.
Human hnRNP A1 is an ∼36 kDa protein composed of tandem N-terminal RNA recognition motifs, collectively known as UP1, and a C-terminal glycine-rich domain.14 The UP1 domain confers nucleic acid binding specificity, whereas the glycine-rich domain facilitates protein–protein contacts and interacts with nucleic acids nonspecifically.15−19 The binding properties of hnRNP A1 have been studied to a greater extent with nucleic acid substrates that contain a low degree of structural complexity, as compared to substrates that adopt defined secondary structures. These studies have revealed hnRNP A1 interacts with nucleic acids over a wide affinity range, preferentially binds single-stranded nucleic acids, and has a higher specificity for purines than for pyrimidines.15−17,20,21 Moreover, in vitro selection experiments showed hnRNP A1 binds a 5′-UAGGGA/U-3′ consensus sequence with nanomolar affinity.22
Structural insights into the determinants of sequence-specific binding have been provided through the crystal structure of UP1 bound to single-stranded telomeric DNA, which contains two copies of a high-affintiy TAGG motif. In the crystal, UP1 forms a dimer in which the relative intra-RRM orientation is antiparallel.23 This arrangement positions RRM1 from one monomer in contact with RRM2 from the symmetry-related monomer, therefore creating an extended nucleic acid binding platform. In the crystal structure, the single-stranded telomeric DNA stretches in a 5′–3′ direction from RRM1 of one monomer to RRM2 of the other monomer. The extended binding platform observed in the UP1 dimer has been used to explain the high-affinity binding properties of target nucleic acid sequences as well as hnRNP A1 functional mechanisms.23 While the UP1–DNA structure offers a wealth of insight into principles of sequence-specific recognition, the structure does not explain how hnRNP A1 recognizes complex RNA secondary structures as observed for the ssA7 SRE locus.
Our previously determined solution NMR structure of SL3ESS3 revealed the ESS3b element (5′-GAUUAG-3′) adopts a quasi-symmetric heptaloop that we reasoned forms a binding surface for hnRNP A1.24 Using titration calorimetry, we determined both RRM domains of UP1 are required to form a 1:1 high-affinity complex with SL3ESS3 in which binding does not disrupt the RNA secondary structure.24 Here, we have further characterized the binding properties of the UP1–SL3ESS3 complex by conducting a systematic mutational and biophysical study. Key findings of this work reveal the SL3ESS3 secondary structure is highly conserved across several HIV-1 subtypes and is an important determinant in forming a high-affinity UP1–SL3ESS3 complex and binding involves multiple contact points primarily with purine bases of the ESS3b loop. Moreover, the data presented here shed light on how UP1 interacts with stable stem loop structures.
Materials and Methods
Phylogenetic Analysis of ESS3 Elements
Nucleotide sequences (excluding recombinants) comprising the ESS3b stem loop region (NL4-3 coordinates) were derived from the Los Alamos HIV sequence database (http://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html) for HIV-1 group M subtypes (12 total) for which at least 10 sequences had been submitted. Sequences for individual subtypes were aligned in Geneious (version 6.1.7) using the ClustalW multiple-sequence alignment algorithm.25 Consensus sequences and logos for individual subtypes were generated also using Geneious (version 6.1.7) based on the majority nucleotide present at each site. Regions of conservation for each subtype were defined on the basis of a threshold frequency (represented as the height of the logo) of 75% within the consensus logo for the corresponding aligned sequences, and nucleotides most representative of these conserved sites were used for subtype-specific secondary structure predictions. A global consensus sequence and logo were also determined using an alignment of the consensus sequences for each subtype using the same method in Geneious.
Likelihood mapping implemented in Tree Puzzle (version 5.2)26 was used to determine if sufficient phylogenetic signal was present for the group M consensus sequence alignment prior to tree reconstruction. This alignment was then used to infer a maximum likelihood (ML) phylogenetic tree (Figure 1) and to calculate the overall mean genetic distance in Mega (version 5.2).27 The tree was outgroup-rooted using the consensus sequence for HIV-1 group O sequences also acquired from the Los Alamos HIV database. The Kimura two-parameter model with invariant sites28 was chosen for both the tree reconstruction and distance calculations based on the likelihood ratio test of log likelihood values provided using the nucleotide model test also implemented in Mega. A series of 2000 bootstrap replicates was performed in addition to a partition analysis in Phylopart (version 2.0)29 based on a percentile distance threshold of 9% and bootstrap support of ≥80%.
Figure 1.
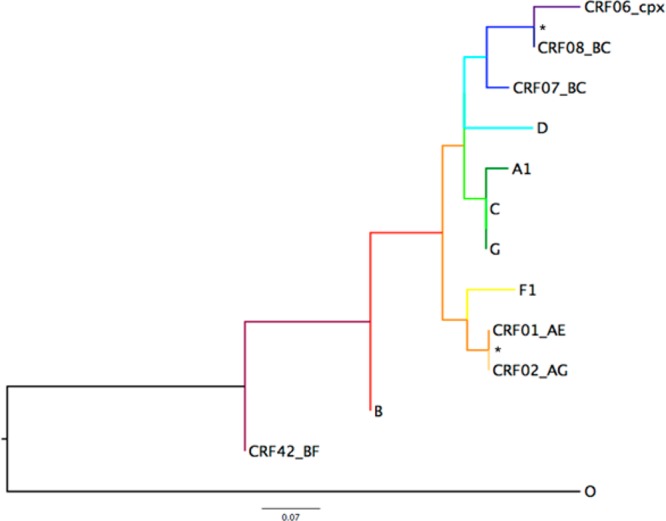
Maximum likelihood (ML) phylogenetic tree inferred from HIV-1 group M subtype-specific consensus sequences. The Kimura two-parameter model with invariant sites was used for ML tree reconstruction and calculation of the mean genetic distance. The HIV-1 group O consensus sequence was used for outgroup rooting of the tree. Asterisks denote bootstrap support of ≥80% for 2000 replicates. Branch lengths are presented in substitutions per site (sub/site) and scaled according to the scale bar depicted at the bottom. The overall mean genetic distance was 0.165 ± 0.041 sub/site.
In Vitro Transcription of SL3ESS3 Constructs
SL3ESS3 and the cytosine-substituted constructs were transcribed in vitro using recombinant T7 RNA polymerase expressed from BL21(DE3) cells. Synthetic DNA templates were purchased from Integrated DNA Technologies (Coralville, IA) and correspond to the NL4-3 HIV-1 subtype. Transcription reactions were conducted using standard procedures30,31 in reaction volumes ranging from 10 to 30 mL and mixtures subsequently purified via urea–polyacrylamide gel electrophoresis (PAGE) and electroelution and washed multiple times in a Millipore Amicon Ultra-4 centrifugal filter device. The RNA was then dried in a Vacufuge Plus (Eppendorf) and resuspended in a buffer consisting of 120 mM KCl, 10 mM K2HPO4, and 0.5 mM EDTA (pH 6.5). NMR samples were dried down in buffer and resuspended in a 90% H2O/10% D2O mixture. Prior to being used, each RNA was heated to 95 °C for 2 min and snap-cooled on ice to ensure a single hairpin conformation, as determined by nondenaturing PAGE. The six-nucleotide ESS3b loop construct was purchased from Thermo Scientific Dharmacon RNAi Technologies and deprotected following the manufacturer’s protocol. Concentrations were determined using theoretical molar extinction coefficients calculated with NanoDrop 2000 (Thermo Fisher).
UP1 Expression and Purification
The N-terminal (His)6-UP1 fusion (used for ITC and size exclusion chromatography) was prepared as previously described.24 For NMR studies, a C-terminal UP1-(His)6 fusion was used because it gave better resolved 1H–15N HSQC spectra. We previously showed UP1 binds SL3ESS3 with identical thermodynamics despite the location of the (His)6 tag.24 Briefly, UP1-(His)6 was transformed into BL21DE3 cells and grown in M9 medium supplemented with either 1 g/L 15NH4Cl for HSQC titrations or 1 g/L 15NH4Cl and 2 g/L [13C]glucose for backbone assignments. The labeled UP1-(His)6 constructs were purified via nickel affinity chromatography using a 1 mL Hi-trap column (GE Biosciences) followed by further purification and buffer exchange using a HiLoad 16/600 Superdex 75 pg (GE Biosciences) gel filtration column. The purity was evaluated via sodium dodecyl sulfate (SDS)–PAGE, and the concentration was determined using a theoretical molar extinction coefficient. Protein stock solutions were kept in a buffer consisting of 120 mM KCl, 10 mM K2HPO4 (pH 6.5), and 0.5 mM EDTA at 4 °C until they were used.
Analytical Size Exclusion Chromatographic Titrations
Chromatographic titrations were performed with a Superdex 200 10/300 GL column (GE Healthcare Life Sciences). Each RNA was heated to 95 °C for 2 min and snap-cooled on ice to ensure a single hairpin conformation. Samples in 120 mM KCl, 10 mM K2HPO4, and 0.5 mM EDTA (pH 6.5) were loaded into a 100 μL loop and run at a flow rate of 0.5 mL/min. Samples were run at 5 μM RNA, and complexes were prepared by increasing the level of UP1 to give molar ratios of 0.5:1, 1:1, and 1.5:1.
Calorimetric Titrations
Titrations were performed at 25 °C using a VP-ITC calorimeter (MicroCal, LLC). The N-terminal (His)6-UP1 construct was used for all titrations. To avoid adding reducing agent to the ITC cell, two Cys-to-Ser point mutations were introduced at positions 43 and 175. The Cys-to-Ser mutant gave identical HSQC spectra, confirming the protein behaves like the wild type. The RNA constructs were prepared by being dried down in water utilizing a Vacufuge Plus (Eppendorf) and resuspended in 120 mM KCl, 10 mM K2HPO4, and 0.5 mM EDTA (pH 6.5). The samples were diluted to concentrations of 2.5–3 μM for the wild type and cytosine-substituted SL3ESS3 constructs. The ESS3b loop construct was prepared at a concentration of 20 μM. Prior to titrations, the samples were annealed by being heated at 95 °C for 2 min and snap-cooled on ice. UP1 was prepared at a concentration of 335 μM for ESS3b loop titrations and 50 μM for SL3ESS3 titrations. UP1 was titrated into ∼1.4 mL of RNA over 35 injections of 8 μL each. Each titration was performed in three replicates. Prior to nonlinear least-squares fitting in Origin version 7.0, the raw data were corrected for dilution by subtracting the average heats from the last few points of the saturated upper asymptotes.
NMR Spectroscopy
RNA secondary structures were confirmed with one-dimensional (1D) 1H NMR of the imino region on a Varian 600 MHz instrument equipped with an HCN room-temperature Bioprobe. Spectra were recorded at 283 K using the Wet pulse sequence with 256 scans at a spectral width of 15000 Hz. Sample concentrations of 300 μM were prepared for SL3ESS3 and the cytosine-substituted constructs. The ESS3b loop was prepared at a concentration of 50 μM. Each sample was annealed and snap-cooled prior to collection.
1H–15N HSQC titrations were performed on a Bruker 900 MHz spectrophotomer (TXI cryoprobe) with the HSQCETFPF3GPSI pulse sequence at 298 K. Titrations of unlabeled SL3ESS3 into 15N-labeled UP1-(His)6 were performed at molar ratios from 0.25 to 1.0 in 120 mM KCl, 10 mM K2HPO4, and 0.5 mM EDTA (pH 6.5) in a 90% H2O/10% D2O mixture. 1H–15N HSQC chemical shift assignments for free UP1 were taken from the BMRB (18728)32 and further confirmed for our construct by running the standard suite of triple-resonance NMR experiments: HNCACB, HNCO, and C(CO)NH. All spectra were recorded at 298 K. All NMR data were processed with NMRPipe/NMRDraw33 and analyzed using NMRView J.34
Saturation Transfer Difference NMR
NMR-STD experiments were conducted at 5 μM UP1 and 100 μM SL3ESS3 for fully protonated RNA and 10 μM UP1 and 200 μM SL3ESS3 for 2H(GC)-labeled SL3ESS3. All samples were prepared in buffer consisting of 120 mM KCl, 10 mM K2HPO4 (pH 6.5), 0.5 mM EDTA, 5 mM DTT, and 99% 2H2O. Spectra were recorded on a Bruker 800 MHz spectrometer at 298 K running a modified version of the zgf2pr pulse sequence. The on-resonance frequency was set to 1.15 ppm with an irradiation time of 1.25 s and a power level of 60 dB.
Results
Evidence of Phylogenetic Conservation of the ESS3 Stem Loop Structure
To determine the level of conservation of the ESS3 stem loop structure (SL3ESS3), we performed alignments of HIV-1 subtypes derived from group M for which there are at least 10 sequences in the Los Alamos HIV database. The mean number of HIV-1 ESS3 sequences analyzed is 2860, with the total number of sequences per subtype provided in Table S1 of the Supporting Information. Maximum likelihood analysis for group M subtype-specific consensus sequences revealed a relatively large amount of nucleotide diversity for the ESS3b stem loop region with a mean genetic distance of 0.165 ± 0.041 substitute per site and a low frequency of statistical support for individual clusters (Figure 1). However, as shown in Figure 2, phylogenetic conservation is observed for particular regions within the sequence for all subtypes analyzed. The most conserved features occur in three subregions: the ESS3b element, a five-nucleotide stretch upstream of ESS3b, and a five-nucleotide stretch downstream of ESS3b. Secondary structure predictions using the most frequently occurring nucleotides of the consensus logos reveal the five-nucleotide stretch regions form stable base pairs in all strains, which universally exposes the ESS3b element at the apex of stem loop structures (Figure S1 of the Supporting Information). Although not predicted, the subtype-specific secondary structures might potentially form noncanonical pairs within the adjacent loop regions. Given the moderate conservation observed for the individual subtypes, a global consensus sequence and logo were determined from subtype-specific alignments (Figure 2B). The consensus sequence determined here for the ESS3 stem loop structure is 5′-GAUCCRUKCGAUUAGUGARCGGAUU-3′, where R corresponds to A or G and K corresponds to G or U. As expected, the global consensus sequence shows a high level of conservation of compensatory base pairing proximal to ESS3b with secondary structure predictions further supporting a conserved stem loop structure (Figure 2C). The consensus secondary structure consists of a 5 bp lower helix, a 4 bp upper helix, and a five-nucleotide single-stranded ESS3b loop. Using the ESS3 stem loop structure derived from NL4-3, we previously showed the predicted GU base pair adjacent to the ESS3b loop does not form, which increases the loop size to seven nucleotides.24 Furthermore, the AC juxtaposition that separates the lower and upper helices forms a pH-dependent AH+C base pair in NL4-3 that enhances the thermodynamic stability of the stem loop structure by ∼1.5 kcal/mol.24 The phylogenetic results presented here are consistent with previously published work using a much smaller data set.5,9 The ability to form a stable stem loop structure that exposes the ESS3b silencer element is a phylogenetically conserved structural motif in group M viruses.
Figure 2.
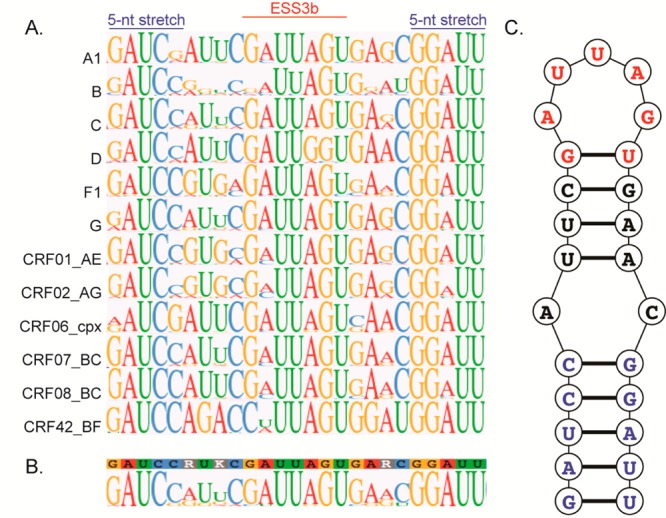
Phylogenetically conserved ESS3 stem loop structure. (A) Consensus logo alignments of the HIV-1 ESS3 stem loop region derived from group M subtypes. The region shown corresponds to residues 8445–8469 using the NL4-3 numbering system. As depicted, the alignments show three subregions with a high level of sequence conservation: the ESS3b loop, a five-nucleotide stretch upstream of ESS3b, and a five-nucleotide stretch downstream of ESS3b. (B) Global consensus logo of the ESS3 stem loop structure derived from subtype-specific alignments. (C) Predicted secondary structure using the most frequently occurring nucleotides of the global consensus logo. As illustrated, ESS3 folds into a phylogenetically conserved stem loop structure that exposes the ESS3b silencer.
The ESS3 Stem Loop Structure Is Required To Form a High-Affinity UP1–SL3ESS3 Complex
The phylogenetic conservation of the ESS3 stem loop structure suggests RNA structure may contribute to its silencer activity. Early biochemical mapping studies with hnRNP A1 yield mixed results regarding the integrity of the stem loop structure within the protein–RNA silencer complex, however.5,9,11,13 Using CD spectroscopy and the UP1 domain of hnRNP A1, we previously showed the ESS3 stem loop structure remains intact within the UP1–SL3ESS3 complex.24 Moreover, calorimetric titrations revealed UP1 binds SL3ESS3 as a high-affinity (Kd = 37.8 ± 1.1 nM) 1:1 complex, where the binding profile is characterized by a large favorable change in enthalpy (ΔH° = −38.8 ± 2.1 kcal/mol) and opposed by an unfavorable change in entropy (−TΔS° = 28.7 ± 2.1 kcal/mol). Repeat calorimetric titrations of the complex performed here under slightly different buffer conditions [140 mM K+ (pH 6.5)] are in excellent agreement with previously published results (Table 1), further validating the thermodynamic signature of this interaction.
Table 1. Thermodynamic Profiles of the Interactions of UP1 with Wild-Type and Cytosine-Substituted SL3ESS3 Constructsa.
| SLESS3 | loop sequence (5′ → 3′) | ΔG (kcal/mol) | ΔH (kcal/mol) | –TΔS (kcal/mol) | Kd (nM) | Fb | nc |
|---|---|---|---|---|---|---|---|
| WT | ..GAUUAG.. | –10.2 ± 0.1 | –44.7 ± 2.2 | 34.5 ± 2.2 | 27.8 ± 0.5 | – | 1.1 |
| G8454C | ..CAUUAG.. | –9.3 ± 0.1 | –32.7 ± 0.7 | 23.4 ± 0.6 | 151.5 ± 28.9 | 5 | 1.2 |
| A8455C | ..GCUUAG.. | –9.7 ± 0.2 | –34.1 ± 1.6 | 24.5 ± 1.8 | 86.8 ± 30.0 | 3 | 1.2 |
| U8456C | ..GACUAG.. | –10.2 ± 0.1 | –46.3 ± 0.5 | 36.1 ± 0.5 | 29.8 ± 4.1 | ∼1 | 1.1 |
| U8457C | ..GAUCAG.. | –9.7 ± 0.2 | –38.8 ± 2.2 | 29.1 ± 2.2 | 83.4 ± 17.7 | 3 | 1.2 |
| A8458C | ..GAUUCG.. | –8.9 ± 0.1 | –25.2 ± 0.5 | 16.3 ± 0.6 | 316.6 ± 64.6 | 11 | 1.2 |
| G8459C | ..GAUUAC.. | –9.4 ± 0.2 | –44.5 ± 2.9 | 35.1 ± 3.1 | 120.1 ± 55.0 | 4 | 1.0 |
| G8454C/G8459C | ..CAUUAC.. | –9.2 ± 0.01 | –32.4 ± 0.9 | 23.2 ± 0.9 | 196.6 ± 27.2 | 7 | 1.1 |
| A8455C/A8458C | ..GCUUCG.. | –8.5 ± 0.1 | –21.0 ± 1.3 | 12.5 ± 1.3 | 556.6 ± 133.0 | 20 | 1.1 |
| U8456C/U8457C | ..GACCAG.. | –9.8 ± 0.2 | –43.1 ± 0.8 | 33.3 ± 0.8 | 64.1 ± 17.7 | 2 | 1.0 |
| ESS3b loop | 5′-GAUUAG-3′ | –7.3 ± 0.06 | –15.3 ± 0.6 | 8.0 ± 0.7 | 4166.4 ± 420.9 | 150 | 0.9 |
Each construct is defined by its mutation in the loop, which is represented in bold in the 5′ to 3′ sequence. The loop construct lacks the stem of SL3ESS3 and consists of a single-stranded 5′-GAUUAG-3′ sequence. The data were collected at 298 K, 140 mM K+, and pH 6.5. The thermodynamic parameters were derived from fits to a single-site binding isotherm (three replicates).
F represents the factor by which the Kd of the mutant SL3ESS3 construct changes relative to that of the wild type.
Standard errors for stoichiometries were less than 5%.
To assess whether the ESS3 sequence alone is sufficient for high-affinity UP1 binding, we conducted calorimetric titrations using a six-nucleotide oligomer that mimics the ESS3b loop sequence. Isolated ESS3b does not contain any detectable secondary structure as determined by 1D 1H NMR spectroscopy (not shown); therefore, thermodynamic measurements using this construct report primarily on sequence determinants of binding. As shown in Figure 3, UP1 binds the ESS3b loop very weakly (Kd = 4.2 ± 0.4 μM) and with a thermodynamic signature markedly different from that of the UP1–SL3ESS3 interaction (Table 1). The UP1–ESS3b loop complex shows large reductions in free energy (ΔΔG° = 3.0 kcal/mol) and enthalpy (ΔΔH° = 29.4 kcal/mol) relative to those of the UP1–SL3ESS3 complex. By contrast, the loss of entropy upon formation of the UP1–ESS3b complex is smaller compared to that observed for the UP1–SL3ESS3 complex (Table 1). Collectively, the thermodynamic results provide clear evidence that UP1 recognizes the ESS3 stem loop structure using a fundamentally different set of interactions compared to how it recognizes the single-stranded isolated ESS3b oligomer.
Figure 3.

ESS3 stem loop structure is a key UP1 binding determinant. (A) Secondary structure of the ESS3 stem loop structure derived from the NL4-3 isolate. The ESS3b loop is colored red where the GU juxtaposition is unpaired as determined by NMR spectroscopy.24 Representative isotherms of UP1 titrated into (B) SL3ESS3 and (C) the ESS3b loop, with the average Kd (three replicates) calculated from nonlinear regression to a single-site isotherm. Changes in binding affinity are illustrated by the slope of the curve, with a lower slope indicating weaker binding. The comparison of the ESS3b loop oligomer (5′-GAUUAG-3′) to SL3ESS3 demonstrates the importance of structured elements in the interaction. Calorimetric titrations were performed at 298 K in pH 6.5 phosphate buffer containing 140 mM K+.
Single-Cytosine Substitutions Reveal the Importance of Conserved ESS3b Nucleotides in Stabilizing the UP1–SL3ESS3 Interaction
Having established the importance of secondary structure in the UP1–SLess3 interaction, we sought to determine the energetic contribution of each ESS3b nucleotide position within the context of the folded RNA. Stoltzfus et al. previously reported the ESS3b loop sequence is conserved across different HIV-1 group M clades, with a determined 5′-G(Y/A)UAG-3′ consensus motif.2 Their results are consistent with the alignments performed here. The ESS3b consensus motif resembles the high-affinity hnRNP A1 winner sequence identified by in vitro selection experiments; however, only a single UAG copy is present in ESS3b, whereas two copies are present in the winner sequence.22 To assess the energetic contribution of the ESS3b motif to high-affinity UP1 binding, we prepared a series of SL3ESS3 constructs in which each position of the 5′-GAUUAG-3′ apical loop was mutated to a cytosine residue. Fluorescence competition assays have demonstrated hnRNP A1 to have lower binding affinities with pyrimidines, particularly cytosine.21 One-dimensional 1H NMR spectra recorded for the mutant constructs show the cytosine substitutions do not affect the global secondary structure of SL3ESS3 as diagnostic imino signals of base-paired regions were observed (Figure S2 of the Supporting Information). An additional imino signal appeared in the spectrum recorded for the G8454C construct, however. On the basis of the chemical shift (∼11.9 ppm) of the additional signal, we predict G8454C forms a new Watson–Crick base pair with G8459.
As a preliminary measure of the binding properties, we monitored stepwise titrations of UP1 with the SL3ESS3 constructs (wild-type and mutant) via analytical size exclusion chromatography. Chromatographic traces of each titration are shown in Figure 4. Consistent with our previous solution NMR studies, free SL3ESS3 elutes from the column as a symmetric peak indicative of a stably folded structure (Figure 4). Upon titration of UP1 to a 0.5:1 molar ratio, the elution profile shows two peaks, of roughly equal A280 intensities, consistent with free and UP1-bound SL3ESS3 species. At a 1:1 molar ratio, the A280 intensity of the free peak is reduced to <20% of its starting A280 intensity, whereas a completely resolved symmetrical peak is observed for the complex. Further increasing the molar ratio to 1.5:1 leads to a complete loss of the free SL3ESS3 peak with no evidence of additional higher-order complexes (Figure 4). These data are in excellent agreement with the 1:1 stoichiometry observed by titration calorimetry (Figure 3) and validate the method as a qualitative tool for assessing UP1–RNA binding properties.
Figure 4.

Cytosine substitutions of conserved ESS3b loop nucleobases affect the stability of the UP1–SL3ESS3 complex. Analytical size exclusion chromatographic titrations of UP1 into wild-type and cytosine-substituted SL3ESS3 constructs. Titrations were performed by incubating a fixed amount (5 μM) of RNA with increasing amounts of UP1 until a final molar ratio of 1:1 was reached. Each complex was resolved at 277 K on a Superdex 200 10/300 GL column (GE Healthcare Life Sciences). Vertical dashed and solid lines correspond to free and UP1-bound SL3ESS3, respectively. The variation in the amount of free RNA remaining at a 1:1 molar ratio is indicative of differences in UP1–SL3ESS3 binding affinity.
As shown in Figure 4, the chromatographic traces of the mutant titrations show UP1 binds the SL3ESS3 cytosine-substituted constructs with similar 1:1 stoichiometries. For each titration event, a well-resolved complex peak is observed. The traces offer insight into the relative binding affinities as clear differences in the amount of complex formed across the titrations are observed. Notably, the data show the G8454C, A8458C, and G8459C constructs bind UP1 with affinities lower than that of wild-type SL3ESS3 because a significant peak for free RNA is observed at a 1:1 molar ratio (Figure 4). By comparison, both U-to-C substitutions exhibit a titration profile very similar to that of the wild-type construct. Taken together, these qualitative results reveal U-to-C substitutions have only minor effects on complex stability, whereas G/A-to-C substitutions measurably destabilize the complex.
A more quantitative description of the ESS3b nucleotides involved in high-affinity UP1 binding is provided via calorimetric titrations with the SL3ESS3 cytosine-substituted constructs (Figure 5). In agreement with the chromatographic titrations, the calorimetry results show the U8456C and U8457C constructs have thermodynamic signatures similar to those of wild-type SL3ESS3 (Table 1). Indeed, U8456C binds UP1 with an identical affinity, whereas U8457C shows an ∼3-fold reduction. For both constructs, a large favorable change in enthalpy is the thermodynamic driving force of complex formation. By comparison, the G/A-to-C constructs show binding affinities reduced from 3- to 11-fold (Table 1). These results are generally consistent with the qualitative binding properties measured by size exclusion chromatography (Figure 4). Relative to wild-type SL3ESS3, the A8455C construct showed the smallest perturbation in binding free energy (ΔΔG° = 0.6 kcal/mol), whereas perturbations for G8454C (ΔΔG° = 1.0 kcal/mol) and G8459C (ΔΔG° = 0.9 kcal/mol) were larger and approximately equal in magnitude. Notably, the 8458C construct displayed the largest reduction in binding free energy (ΔΔG° = 1.4 kcal/mol, corresponding to an ∼10-fold decrease in Kd) and enthalpy (ΔΔH° = 19.5 kcal/mol) relative to those of wild-type SL3ESS3. The collective thermodynamic data clearly show UP1 discriminates less against U-to-C substitutions than against G/A-to-C substitutions within the context of SL3ESS3. Moreover, the data reveal the high-affinity nature of the UP1–SL3ESS3 interaction derives from more than a single-site interaction because the construct with the weakest affinity (A8458C) still binds with a Kd in the high nanomolar range and approximately 13-fold tighter than the ESS3b loop.
Figure 5.

Calorimetric titrations reveal the energetic contribution of ESS3b nucleobases to the stability of the UP1–SL3ESS3 complex. Representative isotherms of UP1 titrated into each respective cytosine-substituted SL3ESS3 construct are illustrated. Differences in binding affinities are reflected by the slope of the curve, with a smaller slope indicative of weaker binding. Titrations were performed at 298 K, 140 mM K+, and pH 6.5. Processed isotherms fitted with a nonlinear 1:1 binding isotherm are shown. Average values and standard deviations of the dissociation constants (three replicates) are provided.
Double-Cytosine Substitutions Reveal UP1 Interacts with Multiple Purine Nucleobases of the ESS3b Loop
The UP1 titrations with single-cytosine-substituted SL3ESS3 constructs revealed a specific preference for G and A residues in the apical ESS3b loop. Therefore, we decided to probe for thermodynamic synergy between pairs of ESS3b residues by measuring binding affinities using double-cytosine-substituted SL3ESS3 constructs. For these studies, three constructs were prepared: G8454C/G8459C, A8455C/A8458C, and U8456C/U8457C. The rationale behind the design of the constructs derives from the approximate sequence symmetry of the ESS3b loop sequence, 5′-GAU⊙UAGU-3′ (where ⊙ represents the axis of mirror symmetry), and from our previously determined SL3ESS3 structure in which we showed the apical loop has quasi-stereochemical symmetry.24 One-dimensional 1H NMR spectra recorded for the double-cytosine-substituted constructs showed that each folds with the expected stem loop structure (Figure S2 of the Supporting Information). The additional imino signal observed for the G8454C construct is missing in the G8454C/G8459C construct, thus supporting the prediction that a new Watson–Crick pair forms between G8454C and G8459C.
As with the single-cytosine-substituted constructs, initial UP1–RNA binding properties were assessed by size exclusion chromatography (Figure 6A). Analysis of the chromatographic traces shows UP1 binds the double-cytosine-substituted constructs with a 1:1 stoichiometry and with a binding affinity trend consistent with the single mutants [Kd(G/A)2-to-(C)2 > Kd(U)2-to-(C)2]. The chromatographic titrations of the double mutants did not show conclusive evidence of a further reduction in binding affinity compared to those of the single mutants, however.
Figure 6.

Double-cytosine substitutions reveal UP1 makes multiple stabilizing contacts with the ESS3b loop. (A) Analytical size exclusion chromatographic titrations of UP1 into double-cytosine-substituted SL3ESS3 constructs performed as described in the legend of Figure 4. The reduced binding affinities of UP1 for the G8454C/G8459C and A8455C/A8458C constructs are evident by the amount of free RNA remaining at a 1:1 molar ratio. (B) Representative isotherms of UP1 titrated into double-cytosine-substituted SL3ESS3 constructs performed as described in the legend of Figure 5. The calorimetric results show a further decrease in binding affinity for the G8454C/G8459C and A8455C/A8458C constructs relative to those of the single-cytosine substitutions.
To gain a more quantitative description of how the double-cytosine substitutions affect UP1 binding affinity, calorimetric titrations were repeated. The calorimetry results are consistent with the binding affinity trend observed via size exclusion chromatography (Figure 6B). As summarized in Table 1, UP1 binds the U8456C/U8457C construct with an affinity comparable to that of wild-type SL3ESS3, corresponding to a small free energy perturbation of 0.5 kcal/mol. This result agrees favorably with the U8456C (ΔΔG° ≈ 0) and U8457C (ΔΔG° = 0.6 kcal/mol) substitutions and further supports the conclusion that UP1 exhibits low-level discrimination between uracil and cytosine nucleobases. The G8454C/G8459C construct also binds with comparable affinity to the corresponding single G-to-C substitutions (Table 1). The free energy perturbation for G8454C/G8459C relative to that of the wild type is 1.1 kcal/mol, whereas the free energy perturbations for G8454C and G8459C are 1.0 and 0.9 kcal/mol, respectively. A reasonable interpretation of these results is that sufficient residual binding energy is provided through favorable interactions with the remaining wild-type bases so that effects of the double G-to-C substitutions are compensated. Consistent with this conclusion, the A8455C/A8458C construct showed a further 2-fold (A8458C) or 6-fold (A8455C) reduction in binding affinity relative to those of the single A-to-C constructs and a 20-fold reduction relative to that of wild-type SL3ESS3, which corresponds to a free energy perturbation of 1.8 kcal/mol. On the basis of these results, we conclude that both GA and AG dinucleotide steps of the apical loop contribute to high-affinity (Kd < 100 nM) UP1 recognition, where the adenosine bases form primary contact points.
NMR Detection of Binding Epitopes
To glean further molecular insight into the UP1–SL3ESS3 binding mechanism, saturation transfer difference NMR (STD-NMR) was used to probe the SL3ESS3 binding epitopes. STD-NMR spectroscopy allows direct detection of protons located at a binding interface by selectively irradiating NMR signals of a receptor molecule while detecting the saturation transfer to its bound ligand via a difference spectrum. Here, NMR signals of UP1 were selectively irradiated, and site-specific binding epitopes were detected via saturation transfer to SL3ESS3 (Figure 7). The SL3ESS3 NMR signals most attenuated by the saturation transfer correspond to a subset of the aromatic and ribose protons. The sites of closest contact were more precisely determined by repeating the STD-NMR experiment with a selectively 2H(GC)-labeled SL3ESS3 construct. As shown in Figure 7B, the difference spectrum reveals the H2 protons of A8455 and A8458 are in the proximity of UP1. In addition, slightly weaker saturation transfer is observed to the H1′ positions of A8455 and A8458 along with the H2 and H5 positions of A8450, U8452, and U8457. Collectively, these results are consistent with the mutagenesis data presented above and further indicate UP1 interacts site-specifically with nucleobases of the SL3ESS3 apical loop.
Figure 7.

Direct detection of the ESS3b binding epitope revealed by STD-NMR. (A) 1D 1H reference spectrum of SL3ESS3 derived from the NL4-3 subtype (top) and 1D 1H STD spectrum revealing site-specific contacts of UP1 with aromatic and ribose protons of SL3ESS3 (bottom). (B) 1D 1H reference spectrum (top) and 1D 1H STD spectrum (bottom) of a 2H(GC)-selectively labeled SL3ESS3 construct. Selective deuteration allows simplified assignment of aromatic and ribose protons in close contact with UP1 (C) Surface representation of the NMR structure of SL3ESS3 (NL4-3) in which nucleobases most attenuated by the saturation transfer are colored red.
Having identified the UP1 binding epitopes on SL3ESS3, we monitored 1H–15N HSQC titrations of 15N-labeled UP1 with wild-type SL3ESS3. Using published chemical shift assignments and our own backbone NMR experiments, the majority of the free UP1 1H–15N correlation peaks were assigned for the construct used here. Because of the large size of the UP1–SL3ESS3 complex (∼35 kDa), de novo backbone chemical shift assignments of the complex have not been completed at this time. Nevertheless, comparing 1H–15N HSQC spectra of the apo and holo forms of UP1 reveals qualitative information about the binding surface and possible sites of conformational change. As shown in Figure 8, large chemical shift perturbations are observed upon addition of SL3ESS3 to 15N-labeled UP1 (final molar ratio of 1:1), with signals of the free form disappearing and reappearing at new positions. Stepwise titrations of SL3ESS3 into 15N-labeled UP1 reveal slow exchange on the NMR chemical shift time scale (Figure 8B), consistent with the nanomolar binding affinity determined by calorimetry (Table 1). The signals undergoing slow exchange map to a broad surface that encompasses RRM1, the inter-RRM linker, and RRM2 (Figure 8C). On RRM1, the amide backbone positions most affected by addition of SL3ESS3 map to β1 and β3, whereas the positions showing the largest perturbations on RRM2 map to β3 and β4. More delocalized perturbations are observed on α1, α2, and loop 1 from RRM1 as well as α2 and loop 2 from RRM2. The broad surface detected here is consistent with our previous results showing both RRM domains of UP1 are necessary to form a high-affinity complex with SL3ESS3.24 Given the clear evidence of binding specificity observed in both the chromatographic and calorimetric titrations along with the specific ESS3b loop epitopes detected by STD-NMR, a reasonable interpretation of these results is that binding of SL3ESS3 induces a conformational change in UP1; however, additional NMR experiments are needed to fully understand the binding profile reported by the 1H–15N HSQC titrations.
Figure 8.

Chemical shift mapping of the UP1–SL3ESS3 binding interface. (A) Superposition of 1H–15N HSQC spectra recorded on a 1:1 UP1–SL3ESS3 complex at 298 K, 140 mM K+, and pH 6.5. The spectrum of apo UP1 is colored black and that of holo UP1 red. Assignments correspond to those peaks that undergo large chemical shift perturbations in the holo form. The boxed region corresponds to those peaks selected to represent the titration. (B) Stepwise 1H–15N HSQC titrations of unlabeled SL3ESS3 into 15N-labeled UP1 reveals slow exchange on the NMR chemical shift time scale. (C) Mapping of the amide peaks showing the largest chemical shift perturbations onto the solution structure of UP1 (Protein Data Bank entry 2LYV).32
Discussion
Balanced expression of HIV-1 proteins requires inefficient splicing of the RNA genome. In addition to suboptimal 3′ acceptor sites, regulation is mediated through a dynamic competition between host hnRNP and SR proteins for viral splicing regulatory elements. Efforts to understand mechanisms of HIV-1 splicing have revealed some splicing regulatory elements are embedded within stable stem loop structures. These observations suggest a functional role for RNA structure as a regulatory factor; however, correlations between RNA structure and HIV-1 splicing have yet to be demonstrated. In this study, we used phylogenetics to show the HIV-1 ESS3 stem loop structure is highly conserved among different group M subtypes. The host hnRNP A1 protein binds ESS3 to repress splicing at the upstream acceptor site A7, and our results reveal the binding affinity of the UP1 domain for SL3ESS3 depends on the integrity of the stem loop structure as well as purine nucleobases within the ESS3b loop. Given these observations, we hypothesize the ESS3 stem loop structure functions as a “structural beacon” to direct site-specific and high-affinity hnRNP A1 assembly over otherwise degenerate YAG genomic sites.
Collectively, the results presented here demonstrate that maintenance of secondary structure within the ESS3 stem loop structure through compensatory substitutions has been more conserved throughout the evolution of HIV-1 than the primary nucleotide sequence. Although phylogenetic analysis of nucleotide sequences is useful for interpreting many aspects of evolutionary relationships, conservation and selective pressure are primarily inferred at the amino acid level, not at the individual nucleotide level. Conservation through maintenance of base pairs within RNA secondary structure implicates another level of selective pressure and demonstrates the power of combining phylogenetic analysis with methods for analyzing RNA structure.
Although the ESS3 stem loop structure is conserved, subtype-specific differences in folding patterns and predicted stabilities are seen (Figure S1 of the Supporting Information). Of note, the structural predictions shown in Figure S1 of the Supporting Information do not account for the possibility of noncanonical base pair interactions within the loop regions. Five of the 12 subtypes fold with secondary structures identical to the global consensus structure shown in Figure 2C, whereas the remaining subtypes show minor structural variations. ESS3 elements derived from subtypes B, CRF1, and CRF2 fold into stem loop structures that contain either one or two purine-rich bulges in the adjacent helical regions. The average predicted thermodynamic stability for these stem loop structures is −7.3 kcal/mol. By comparison, the ESS3 stem loop structures derived from subtypes A1, CRF6, and CRF42 fold with either 2 × 1 or 2 × 2 internal loops and with a lower average thermodynamic stability (−4.6 kcal/mol). The internal loops of these secondary structures are purine-rich and could potentially form noncanonical base pairs as observed in model RNA oligomers.35−37 The F1 subtype shows the most divergent ESS3 stem loop fold. Its helix is perfectly base paired, and the apical ESS3b loop is expanded to 11 nucleotides. The four additional nucleotides are all purines with an alternating GA/AG juxtaposition. The common feature among all subtypes is an exposed ESS3b loop with a global consensus 5′-GAUUAGU-3′ motif (Figure 2B). Low-frequency substitutions are observed at positions 1, 2, and 5, however. Interestingly, in the SL3ESS3 structure derived from NL4-3, the uracils in positions 3 and 4 form part of a U-turn-like motif and the uracil in the seventh position is flipped out. The conservation of uracils at positions 3, 4, and 7 may reflect pressure to maintain a particular ESS3b stereochemical geometry.
As reported here, the binding affinity of UP1 for ESS3 shows a striking dependence on the stem loop structure (Figure 3 and Table 1). There is an ∼150-fold decrease in binding affinity (nanomolar to micromolar) when the apical sequence is removed from the stem loop context. The large reduction in stability is accompanied by a marked decrease in binding enthalpy (ΔΔH° = 29.4 kcal/mol). The magnitude of ΔΔH° is comparable to binding enthalpies reported for some single RRM domains bound to short (<8 nucleotides) model RNA oligomers.38−41 This observation indicates the stability of the UP1–SL3ESS3 complex derives in part from intermolecular contacts that depend on RNA structure. Specific UP1 recognition of the ESS3b loop embedded within a conserved RNA structure may explain regulatory properties of hnRNP A1 on splice site A7, as well as other 3′ acceptor sites.42 The large binding enthalpy of the UP1–SL3ESS3 complex is consistent with enthalpies reported for other tandem RRM–RNA interactions where the RNA strands are long enough to traverse both RRM domains.43,44 We previously showed the tandem RRMs of UP1 are required for high-affinity SL3ESS3 recognition.24 In light of the data presented here, we predict the ESS3 stem loop structure facilitates contact with both RRM domains of UP1, an interaction that obviously does not take place with the isolated ESS3b loop sequence.
The single-cytosine substitutions provide further insight into the role of conserved ESS3b nucleobases. The thermodynamic signatures reveal UP1 has a preference for purines over pyrimidines, where A8458 and G8459 likely form sites of primary contact. The further reductions in binding affinity observed for G8454C/G8459C and A8455C/A8458C suggest partial synergy between pairs of purine nucleobases, although other effects such as induced changes in the ESS3b loop geometry cannot be ruled out. In general, these results are consistent with fluorescence competition experiments that showed UP1 binds purines with higher affinity than pyrimidines.21 The data are also in agreement with available UP1–DNA crystal structures where stacking interactions between signature aromatic residues (of the RNP1 and RNP2 motifs) and AG dinucleotides are observed.23 In line with the available UP1–DNA structures and consistent with our mutational studies, the STD-NMR spectra reveal UP1 makes close contact with A8455 and A8459 (Figure 7B). Furthermore, binding of SL3ESS3 to UP1 induces changes in the 1H–15N correlation peaks of both RRM domains and the inter-RRM linker. Collectively, the data presented here describe a UP1 binding mechanism that is likely very different from existing models used to interpret the alternative splicing activity of hnRNP A1.23
In summary, we have shown the ESS3 stem loop structure is phylogenetically conserved across different HIV-1 group M subtypes. Our results clearly show the structure and ESS3b nucleobases are key UP1 binding determinants. On the basis of the collective work presented here, we propose HIV-1 uses RNA structural beacons to direct recruitment of hnRNP A1 to specific genomic splice sites. It will be of interest to see if the ESS3 structural variants observed for different HIV-1 subtypes correlate with changes in UP1 binding affinities and levels of ssA7 usage.
Acknowledgments
We thank Dr. Xian Mao of the Cleveland Center for Membrane and Structural Biology for assistance in optimizing the STD-NMR pulse sequence and parameters.
Glossary
Abbreviations
- hnRNP A1
heterogeneous nuclear ribonucleoprotein A1
- ESS3
exon splicing silencer 3
- ITC
isothermal titration calorimetry
- STD
saturation transfer difference
- HSQC
heteronuclear single-quantum coherence.
Supporting Information Available
Predicted secondary structures for subtype-specific ESS3 stem loop structures (Figure S1), one-dimensional 1H NMR spectra for each cytosine-substituted construct (Figure S2), and a complete list of group M subtypes aligned for phylogenetic comparisons (Table S1). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by National Institutes of Health Grants R01 GM101979 and P50 GM103297 (to B.S.T. and M.S.).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Stoltzfus C. M. (2009) Chapter 1. Regulation of HIV-1 alternative RNA splicing and its role in virus replication. Adv. Virus Res. 74, 1–40. [DOI] [PubMed] [Google Scholar]
- Stoltzfus C. M.; Madsen J. M. (2006) Role of viral splicing elements and cellular RNA binding proteins in regulation of HIV-1 alternative RNA splicing. Curr. HIV Res. 4, 43–55. [DOI] [PubMed] [Google Scholar]
- Purcell D. F.; Martin M. A. (1993) Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J. Virol. 67, 6365–6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocwieja K. E.; Sherrill-Mix S.; Mukherjee R.; Custers-Allen R.; David P.; Brown M.; Wang S.; Link D. R.; Olson J.; Travers K.; Schadt E.; Bushman F. D. (2012) Dynamic regulation of HIV-1 mRNA populations analyzed by single-molecule enrichment and long-read sequencing. Nucleic Acids Res. 40, 10345–10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard C. K.; Tange T. O.; Kjems J. (2002) hnRNP A1 controls HIV-1 mRNA splicing through cooperative binding to intron and exon splicing silencers in the context of a conserved secondary structure. RNA 8, 1401–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tange T. O.; Damgaard C. K.; Guth S.; Valcarcel J.; Kjems J. (2001) The hnRNP A1 protein regulates HIV-1 tat splicing via a novel intron silencer element. EMBO J. 20, 5748–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tange T. O.; Kjems J. (2001) SF2/ASF binds to a splicing enhancer in the third HIV-1 tat exon and stimulates U2AF binding independently of the RS domain. J. Mol. Biol. 312, 649–662. [DOI] [PubMed] [Google Scholar]
- Marchand V.; Santerre M.; Aigueperse C.; Fouillen L.; Saliou J. M.; Van Dorsselaer A.; Sanglier-Cianferani S.; Branlant C.; Motorin Y. (2011) Identification of protein partners of the human immunodeficiency virus 1 tat/rev exon 3 leads to the discovery of a new HIV-1 splicing regulator, protein hnRNP K. RNA Biol. 8, 325–342. [DOI] [PubMed] [Google Scholar]
- Marchand V.; Mereau A.; Jacquenet S.; Thomas D.; Mougin A.; Gattoni R.; Stevenin J.; Branlant C. (2002) A Janus splicing regulatory element modulates HIV-1 tat and rev mRNA production by coordination of hnRNP A1 cooperative binding. J. Mol. Biol. 323, 629–652. [DOI] [PubMed] [Google Scholar]
- Asai K.; Platt C.; Cochrane A. (2003) Control of HIV-1 env RNA splicing and transport: Investigating the role of hnRNP A1 in exon splicing silencer (ESS3a) function. Virology 314, 229–242. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Mayeda A.; Krainer A. R. (2001) Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol. Cell 8, 1351–1361. [DOI] [PubMed] [Google Scholar]
- Si Z. H.; Rauch D.; Stoltzfus C. M. (1998) The exon splicing silencer in human immunodeficiency virus type 1 Tat exon 3 is bipartite and acts early in spliceosome assembly. Mol. Cell. Biol. 18, 5404–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okunola H. L.; Krainer A. R. (2009) Cooperative-binding and splicing-repressive properties of hnRNP A1. Mol. Cell. Biol. 29, 5620–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Smith R. (2009) Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell. Mol. Life Sci. 66, 1239–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeda A.; Munroe S. H.; Caceres J. F.; Krainer A. R. (1994) Function of conserved domains of hnRNP A1 and other hnRNP A/B proteins. EMBO J. 13, 5483–5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamoo Y.; Abdul-Manan N.; Patten A. M.; Crawford J. K.; Pellegrini M. C.; Williams K. R. (1994) Both RNA-binding domains in heterogenous nuclear ribonucleoprotein A1 contribute toward single-stranded-RNA binding. Biochemistry 33, 8272–8281. [DOI] [PubMed] [Google Scholar]
- Abdul-Manan N.; O’Malley S. M.; Williams K. R. (1996) Origins of binding specificity of the A1 heterogeneous nuclear ribonucleoprotein. Biochemistry 35, 3545–3554. [DOI] [PubMed] [Google Scholar]
- Cartegni L.; Maconi M.; Morandi E.; Cobianchi F.; Riva S.; Biamonti G. (1996) hnRNP A1 selectively interacts through its Gly-rich domain with different RNA-binding proteins. J. Mol. Biol. 259, 337–348. [DOI] [PubMed] [Google Scholar]
- Fisette J. F.; Toutant J.; Dugre-Brisson S.; Desgroseillers L.; Chabot B. (2010) hnRNP A1 and hnRNP H can collaborate to modulate 5′ splice site selection. RNA 16, 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler S. G.; Merrill B. M.; Roberts W. J.; Keating K. M.; Lisbin M. J.; Barnett S. F.; Wilson S. H.; Williams K. R. (1991) Interactions of the A1 heterogeneous nuclear ribonucleoprotein and its proteolytic derivative, UP1, with RNA and DNA: Evidence for multiple RNA binding domains and salt-dependent binding mode transitions. Biochemistry 30, 2968–2976. [DOI] [PubMed] [Google Scholar]
- Abdul-Manan N.; Williams K. R. (1996) hnRNP A1 binds promiscuously to oligoribonucleotides: Utilization of random and homo-oligonucleotides to discriminate sequence from base-specific binding. Nucleic Acids Res. 24, 4063–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd C. G.; Dreyfuss G. (1994) RNA binding specificity of hnRNP A1: Significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J. 13, 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J.; Hayashi M. K.; Zhang Y.; Manche L.; Krainer A. R.; Xu R. M. (1999) Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev. 13, 1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levengood J. D.; Rollins C.; Mishler C. H.; Johnson C. A.; Miner G.; Rajan P.; Znosko B. M.; Tolbert B. S. (2012) Solution Structure of the HIV-1 Exon Splicing Silencer 3. J. Mol. Biol. 15, 680–698. [DOI] [PubMed] [Google Scholar]
- Thompson J. D.; Higgins D. G.; Gibson T. J. (1994) CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H. A.; Strimmer K.; Vingron M.; von Haeseler A. (2002) TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18, 502–504. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Nei M.; Dudley J.; Tamura K. (2008) MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Briefings Bioinf. 9, 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. [DOI] [PubMed] [Google Scholar]
- Prosperi M. C., Ciccozzi M., Fanti I., Saladini F., Pecorari M., Borghi V., Di Giambenedetto S., Bruzzone B., Capetti A., Vivarelli A., Rusconi S., Re M. C., Gismondo M. R., Sighinolfi L., Gray R. R., Salemi M., Zazzi M., De Luca A., and . ARCA collaborative group (2011) A novel methodology for large-scale phylogeny partition. Nat. Commun. 2, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan J. F.; Groebe D. R.; Witherell G. W.; Uhlenbeck O. C. (1987) Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 15, 8783–9798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan J. F.; Uhlenbeck O. C. (1989) Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 180, 51–62. [DOI] [PubMed] [Google Scholar]
- Barraud P.; Allain F. H. (2013) Solution structure of the two RNA recognition motifs of hnRNP A1 using segmental isotope labeling: How the relative orientation between RRMs influences the nucleic acid binding topology. J. Biomol. NMR 55, 119–138. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Johnson B. A.; Blevins R. A. (1994) NMRview: A Computer Program for the Visualization and Analysis of NMR Data. J. Biomol. NMR 4, 603–614. [DOI] [PubMed] [Google Scholar]
- Tolbert B. S.; Kennedy S. D.; Schroeder S. J.; Krugh T. R.; Turner D. H. (2007) NMR structures of (rGCUGAGGCU)2 and (rGCGGAUGCU)2: Probing the structural features that shape the thermodynamic stability of GA pairs. Biochemistry 46, 1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond N. B.; Tolbert B. S.; Kierzek R.; Turner D. H.; Kennedy S. D. (2010) RNA internal loops with tandem AG pairs: The structure of the 5′GAGU/3′UGAG loop can be dramatically different from others, including 5′AAGU/3′UGAA. Biochemistry 49, 5817–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.; Kennedy S. D.; Turner D. H. (2009) A CA(+) pair adjacent to a sheared GA or AA pair stabilizes size-symmetric RNA internal loops. Biochemistry 48, 5738–5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan M. M.; Goult B. T.; Clayton J. C.; Hautbergue G. M.; Wilson S. A.; Lian L. Y. (2012) The structure and selectivity of the SR protein SRSF2 RRM domain with RNA. Nucleic Acids Res. 40, 3232–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clery A.; Jayne S.; Benderska N.; Dominguez C.; Stamm S.; Allain F. H. (2011) Molecular basis of purine-rich RNA recognition by the human SR-like protein Tra2-beta1. Nat. Struct. Mol. Biol. 18, 443–450. [DOI] [PubMed] [Google Scholar]
- Clery A.; Sinha R.; Anczukow O.; Corrionero A.; Moursy A.; Daubner G. M.; Valcarcel J.; Krainer A. R.; Allain F. H. (2013) Isolated pseudo-RNA-recognition motifs of SR proteins can regulate splicing using a noncanonical mode of RNA recognition. Proc. Natl. Acad. Sci. U.S.A. 110, E2802–E2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubner G. M.; Clery A.; Jayne S.; Stevenin J.; Allain F. H. (2012) A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 31, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliou J. M.; Bourgeois C. F.; Ayadi-Ben Mena L.; Ropers D.; Jacquenet S.; Marchand V.; Stevenin J.; Branlant C. (2009) Role of RNA structure and protein factors in the control of HIV-1 splicing. Front. Biosci. 14, 2714–2729. [DOI] [PubMed] [Google Scholar]
- McLaughlin K. J.; Jenkins J. L.; Kielkopf C. L. (2011) Large favorable enthalpy changes drive specific RNA recognition by RNA recognition motif proteins. Biochemistry 50, 1429–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukavsky P. J.; Daujotyte D.; Tollervey J. R.; Ule J.; Stuani C.; Buratti E.; Baralle F. E.; Damberger F. F.; Allain F. H. (2013) Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20, 1443–1449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.