

Abstract
Background
Glucocorticoids play a critical role in normative regulation of fetal brain development. Exposure to excessive levels may have detrimental consequences and disrupt maturational processes. This may especially be true when synthetic glucocorticoids are administered during the fetal period, as they are to women in preterm labor. The present study investigated the consequences for brain development and affective problems of fetal exposure to synthetic glucocorticoids.
Methods
Brain development and affective problems were evaluated in fifty-four children (56% female), ages 6 to 10, who were full term at birth. Children were recruited into two groups: those with and without fetal exposure to synthetic glucocorticoids. Structural magnetic resonance imaging (MRI) scans were acquired and cortical thickness was determined. Child affective problems were assessed using the Child Behavior Checklist.
Results
Children in the fetal glucocorticoid exposure group showed significant and bilateral cortical thinning. The largest group differences were in the rostral anterior cingulate cortex (rACC). Over 30% of the rACC was thinner among children with fetal glucocorticoid exposure. Further, children with more affective problems had a thinner left rACC.
Conclusions
Fetal exposure to synthetic glucocorticoids has neurological consequences that persist for at least 6 to 10 years. Children with fetal glucocorticoid exposure had a thinner cortex primarily in the rACC. Our data indicating that the rACC is associated with affective problems in conjunction with evidence that this region is involved in affective disorders raises the possibility that glucocorticoid associated neurological changes increase vulnerability to mental health problems.
Keywords: Glucocorticoid, stress, prenatal, MRI, development, fetal programming
The origins of mental illness often begin early in life (1-4) and it is thought that developmental alterations in brain neuroanatomy underlie vulnerability to psychopathology. Although existing research primarily has focused on postnatal stress exposure (5-9), it is becoming evident that exposure to stress and stress hormones during the prenatal period exert long lasting consequences on risk for mental health problems including anxiety and depression (10-14). Because of the rapid neurological advances during the prenatal period including neurogenesis, migration, cell neuronal differentiation, dendritic arborization, axonal elongation, synapse formation, collateralization, pruning and myelination (15-18) the fetus is susceptible to environmental influences.
The purpose of the present study is to investigate the programming influence of prenatal exposure to excess glucocorticoids on the developing central nervous system during preadolescence. For a number of reasons, glucocorticoids are a candidate for fetal programming of brain development. Glucocorticoids exert a wider array of key metabolic, endocrine and immune effects on most cells than any other biological ligand (19, 20). Further, glucocorticoids pass through the blood brain barrier, target receptors throughout the central nervous system, and play a central role in normal brain development (21, 22). Although glucocorticoids are necessary for normative fetal brain development, exposure to excessive levels may disrupt basic neurodevelopmental processes and excessive exposures are associated with increased risk for affective problems (12, 23). The risk may be even greater when glucocorticoids are administered exogenously, as they are to women in preterm labor, thus exposing the fetus to supraphysiological levels.
Synthetic glucocorticoids, such as betamethasone, are widely used during pregnancy to prevent respiratory distress syndrome in preterm infants (24, 25). The synthetic glucocorticoids used for fetal lung maturation cross the placenta more readily than endogenous maternal glucocorticoids (i.e., cortisol) because they are not metabolized and inactivated by placental enzymes and act directly on the developing fetus (26, 27). The pulmonary benefits are undisputed. It is, however, plausible that exposure to excess glucocorticoids has consequences for the developing fetal brain (28, 29). Experimental animal research demonstrates that exposure to excess glucocorticoids reduces brain weight, decreases cell proliferation and dendritic branching, disrupts myelination and alters neural activity across several species including rodents, sheep and non-human primates (30-35). Studies in human neonates indicate that antenatal glucocorticoid treatment is associated with decreased cortical volume and complexity of cortical folding (36, 37). Adult human and animal research indicates that limbic and prefrontal regions are particularly affected by excess glucocorticoids due to the abundance of glucocorticoid receptors in these brain regions (9, 38-41).
The specific effects of prenatal synthetic glucocorticoids treatment may be observed best among healthy children born at term who are not at risk for the neurological impairments associated with preterm delivery (42, 43). The purpose of the present investigation was to 1) determine the long-term influence of fetal exposure to synthetic glucocorticoids and 2) determine whether cortical changes are associated with affective problems among 6 to 10 year old preadolescent children.
Methods
Participants
Participants included fifty-four children, ages 6 to 10, who were full term at birth and their mothers. Three additional children were recruited into the MRI protocol, but not included in this manuscript because adequate imaging data could not be collected due to child refusal or motion artifact. Children were born at either the University of California Irvine Medical Center or Long Beach Memorial Medical Center/Miller Children’s Hospital, a community hospital affiliate of the university. The Institutional Review Boards for protection of human subjects at both institutions approved the study protocol. Written and informed consent from the mother and informed assent from the child were obtained prior to study enrollment.
Inclusion criteria were birth at term (gestational age at birth greater than 37 weeks based on ACOG dating criteria) (44), appropriate weight for gestational age at birth and singleton status. Exclusion criteria were chromosomal or other congenital anomalies (e.g., trisomy 21), postnatal steroid administration and major neonatal illness (e.g., sepsis), maternal preeclampsia or HELLP syndrome, maternal drug use, as well as maternal disorders during pregnancy requiring corticosteroid treatment or thyroid medication. Subjects that met inclusion and exclusion criteria were recruited into two groups: those with and without antenatal exposure to synthetic glucocorticoids. The glucocorticoid group included 18 children (10 female and 8 male) whose mothers were at risk for preterm delivery and received the glucocorticoid betamethasone for fetal lung maturation. Betamethasone is administered in 2 doses (12 mg intramuscularly, 24 hours apart). The primary indication for prenatal glucocorticoid administration was preterm labor (72%). Preterm labor was diagnosed by the attending obstetrician based on the following factors: cervical change over time, bloody show (spotting that occurs as the cervix changes shape prior to or early in labor), cervical effacement and/or dilation, and rupture of membranes. Other associated factors included placenta previa and prolonged premature rupture of membranes. In this cohort the first dose of betamethasone was given between 24 and 34 weeks’ GA [mean GA at administration = 29.3 (3.2) weeks] and was between 29 and 107 days prior to delivery [mean days = 65.1 (21.5)]. Although, betamethasone is administered because of risk for preterm delivery, 25-30% of women who receive glucocorticoid treatment deliver full term (42). Notably, all children in the present investigation were full term at birth. To create a more stable characterization of child brain development among unexposed infants, two children without antenatal glucocorticoids exposure were matched by gestational age at birth and sex to each subject in the prenatal glucocorticoid treated group. Thus, the comparison group consisted of 36 children born at term without prenatal glucocorticoid treatment (20 female, 16 male).
None of the participants in the glucocorticoids treatment group or the comparison group had evidence of intraventricular hemorrhage (determined by ultrasound), periventricular leukomalacia, and/or low-pressure ventriculomegaly in the newborn period and all participants had normal neurologic findings (determined by neuroradiological review of MRI scans), including normal ventricle size, at assessment. Further, at 6 to 10 years of age, no emotional or physical problems were reported in a structured interview using the MacArthur Health and Behavior Questionnaire (45).
Background Information
Sociodemographic characteristics were determined at the time of study entry by standardized maternal interview. Maternal intellectual performance was determined by the Perceptual Reasoning Scale of the Wechsler Adult Intelligence Scale (46). Maternal depression was evaluated with the Beck Depression Inventory (47). Neonatal and maternal medical characteristics including birth outcome data were determined by medical record abstraction.
Child Behavioral Problems
Child affective problems were measured using the Achenbach System of Empirically Based Assessment (ASEBA), which offers a comprehensive approach to assessing adaptive and maladaptive functioning (48). The ASEBA is a reliable and valid measure that is widely used in research and clinical practice with children. The parent report form, the Child Behavior Checklist (CBCL), was administered to mothers by a trained interviewer who was directly supervised by a clinical psychologist. The CBCL contains 113 items representing a broad scope of behaviors. It has high test-retest stability and good internal consistency. The Affective Problems subscale was used because it is consistent with the Diagnostic and Statistical Manual of Mental Disorders-IV evaluation affective problems and is a reliable screening instrument (48). The affective problems scale consists of six statements. Responses were made on a 3- point Likert scale ranging from 0 (not true) to 2 (very true). The raw sum scores were transferred to T-scores based on the sex-specific reference tables (48).
MRI Acquisition
Structural MRI scans were acquired on a 3-T Philips Achieva system. To minimize head motion, padding was placed around the head. Ear protection was given to all children. To further increase compliance and reduce motion, children were fitted with headphones and allowed to watch a movie of their choice while in the scanner. Following the scanner calibration and pilot scans, a high resolution T1 anatomical scan was acquired in the sagittal plane with 1mm3 isotropic voxel dimensions. An Inversion-Recovery Spoiled Gradient Recalled Acquisition (IR-SPGR) sequence with the following parameters were applied: Repetition rate (TR)= 11ms, Echo Time (TE)= 3.3ms, Inversion Time (TI)= 1100ms, Turbo Field Echo factor (TFE)= 192, Number of slices: 150, no SENSE acceleration, Flip angle=18°. Acquisition time for this protocol was seven minutes.
Image processing
Cortical surface reconstruction was performed with the FreeSurfer image analysis software suite (http://surfer.nmr.mgh.harvard.edu/). Streamlined image processing procedures are initiated by applying intensity normalization prior to segmentation to minimize errors in identifying the boundaries (49). This is followed by removal of non-brain tissues (50) and then, the images are transformed into the Talairach space for the segmentation of subcortical white matter and subcortical gray matter (51, 52). Pial and white matter surfaces were located by finding the highest intensity gradient, which defines the transition from one tissue class to the other (53, 54). Once the preprocessing steps were completed, surface inflation was applied to each individual brain (55) and the inflated brains were registered to a spherical atlas. This procedure utilized individual cortical folding patterns to achieve accurate registration of cortical geometry across subjects (56). Cortical thickness was calculated as the closest distance from the gray matter / white matter surface to the pial surface at each vertex on the tessellated surface (54). Procedures for the measurement of cortical thickness have been validated with histological analysis (57) and manual measurements (58, 59). The cortical surface images generated by the FreeSurfer software were visually inspected for errors in segmentation and corrections were made as needed.
After FDR corrections, the number of vertices was determined in each significant area in each region of the brain. The number of significant vertices for each area was added and divided by the total number of vertices (X 100) in that area to provide the percentage of the vertices that were significantly thinner in the glucocorticoid group as compared to the comparison group. The same procedure was computed for the number of significant vertices in each lobe. For hemispheric and whole brain percentages, the procedure was the same except the total number of subcortical vertices was subtracted from the total.
Data Analysis
Preliminary analyses were performed using chi square and t-tests to determine whether sociodemographic (i.e., race/ethnicity, maternal marital status, maternal education, and household income), maternal (i.e., intelligence, depression), neonatal (i.e., birth weight, birth order) or child (i.e., age at assessment, affective problems) variables differed by group.
Differences between groups in cortical thickness were analyzed at each node on the cortical surface using the Monte Carlo method. This technique is based on testing statistical significance of cluster-wise differences between the two groups for correct labeling of the data against random relabeling (permutations) of the same data. 10,000 permutations were tested for this study. Spatially normalized cortical thickness maps of each subject were entered into a regression model. Associations were considered to be statistically significant at p<0.05 after False Discovery Rate (FDR) correction for multiple comparisons as recommended by Genovese and colleagues (60).
Primary regions in which the prenatal glucocorticoid treatment group and the comparison group significantly differed were further evaluated as regions of interest to determine associations with child behavior (affective problems) and timing of glucocorticoid exposure. For each region of interest the average thickness in mm was extracted for each subject from the statistical cortical parcellation file was created by FreeSurfer during the segmentation process. This file contains the average thickness in mm of the distance between the white matter and the pial surface. Parcellation is based on the Desiken/Killiany Atlas (61).
Results
Demographic and Clinical Data
Tables 1 and 2 display descriptive information for the study sample. Groups were matched for gestational age at birth and sex. The children in the prenatal glucocorticoid treatment and comparison groups did not significantly differ in birth weight, Apgar scores, race, age at assessment, child affective problems or in total grey matter volume. Maternal education, marital status, household income, intelligence and depression did not significantly differ between the treatment and comparison groups.
Table 1.
Descriptive information for children in the study sample
| Glucocorticoid Treatment Group (N = 18) | Comparison Group (N = 36) | |
|---|---|---|
| Gestational age at birth (weeks) | 38.5 (1.1) | 39.0 (1.0) |
| Range: 37.1 – 41.3 | Range: 37.0 – 41.3 | |
| Birth weight (grams) | 3410 (392.8) | 3358 (447.3) |
| Range: 2896 - 4561 | Range: 2280 - 4490 | |
| Sex (% female) | 56 | 56 |
| Apgar score at 5-minutes | 8.8 (0.5) | 8.9 (0.5) |
| Range: 7 - 9 | Range: 7 - 10 | |
| Race/Ethnicity (%) | ||
| Hispanic | 39 | 53 |
| Non-Hispanic White | 28 | 28 |
| African American | 6 | 6 |
| Multi-ethnic | 28 | 14 |
| Child Age at MRI (years) | 8.5 (1.3) | 8.1 (1.1) |
| Range: 6.3 – 10.4 | Range: 6.1 – 10.5 | |
| GA at first dose (weeks) | 29.3 (3.2) | N/A |
| Range: 23.7 – 34.1 | ||
| Days between first dose and delivery | 65.1 (21.5) | N/A |
| Range: 29 - 107 | ||
| Received tocolytics prenatally (%) | 94% | 0% |
| Birth order (% firstborn) | 39 | 19 |
| Total grey matter volume (cc) | 668609 | 660740 |
| Affective Problems T-score | 52.4 (3.2) | 53.9 (4.4) |
Table 2.
Descriptive information for mothers in the study sample
| Glucocorticoid Treatment Group (N = 18) | Comparison Group (N = 36) | |
|---|---|---|
| Maternal Age at Assessment (years) | 36.9 (6.2) | 36.3 (6.4) |
| Range: 26.1 – 48.8 | Range: 24.4 – 48.8 | |
| Married or Cohabitating (%) | 78 | 83 |
| Education (%) | ||
| Primary, elementary, or middle school | 0 | 19 |
| High school or equivalent | 17 | 39 |
| Associates or vocational | 28 | 14 |
| Bachelors degree | 28 | 19 |
| Graduate degree | 11 | 8 |
| Annual Household Income (%) | ||
| $0 - $30,000 | 6 | 31 |
| $30,001 - $60,000 | 39 | 17 |
| $60,001 - $100,000 | 39 | 20 |
| Over $100,000 | 17 | 31 |
| Beck Depression Inventory Score | 7.7 (6.0) | 8.1 (8.7) |
| Range: 0-19 | Range 0-32 | |
| WAIS: POI Index Score | 97.8 (13.7) | 95.4 (16.0) |
| Range: 65 - 125 | Range: 67 - 128 |
Study groups did not significantly differ on any of these measures. All p’s >0.2.
Is prenatal glucocorticoid treatment associated with cortical thickness in preadolescent children?
Widespread and predominantly bilateral differences in cortical thickness were observed between groups. Four percent of the cortex was thinner among children who were exposed to synthetic glucocorticoids during the fetal period. Regionally specific consequences of fetal exposure to synthetic glucocorticoids were observed (See Table 3). Regions where group differences achieved statistical significance (p<0.05) after FDR correction are shown in Figure 1 (red overlays).
Table 3.
Percentage of whole brain and major areas that are significantly thinner after correction for multiple comparisons in children who were exposed as fetuses to treatment with synthetic glucocorticoids.
| Total | Left Hemisphere | Right Hemisphere | |
|---|---|---|---|
| Whole Brain | 4 | 4 | 4 |
|
| |||
| Cingulate/Limbic | 13 | 13 | 13 |
| Rostral Anterior Cingulate | 33 | 28 | 37 |
| Caudal Anterior Cingulate | 9 | 10 | 8 |
| Posterior Cingulate | 3 | 6 | 0 |
| Isthmus Cingulate | 20 | 15 | 26 |
|
| |||
| Frontal Cortex | 4 | 3 | 5 |
| Superior Frontal | 3 | 2 | 3 |
| Rostral Middle Frontal | 3 | 1 | 4 |
| Lateral Orbital Frontal | 3 | 2 | 4 |
| Medial Orbital Frontal | 8 | 0 | 15 |
| Caudal Middle Frontal | 7 | 1 | 13 |
| Precentral Gyrus | 8 | 5 | 10 |
| Insula | 3 | 4 | 1 |
|
| |||
| Parietal Cortex | 5 | 7 | 3 |
| Supramarginal | 4 | 8 | 0 |
| Superior Parietal | 8 | 12 | 5 |
|
| |||
| Temporal Cortex | 3 | 1 | 4 |
| Transverse Temporal | 5 | 11 | 0 |
| Inferior Temporal | 10 | 0 | 21 |
|
| |||
| Occipital Cortex | >1 | 1 | 0 |
Figure 1.
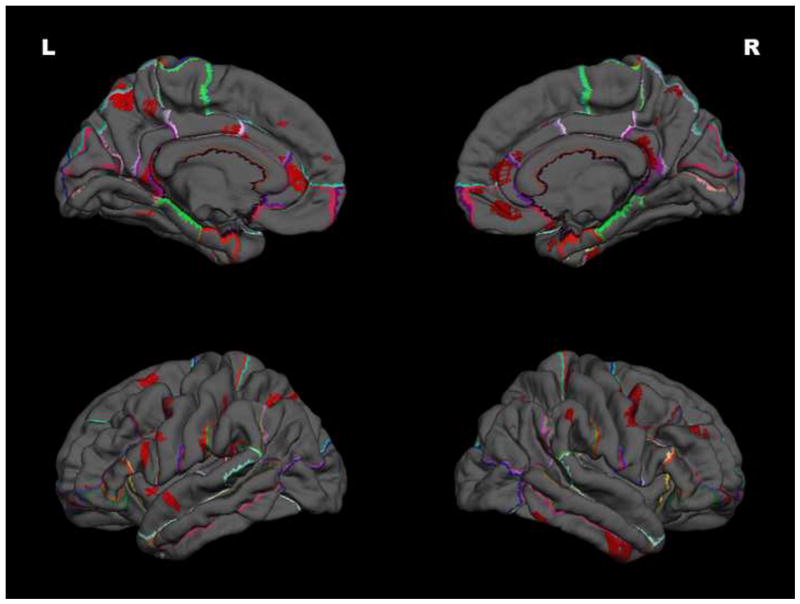
Children exposed as fetuses to glucocorticoid treatment have a significantly thinner cortex, primarily in the anterior cingulate.
Note: Red overlays indicate areas where the cortex is significantly thinner after correction for multiple comparisons in the children with fetal exposure to glucocorticoid treatment in reference to a comparison group.
The most prominent group differences were seen bilaterally in areas of the cingulate cortex including the rostral and caudal anterior cingulate, the isthmus cingulate and the left posterior cingulate. The cortex was additionally significantly thinner in frontal regions including the superior frontal cortex, the lateral orbital frontal cortex and the precentral gyrus as well as the right medial orbital frontal cortex and rostral and caudal middle frontal cortex (dorsolateral prefrontal cortex), and the left pars triangularis and parsopercularis. Areas of significant bilateral cortical thinness among the glucocorticoid-exposed group were observed in the superior parietal cortex. Unilateral regions of thinning were observed in the left insula, left supramarginal gyrus, and the left transverse temporal cortex as well as the right inferior temporal cortex. As shown in Table 3 and Figure 2, by far the area most strongly associated with prenatal glucocorticoid treatment was the rostral anterior cingulate cortex (rACC). Over 30% of the rACC was thinner in the GC group. Further, the magnitude of the effect was substantial; the rACC was 8% and 9% thinner for the left and right hemisphere respectively. We investigated this region of interest to determine whether thickness was associated with affective problems or timing of exposure. Although groups did not differ in affective problems it is plausible that thickness of the rACC is associated with affective problems suggesting that reduced rACC thickness is a prodromal risk factor for affective problems.
Figure 2.
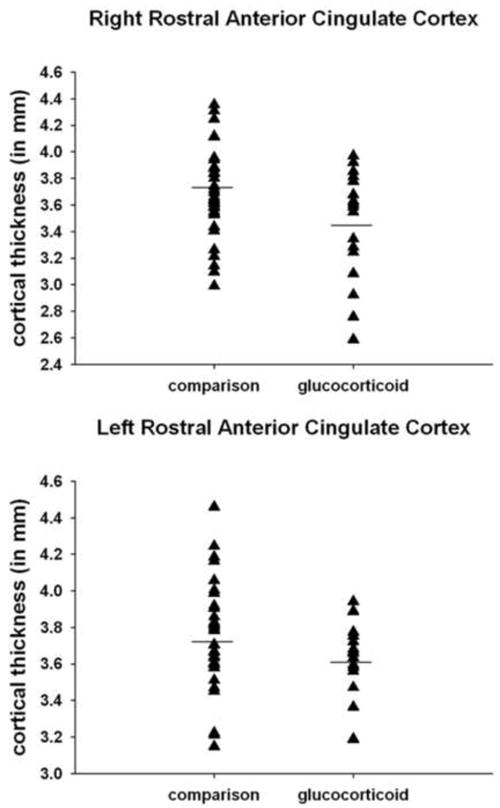
Prenatal glucocorticoid treatment is associated with a thinner rostral anterior cingulate cortex.
Is thickness of the rACC associated with affective problems?
Hierarchical linear regression, covarying group status, indicates that a thinner left rACC cortex is significantly associated with a higher level of child affective problems; ΔR2 = .12, Beta = -.36, t = -2.7, p < .01. As shown in Figure 3, these data suggest that thinning in this region is associated with risk for affective problems. The association was not significant for the right rACC; ΔR2 = .003, Beta = .06, t = .4, p =0.7. Evaluation of the association between rACC and affective problems within each group indicated a significant association within the comparison group (r(36) = -.45, p< .01), but not for the glucocorticoid group (r(18) = -.1, ns).
Figure 3.
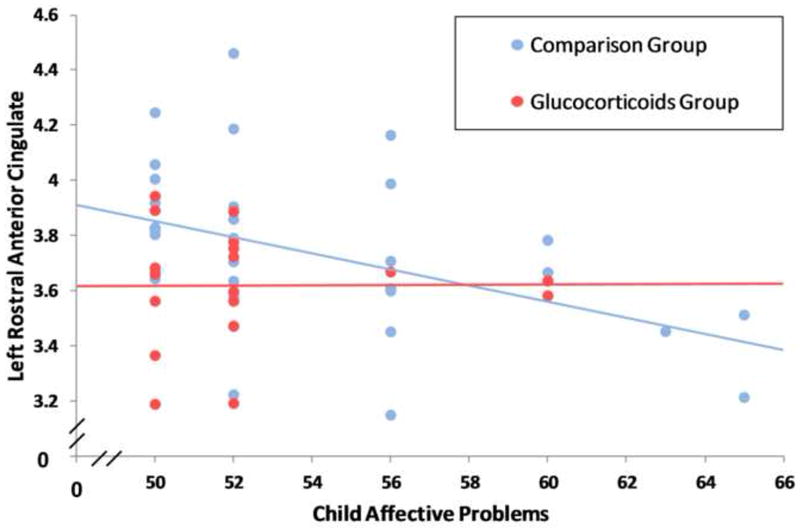
A thinner left rostral anterior cingulate cortex is associated with a higher level of affective problems.
Is timing of exposure associated with cortical thickness?
Within the range of glucocorticoid administration (24 to 34 gestational weeks), timing of administration was not significantly associated with thickness of the rACC (p’s >0.1).
Discussion
To the best of our knowledge, these data provide the first evidence that prenatal treatment with glucocorticoids alters the trajectory of fetal brain development with neurological consequences that persist into the preadolescent period. Children who were exposed as fetuses to glucocorticoid treatment have significantly thinner cortices, primarily in the rACC, a region that plays a critical role in stress and emotional regulation (62, 63). Not only is over 30% of the rACC thinner among children with fetal exposure to glucocorticoid treatment, but also the magnitude of the effect is substantial. The rACC is 8-9% thinner among glucocorticoid treated children. The possible clinical significance of this association is underscored by the observation that there is a 10-14% reduction in the rACC among children with depressive symptomotology (64). Consistent with the possibility that these neurological changes may indicate prodromal risk for mental health problems, we show that a thinner rACC predicts increased child affective problems. Notably the significant associations are observed among children who are healthy and born at term, and these findings cannot be explained by sociodemographic (income or education) or maternal (depression, IQ) factors.
Recent studies provide evidence that fetal exposure to elevated levels of glucocorticoids has consequences for stress and emotional regulation including HPA axis dysregulation (65-67), fearful temperament (68-70), anxiety (12) and affective problems (23) during infancy and childhood. The present study indicates fetal glucocorticoid exposure alters the development of the ACC, and represents one pathway by which fetal stress exposures may contribute to affective problems. Our finding that the ACC is particularly vulnerable is consistent with several lines of evidence. First, the ACC is rich in glucocorticoid receptors and susceptible to damage resulting from excess exposure (71-73). Second, postnatal exposure to stress or trauma is associated with a decrease in ACC volume (5, 74, 75). Third, birth weight is associated with thickness of the rACC indicating that development of this region is affected by fetal experiences (76).
Because of the massive developmental changes occurring during gestation, the fetal brain is vulnerable to exposures including elevated glucocorticoids (77), stress (78), infection (79, 80), poor nutrition (81) and environmental toxins (82). Although glucocorticoids play an organizational role in normative fetal maturational processes (21, 22, 83), exposure to excessive levels of glucocorticoids during sensitive periods has neurotoxic effects and may lead to dysfunctional developmental trajectories (23). Rodent studies have documented that prenatal glucocorticoid exposure affects postnatal brain cell proliferation and levels of glucocorticoid receptor mRNA (84-86). In sheep, antenatal glucocorticoid treatment leads to acute changes in neuronal activity (87, 88), decreased cerebral blood flow (89), disrupted myelination of white matter tracts (90) and decreased brain weight persisting through adulthood (91), with repeated doses having a more profound effect (90, 92). Primate studies provide further evidence for a persisting influence of prenatal glucocorticoid treatment on the brain (93). A single course of betamethasone decreased expression of neuronal cytoskeletal proteins and of the presynaptic marker synaptophysin, proteins that are necessary for brain development and neuronal functioning (94). The few human studies of brain development have evaluated the fetus or neonate and have shown that fetal exposure to glucocorticoid administration is associated with reduced neonatal cerebral cortical gray matter volume among preterm infants (36), decreased complexity of cortical folding and brain surface area among late preterm infants (37) as well as acute changes in fetal cortical functioning between 29 and 34 gestational weeks (95).
Our current findings extend the existing literature by evaluating children born healthy and full term and exposed to a single course of betamethasone and show that fetal glucocorticoid treatment is associated with cortical thinning that persists until at least 6 to 10 years of age. We have focused on cortical thickness because it has been suggested that cortical thickness provides an index of the integrity of cortical cytoarchitecture (96) and as such may be more sensitive to neurodegenerative processes than cortical volumes (97, 98). Thinning of the cerebral cortex during the preadolescent period is a normative developmental process likely associated with synaptic pruning (99). Because age did not significantly differ between groups and was statistically covaried in the present analyses, it is unlikely that cortical thinness observed here is related to maturational processes. Thus, the present findings raise the possibility that exposure to antenatal glucocorticoids is associated with an acceleration of this maturational process. Alternatively, it is plausible that these children already had a thinner cortex even prior to the synaptic pruning associated with preadolescence and adolescence. These possibilities are consistent with rodent research demonstrating that glucocorticoid exposure causes morphological rearrangements in the apical dendrites in the ACC (71), which may underlie the observed associations with cortical thickness.
The ACC, and most predominantly the rACC has been associated with mood disorders including depression, anxiety, and bipolar disorders (100-102) as well as HPA axis disregulation (103). Interestingly, the association between rACC volume and depressive mood has been observed even among children with subclinical symptomatology (64) suggesting that this may be a prodromal risk factor for mental illness. Our data further indicate that thickness of the left rACC is associated with affective problems in children. The observation that this association is only present for the left cingulate is in accordance with recent evidence indicating greater left cingulate vulnerability in affective disorders (101) and in association with HPA axis disregulation (103) although future work is needed to evaluate the laterality of this association.
Although children with fetal exposure to glucocorticoid treatment had cortical thinning in the rACC, this group did not have a significantly higher level of affective problems. Interestingly, our observation is similar to a recent non-human primate study assessing the consequences of postnatal stress exposure in which group differences in brain development were observed despite the absence of group differences in behavior (104). Cortical thickness may be a more sensitive measure than behavioral observations and may detect prodromal risk for behavioral dysfunction at later ages. It is plausible that group differences will emerge during the pubertal transition, a time when affective problems often emerge. Evaluation of the treatment and comparison groups in our study indicates that one consequence of prenatal glucocorticoid treatment is restricted range of cortical thickness in the left rACC. As shown in Figure 2, 20% of the children in the comparison group had a rACC with a thickness of 4mm or greater. In contrast, none of the children in the glucocorticoid group had a rACC with a thickness of 4mm or greater. Our data indicate that a thinner rACC was associated with risk for affective problems regardless of gestational exposure and that prenatal glucocorticoid treatment increases the probability of having a thinner rACC.
Strengths and Limitations
A primary limitation is that participants were not randomly assigned to treatment and thus it is plausible that preexisting fetal differences contribute to the current observations. Because of the benefits of glucocorticoid treatment for survival among children born preterm, it is not ethical to randomly assign women in preterm labor to glucocorticoid treatment. A strength of the current investigation is the inclusion of children who were born full term. Existing published research is complicated by the fact that studies of children who have been exposed to glucocorticoid treatment primarily include children born preterm many of whom were quite ill and thus were already at risk for developmental delays. It is plausible that risk factors associated with prematurity mask the consequences of glucocorticoids (66). In the present investigation, the observed association between fetal exposure to glucocorticoids and child brain development were observed among children born at term and thus cannot be attributed to illness or differences in physiological regulation associated with shortened gestation. It is plausible, however, that fetal or maternal conditions (e.g., prenatal maternal stress hormones) that differed between the two groups contributed to the association between glucocorticoid treatment and cortical development. As illustrated in Table 1, group differences are not observed on a number of clinical and demographic factors including maternal intelligence, psychological state or SES.
Implications
There is growing recognition that early experience is a primary factor contributing to mental illness. It has been recently estimated that exposure to early adversity may explain over 30% of the risk for developing mental illness (4). Glucocorticoids are powerful regulators of neural differentiation and maturation and may play a salient role in neurodevelopment and risk for mental disease. Further, recent animal work has demonstrated that prenatal exposure to elevated glucocorticoids results in epigenetic changes with both lifespan and intergenerational consequences (105-108). We show that prenatal administration of glucocorticoids results in a pattern of cortical thinning that may be an indication of increased vulnerability to mental impairments. Regions such as the ACC that are associated with prenatal glucocorticoid treatment are ones that are implicated in risk for mental health problems including affective problems. Greater understanding of the developmental origins of mental illness is a critical step for the development of new diagnostic methods and improved treatments. The current findings indicate that prenatal glucocorticoids shape the construction of the fetal nervous system with consequences for the developing brain that persist into the preadolescent period.
Acknowledgments
This research was supported by grants from the NIH (HD050662 and HD065823 to EPD). The authors wish to thank the families who participated in this project. The assistance of Megan Faulkner and Natalie Hernandez is gratefully acknowledged.
Footnotes
Financial Disclosures
The authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Bellis MD, Baum AS, Birmaher B, Keshavan MS, Eccard CH, Boring AM, et al. A.E. Bennett Research Award. Developmental traumatology. Part I: Biological stress systems. Biol Psychiatry. 1999;45:1259–1270. doi: 10.1016/s0006-3223(99)00044-x. [DOI] [PubMed] [Google Scholar]
- 2.Nemeroff CB. Early-life adversity, CRF dysregulation, and vulnerability to mood and anxiety disorders. Psychopharmacol Bull. 2004;38:14–20. [PubMed] [Google Scholar]
- 3.Gunnar MR, Quevedo KM. Early care experiences and HPA axis regulation in children: a mechanism for later trauma vulnerability. Progress in Brain Research. 2008;167:137–149. doi: 10.1016/S0079-6123(07)67010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green JG, McLaughlin KA, Berglund PA, Gruber MJ, Sampson NA, Zaslavsky AM, et al. Childhood adversities and adult psychiatric disorders in the national comorbidity survey replication I: associations with first onset of DSM-IV disorders. Arch Gen Psychiatry. 2010;67:113–123. doi: 10.1001/archgenpsychiatry.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ansell EB, Rando K, Tuit K, Guarnaccia J, Sinha R. Cumulative adversity and smaller gray matter volume in medial prefrontal, anterior cingulate, and insula regions. Biol Psychiatry. 2012;72:57–64. doi: 10.1016/j.biopsych.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 7.Tottenham N. Risk and developmental heterogeneity in previously institutionalized children. J Adolesc Health. 2012;51:S29–33. doi: 10.1016/j.jadohealth.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tottenham N, Hare TA, Millner A, Gilhooly T, Zevin JD, Casey BJ. Elevated amygdala response to faces following early deprivation. Dev Sci. 2011;14:190–204. doi: 10.1111/j.1467-7687.2010.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel PD, Katz M, Karssen AM, Lyons DM. Stress-induced changes in corticosteroid receptor expression in primate hippocampus and prefrontal cortex. Psychoneuroendocrinology. 2008;33:360–367. doi: 10.1016/j.psyneuen.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van den Bergh BR, Van Calster B, Smits T, Van Huffel S, Lagae L. Antenatal maternal anxiety is related to HPA-axis dysregulation and self-reported depressive symptoms in adolescence: a prospective study on the fetal origins of depressed mood. Neuropsychopharmacology. 2008;33:536–545. doi: 10.1038/sj.npp.1301450. [DOI] [PubMed] [Google Scholar]
- 11.Van den Bergh BRH, Marcoen A. High antenatal maternal anxiety is related to ADHD symptoms, externalizing problems, and anxiety in 8/9-year-olds. Child Development. 2004;75:1085–1097. doi: 10.1111/j.1467-8624.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- 12.Davis EP, Sandman CA. Prenatal psychobiological predictors of anxiety risk in preadolescent children. Psychoneuroendocrinology. 2012;37:1224–1233. doi: 10.1016/j.psyneuen.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huizink AC, Dick DM, Sihvola E, Pulkkinen L, Rose RJ, Kaprio J. Chernobyl exposure as stressor during pregnancy and behaviour in adolescent offspring. Acta Psychiatr Scand. 2007;116:438–446. doi: 10.1111/j.1600-0447.2007.01050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Connor TG, Heron J, Golding J, Glover V. Maternal antenatal anxiety and behavioural/emotional problems in children: a test of a programming hypothesis. Journal of Child Psychology and Psychiatry. 2003;44:1025–1036. doi: 10.1111/1469-7610.00187. [DOI] [PubMed] [Google Scholar]
- 15.Huttenlocher PR. Synaptogenesis, synapse elimination, and neural plasticity in human cerebral cortex. In: Nelson CA, editor. Threats to Optimal Development: Integrating Biological, Psychological, and Social Risk Factors. Mahwah, NJ: Lawrence Erlbaum; 1994. pp. 35–54. [Google Scholar]
- 16.Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997;387:167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 17.Bourgeois JP. Synaptogenesis, heterochrony and epigenesis in the mammalian neocortex. Acta Paediatr Suppl. 1997;422:27–33. doi: 10.1111/j.1651-2227.1997.tb18340.x. [DOI] [PubMed] [Google Scholar]
- 18.Levitt P. Structural and functional maturation of the developing primate brain. J Pediatr. 2003;143:S35–45. doi: 10.1067/s0022-3476(03)00400-1. [DOI] [PubMed] [Google Scholar]
- 19.Chrousos GP, Kino T. Glucocorticoid signaling in the cell. Expanding clinical implications to complex human behavioral and somatic disorders. Ann N Y Acad Sci. 2009;1179:153–166. doi: 10.1111/j.1749-6632.2009.04988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chrousos GP, Gold PW. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. Jama. 1992;267:1244–1252. [PubMed] [Google Scholar]
- 21.Harris A, Seckl J. Glucocorticoids, prenatal stress and the programming of disease. Hormones and behavior. 2011:279–289. doi: 10.1016/j.yhbeh.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson L, Sapolsky R. The role of the hippocampus in feedback regulation of the hypothalamic pituitary adrenocortical axis. The Endocrine Reviews. 1991;12:118–134. doi: 10.1210/edrv-12-2-118. [DOI] [PubMed] [Google Scholar]
- 23.Buss C, Davis EP, Shahbaba B, Pruessner JC, Head K, Sandman CA. Maternal cortisol over the course of pregnancy and subsequent child amygdala and hippocampus volumes and affective problems. Proc Natl Acad Sci U S A. 2012;109:E1312–1319. doi: 10.1073/pnas.1201295109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crowley P. Antenatal corticosteroid therapy: A meta-analysis of the randomized trials, 1972-1994. American Journal of Obstetrics & Gynecology. 1995;173:322–355. doi: 10.1016/0002-9378(95)90222-8. [DOI] [PubMed] [Google Scholar]
- 25.Gilstrap LC, Christensen R, Clewell WH, et al. Effect of corticosteroids for fetal maturation on perinatal outcomes. NIH Consensus Development Panel on the Effect of Corticosteroids for Fetal Maturation on Perinatal Outcomes. JAMA. 1995;273:413–418. doi: 10.1001/jama.1995.03520290065031. [DOI] [PubMed] [Google Scholar]
- 26.Kajantie E, Raivio T, Janne OA, Hovi P, Dunkel L, Andersson S. Circulating glucocorticoid bioactivity in the preterm newborn after antenatal betamethasone treatment. Journal of Clinical Endocrinology and Metabolism. 2004;89:3999–4003. doi: 10.1210/jc.2004-0013. [DOI] [PubMed] [Google Scholar]
- 27.Albiston AL, Obeyesekere VR, Smith RE, Krozowski ZS. Cloning and tissue distribution of the human 11ß-hydroxysteroid dehydrogenase type 2 enzyme. Mol Cell Endo. 1994;105:R11–R17. doi: 10.1016/0303-7207(94)90176-7. [DOI] [PubMed] [Google Scholar]
- 28.Sandman CA, Davis EP. Neurobehavioral risk is associated with gestational exposure to stress hormones. Expert Rev Endocrinol Metab. 2012;7:445–459. doi: 10.1586/eem.12.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dammann O, Matthews SG. Repeated antenatal glucocorticoid exposure and the developing brain. Pediatric Research. 2001;50:563–564. doi: 10.1203/00006450-200111000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Seckl JR. Glucocorticoids, developmental ‘programming’ and the risk of affective dysfunction. Prog Brain Res. 2008;167:17–34. doi: 10.1016/S0079-6123(07)67002-2. [DOI] [PubMed] [Google Scholar]
- 31.Trejo JL, Cuchillo I, Machin C, Rua C. Maternal adrenalectomy at the early onset of gestation impairs the postnatal development of the rat hippocampal formation: effects on cell numbers and differentiation, connectivity and calbindin-D28K immunoreactivity. Journal of Neuroscience Research. 2000;62:644–667. doi: 10.1002/1097-4547(20001201)62:5<644::AID-JNR4>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 32.Welberg LA, Seckl J. Prenatal stress, glucocorticoids and the programming of the brain. Journal of Neuroendocrinology. 2001;13:113–128. doi: 10.1046/j.1365-2826.2001.00601.x. [DOI] [PubMed] [Google Scholar]
- 33.Weinstock M. Alterations induced by gestational stress in brain morphology and behaviour of the offspring. Progressin Neurobiology. 2001;65:427–451. doi: 10.1016/s0301-0082(01)00018-1. [DOI] [PubMed] [Google Scholar]
- 34.Korte C, Styne D, Merritt AT, Mayes D, Wertz A, Helbock HJ. Adrenocortical function in the very low birthweight infants: Improved testing for sensitivity and association with neonatal outcome. The Journal of Pediatrics. 1996;128:257–263. doi: 10.1016/s0022-3476(96)70404-3. [DOI] [PubMed] [Google Scholar]
- 35.Meaney MJ, Diorio J, Francis D, Widdowson J, La Plante P, Caldui C, et al. Early environmental regulation of forbrain glucocorticoid gene expression: Implications for adrenocortical response to stress. Developmental Neuroscience. 1996;18 doi: 10.1159/000111395. [DOI] [PubMed] [Google Scholar]
- 36.Murphy BP, Inder TE, Huppi PS, Warfield S, Zientara GP, Kikinis R, et al. Impaired cerebral cortical gray matter growth after treatment with dexamethasone for neonatal chronic lung disease. Pediatrics. 2001;107:217–221. doi: 10.1542/peds.107.2.217. [DOI] [PubMed] [Google Scholar]
- 37.Modi N, Lewis H, Al-Naqeeb N, Ajayi-Obe M, Dore CJ, Rutherford M. The effects of repeated antenatal glucocorticoid therapy on the developing brain. Pediatr Res. 2001;50:581–585. doi: 10.1203/00006450-200111000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Uno H, Eisele S, Sakai A, Shelton S, Baker E, DeJesus O, et al. Neurotoxicity of glucocorticoids in the primate brain. Hormone and Behavior. 1994;28:336–348. doi: 10.1006/hbeh.1994.1030. [DOI] [PubMed] [Google Scholar]
- 39.Uno H, Lohmiller L, Thieme C, Kemnitz JW, Engle MJ, Roecker EB, et al. Brain damage induced by prenatal exposure to dexamethasone in fetal macaques. I. Hippocampus. Brain Research Developmental Brain Research. 1990;53:157–167. doi: 10.1016/0165-3806(90)90002-g. [DOI] [PubMed] [Google Scholar]
- 40.Diaz Heijtz R, Fuchs E, Feldon J, Pryce CR, Forssberg H. Effects of antenatal dexamethasone treatment on glucocorticoid receptor and calcyon gene expression in the prefrontal cortex of neonatal and adult common marmoset monkeys. Behav Brain Funct. 2010;6:18. doi: 10.1186/1744-9081-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanchez MM, Young LJ, Plotsky PM, Insel TR. Distribution of corticosteroid receptors in the rhesus brain: Relative absence of glucocorticoid receptors in the hippocampal formation. The Journal of Neuroscience. 2000;20:4657–4668. doi: 10.1523/JNEUROSCI.20-12-04657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis EP, Waffarn F, Uy C, Hobel CJ, Glynn LM, Sandman CA. Effect of prenatal glucocorticoid treatment on size at birth among infants born at term gestation. J Perinatol. 2009;29:731–737. doi: 10.1038/jp.2009.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waffarn F, Davis EP. Effects of antenatal corticosteroids on the hypothalamic-pituitary-adrenocortical axis of the fetus and newborn: experimental findings and clinical considerations. Am J Obstet Gynecol. 2012;207:446–454. doi: 10.1016/j.ajog.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 101: Ultrasonography in pregnancy. Obstet Gynecol. 2009;113:451–461. doi: 10.1097/AOG.0b013e31819930b0. [DOI] [PubMed] [Google Scholar]
- 45.Armstrong JM, Goldstein LH. Manual for the MacArthur Health and Behavior Questionnaire (HBQ 1.0) MacArthur Foundation Research Network on Psychopathology and Development University of Pittsburgh 2003 [Google Scholar]
- 46.Wechsler D. Wechsler Adult Intelligence Scale-Third Edition. San Antonio: The Psychological Corporation; 1997. [Google Scholar]
- 47.Beck AT, Steer RA, Brown GK. Manual for the Beck Depression Inventory-II. San Antonio, TX: Psychological Corporation; 1996. [Google Scholar]
- 48.Achenbach TM, Rescorla LA. Manual for the ASEBA School-Age Forms and Profiles: An Integtated System of Multi-Informant Assessment. Burlington, VT: University of Vermont, Reaserch Center for Children, Youth, & Families; 2001. [Google Scholar]
- 49.Sled JG, Zijdenbos AP, Evans AC. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging. 1998;17:87–97. doi: 10.1109/42.668698. [DOI] [PubMed] [Google Scholar]
- 50.Segonne F, Dale AM, Busa E, Glessner M, Salat D, Hahn HK, et al. A hybrid approach to the skull stripping problem in MRI. Neuroimage. 2004;22:1060–1075. doi: 10.1016/j.neuroimage.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 51.Fischl B, Salat D, Busa E, Albert M, Dieterich M, Haselgrove C, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 52.Fischl B, Salat DH, van der Kouwe AJ, Makris N, Segonne F, Quinn BT, et al. Sequence-independent segmentation of magnetic resonance images. Neuroimage. 2004;23(Suppl 1):S69–84. doi: 10.1016/j.neuroimage.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 53.Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage. 1999;9:179–194. doi: 10.1006/nimg.1998.0395. [DOI] [PubMed] [Google Scholar]
- 54.Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A. 2000;97:11050–11055. doi: 10.1073/pnas.200033797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis. II: Inflation, flattening, and a surface-based coordinate system. Neuroimage. 1999;9:195–207. doi: 10.1006/nimg.1998.0396. [DOI] [PubMed] [Google Scholar]
- 56.Fischl B, Sereno MI, Tootell RB, Dale AM. High-resolution intersubject averaging and a coordinate system for the cortical surface. Hum Brain Mapp. 1999;8:272–284. doi: 10.1002/(SICI)1097-0193(1999)8:4<272::AID-HBM10>3.0.CO;2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosas HD, Liu AK, Hersch S, Glessner M, Ferrante RJ, Salat DH, et al. Regional and progressive thinning of the cortical ribbon in Huntington’s disease. Neurology. 2002;58:695–701. doi: 10.1212/wnl.58.5.695. [DOI] [PubMed] [Google Scholar]
- 58.Kuperberg GR, Broome MR, McGuire PK, David AS, Eddy M, Ozawa F, et al. Regionally localized thinning of the cerebral cortex in schizophrenia. Arch Gen Psychiatry. 2003;60:878–888. doi: 10.1001/archpsyc.60.9.878. [DOI] [PubMed] [Google Scholar]
- 59.Salat DH, Buckner RL, Snyder AZ, Greve DN, Desikan RS, Busa E, et al. Thinning of the cerebral cortex in aging. Cereb Cortex. 2004;14:721–730. doi: 10.1093/cercor/bhh032. [DOI] [PubMed] [Google Scholar]
- 60.Genovese CR, L N, Nichols T. Thresholding of statistical maps in functional neuroimaging use the false discovery rate. Neuroimage. 2002:870–878. doi: 10.1006/nimg.2001.1037. [DOI] [PubMed] [Google Scholar]
- 61.Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 62.Blair KS, Geraci M, Smith BW, Hollon N, DeVido J, Otero M, et al. Reduced dorsal anterior cingulate cortical activity during emotional regulation and top-down attentional control in generalized social phobia, generalized anxiety disorder, and comorbid generalized social phobia/generalized anxiety disorder. Biol Psychiatry. 2012;72:476–482. doi: 10.1016/j.biopsych.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Etkin A, Schatzberg AF. Common abnormalities and disorder-specific compensation during implicit regulation of emotional processing in generalized anxiety and major depressive disorders. Am J Psychiatry. 2011;168:968–978. doi: 10.1176/appi.ajp.2011.10091290. [DOI] [PubMed] [Google Scholar]
- 64.Boes AD, McCormick LM, Coryell WH, Nopoulos P. Rostral anterior cingulate cortex volume correlates with depressed mood in normal healthy children. Biol Psychiatry. 2008;63:391–397. doi: 10.1016/j.biopsych.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davis EP, Glynn LM, Waffarn F, Sandman CA. Prenatal maternal stress programs infant stress regulation. J Child Psychol Psychiatry. 2011;52:119–129. doi: 10.1111/j.1469-7610.2010.02314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davis EP, Waffarn F, Sandman CA. Prenatal treatment with glucocorticoids sensitizes the hpa axis response to stress among full-term infants. Dev Psychobiol. 2011;53:175–183. doi: 10.1002/dev.20510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schaffer L, Luzi F, Burkhardt T, Rauh M, Beinder E. Antenatal betamethasone administration alters stress physiology in healthy neonates. Obstet Gynecol. 2009;113:1082–1088. doi: 10.1097/AOG.0b013e3181a1f0e6. [DOI] [PubMed] [Google Scholar]
- 68.Werner E, Zhao Y, Evans L, Kinsella M, Kurzius L, Altincatal A, et al. Higher maternal prenatal cortisol and younger age predict greater infant reactivity to novelty at 4 months: An observation-based study. Dev Psychobiol. 2012 doi: 10.1002/dev.21066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davis EP, Glynn LM, Schetter CD, Hobel C, Chicz-Demet A, Sandman CA. Prenatal exposure to maternal depression and cortisol influences infant temperament. J Am Acad Child Adolesc Psychiatry. 2007;46:737–746. doi: 10.1097/chi.0b013e318047b775. [DOI] [PubMed] [Google Scholar]
- 70.de Weerth C, van Hees Y, Buitelaar J. Prenatal maternal cortisol levels and infant behavior during the first 5 months. Early Human Development. 2003;74:139–151. doi: 10.1016/s0378-3782(03)00088-4. [DOI] [PubMed] [Google Scholar]
- 71.Cerqueira JJ, Taipa R, Uylings HB, Almeida OF, Sousa N. Specific configuration of dendritic degeneration in pyramidal neurons of the medial prefrontal cortex induced by differing corticosteroid regimens. Cereb Cortex. 2007;17:1998–2006. doi: 10.1093/cercor/bhl108. [DOI] [PubMed] [Google Scholar]
- 72.Herman JP, Ostrander MM, Mueller NK, Figueiredo H. Limbic system mechanisms of stress regulation: hypothalamo-pituitary-adrenocortical axis. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1201–1213. doi: 10.1016/j.pnpbp.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 73.Diorio D, Viau V, Meaney MJ. The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J Neurosci. 1993;13:3839–3847. doi: 10.1523/JNEUROSCI.13-09-03839.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cohen RA, Grieve S, Hoth KF, Paul RH, Sweet L, Tate D, et al. Early life stress and morphometry of the adult anterior cingulate cortex and caudate nuclei. Biol Psychiatry. 2006;59:975–982. doi: 10.1016/j.biopsych.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 75.Bremner JD. Stress and brain atrophy. CNS Neurol Disord Drug Targets. 2006;5:503–512. doi: 10.2174/187152706778559309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walhovd KB, Fjell AM, Brown TT, Kuperman JM, Chung Y, Hagler DJ, Jr, et al. Long-term influence of normal variation in neonatal characteristics on human brain development. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1208180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dammann O, Matthews SG. Repeated antenatal glucocorticoid exposure and the developing brain. Pediatr Res. 2001;50:563–564. doi: 10.1203/00006450-200111000-00004. [DOI] [PubMed] [Google Scholar]
- 78.Sandman CA, Davis EP, Buss C, Glynn LM. Prenatal programming of human neurological function. Int J Pept. 2011;2011:837596. doi: 10.1155/2011/837596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coussons-Read ME, Lobel M, Carey JC, Kreither MO, D’Anna K, Argys L, et al. The occurrence of preterm delivery is linked to pregnancy-specific distress and elevated inflammatory markers across gestation. Brain, Behavior, and Immunity. 2012;26:650–659. doi: 10.1016/j.bbi.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hatfield T, Wing DA, Buss C, Head K, Muftuler LT, Davis EP. Magnetic resonance imaging demonstrates long-term changes in brain structure in children born preterm and exposed to chorioamnionitis. Am J Obstet Gynecol. 2011;205384:e381–384. e388. doi: 10.1016/j.ajog.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Monk C, Georgieff MK, Osterholm EA. Research Review: Maternal prenatal distress and poor nutrition - mutually influencing risk factors affecting infant neurocognitive development. J Child Psychol Psychiatry. 2012 doi: 10.1111/jcpp.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bublitz MH, Stroud LR. Maternal Smoking During Pregnancy and Offspring Brain Structure and Function: Review and Agenda for Future Research. Nicotine & Tobacco Research. 2012;14:388–397. doi: 10.1093/ntr/ntr191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis EP, Sandman CA. The timing of prenatal exposure to maternal cortisol and psychosocial stress is associated with human infant cognitive development. Child Development. 2010;81:131–148. doi: 10.1111/j.1467-8624.2009.01385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scheepens A, van de Waarenburg M, van den Hove D, Blanco CE. A single course of prenatal betamethasone in the rat alters postnatal brain cell proliferation but not apoptosis. J Physiol. 2003;552:163–175. doi: 10.1113/jphysiol.2003.043414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dean F, Matthews SG. Maternal dexamethasone treatment in late gestation alters glucocorticoid and mineralocorticoid receptor mRNA in the fetal guinea pig brain. Brain Res. 1999;846:253–259. doi: 10.1016/s0006-8993(99)02064-8. [DOI] [PubMed] [Google Scholar]
- 86.Dunn E, Kapoor A, Leen J, Matthews SG. Prenatal synthetic glucocorticoid exposure alters hypothalamic-pituitary-adrenal regulation and pregnancy outcomes in mature female guinea pigs. J Physiol. 2010;588:887–899. doi: 10.1113/jphysiol.2009.182139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Antonow-Schlorke I, Kuhn B, Muller T, Schubert H, Sliwka U, Nathanielsz PW, et al. Antenatal betamethasone treatment reduces synaptophysin immunoreactivity in presynaptic terminals in the fetal sheep brain. Neuroscience Letters. 2001;297:147–150. doi: 10.1016/s0304-3940(00)01605-0. [DOI] [PubMed] [Google Scholar]
- 88.Schwab M, Schmidt K, Roedel MA, MA, Muller T, Schubert H, Buchwalder LF, et al. Non-linear changes of electrocortial activity after antenatal betamethasone treatment in fetal sheep. Journal of Physiology. 2001;531:535–543. doi: 10.1111/j.1469-7793.2001.0535i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lohle M, Muller T, Wicher C, Roedel M, Schubert H, Witte OW, et al. Betamethasone effects on fetal sheep cerebral blood flow are not dependent on maturation of cerebrovascular system and pituitary-adrenal axis. J Physiol. 2005;564:575–588. doi: 10.1113/jphysiol.2004.077537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Antonow-Schlorke I, Helgert A, Gey C, Coksaygan T, Schubert H, Nathanielsz PW, et al. Adverse effects of antenatal glucocorticoids on cerebral myelination in sheep. Obstet Gynecol. 2009;113:142–151. doi: 10.1097/AOG.0b013e3181924d3b. [DOI] [PubMed] [Google Scholar]
- 91.Moss TJ, Doherty DA, Nitsos I, Sloboda DM, Harding R, Newnham JP. Effects into adulthood of single or repeated antenatal corticosteroids in sheep. American Journal of Obstetetrics and Gynecology. 2005;192:146–152. doi: 10.1016/j.ajog.2004.06.065. [DOI] [PubMed] [Google Scholar]
- 92.Huang WL, Beazley LD, Quinlivan JA, Evans SF, Newnham JP, Dunlop SA. Effect of corticosteroids on brain growth in fetal sheep. Obstetrics and Gynecology. 1999;94:213–218. doi: 10.1016/s0029-7844(99)00265-3. [DOI] [PubMed] [Google Scholar]
- 93.Coe CL, Lubach GR. Developmental consequences of antenatal dexamethasone treatment in nonhuman primates. Neurosci Biobehav Rev. 2005;29:227–235. doi: 10.1016/j.neubiorev.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 94.Antonow-Schlorke I, Schwab M, Li C, Nathanielsz PW. Glucocorticoid exposure at the dose used clinically alters cytoskeletal proteins and presynaptic terminals in the fetal baboon brain. Journal of Physiology. 2003;547:117–123. doi: 10.1113/jphysiol.2002.025700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schneider U, Arnscheidt C, Schwab M, Haueisen J, Seewald HJ, Schleussner E. Steroids that induce lung maturation acutely affect higher cortical function: a fetal magnetoencephalography study. Reprod Sci. 2011;18:99–106. doi: 10.1177/1933719110381140. [DOI] [PubMed] [Google Scholar]
- 96.Makris N, Biederman J, Valera EM, Bush G, Kaiser J, Kennedy DN, et al. Cortical thinning of the attention and executive function networks in adults with attention-deficit/hyperactivity disorder. Cereb Cortex. 2007;17:1364–1375. doi: 10.1093/cercor/bhl047. [DOI] [PubMed] [Google Scholar]
- 97.Hutton C, Draganski B, Ashburner J, Weiskopf N. A comparison between voxel-based cortical thickness and voxel-based morphometry in normal aging. Neuroimage. 2009;48:371–380. doi: 10.1016/j.neuroimage.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sowell ER, Thompson PM, Leonard CM, Welcome SE, Kan E, Toga AW. Longitudinal mapping of cortical thickness and brain growth in normal children. J Neurosci. 2004;24:8223–8231. doi: 10.1523/JNEUROSCI.1798-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Muftuler LT, Davis EP, Buss C, Head K, Hasso AN, Sandman CA. Cortical and subcortical changes in typically developing preadolescent children. Brain Res. 2011;1399:15–24. doi: 10.1016/j.brainres.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Treadway MT, Grant MM, Ding Z, Hollon SD, Gore JC, Shelton RC. Early adverse events, HPA activity and rostral anterior cingulate volume in MDD. PLoS One. 2009;4:e4887. doi: 10.1371/journal.pone.0004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sassi RB, Brambilla P, Hatch JP, Nicoletti MA, Mallinger AG, Frank E, et al. Reduced left anterior cingulate volumes in untreated bipolar patients. Biol Psychiatry. 2004;56:467–475. doi: 10.1016/j.biopsych.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 102.Drevets WC, Savitz J, Trimble M. The subgenual anterior cingulate cortex in mood disorders. CNS Spectr. 2008;13:663–681. doi: 10.1017/s1092852900013754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.MacLullich AM, Ferguson KJ, Wardlaw JM, Starr JM, Deary IJ, Seckl JR. Smaller left anterior cingulate cortex volumes are associated with impaired hypothalamic-pituitary-adrenal axis regulation in healthy elderly men. J Clin Endocrinol Metab. 2006;91:1591–1594. doi: 10.1210/jc.2005-2610. [DOI] [PubMed] [Google Scholar]
- 104.Parr LA, Boudreau M, Hecht E, Winslow JT, Nemeroff CB, Sanchez MM. Early life stress affects cerebral glucose metabolism in adult rhesus monkeys (Macaca mulatta) Dev Cogn Neurosci. 2012;2:181–193. doi: 10.1016/j.dcn.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Crudo A, Petropoulos S, Moisiadis VG, Iqbal M, Kostaki A, Machnes Z, et al. Prenatal synthetic glucocorticoid treatment changes DNA methylation states in male organ systems: multigenerational effects. Endocrinology. 2012;153:3269–3283. doi: 10.1210/en.2011-2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Iqbal M, Moisiadis VG, Kostaki A, Matthews SG. Transgenerational effects of prenatal synthetic glucocorticoids on hypothalamic-pituitary-adrenal function. Endocrinology. 2012;153:3295–3307. doi: 10.1210/en.2012-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bale TL. Sex differences in prenatal epigenetic programming of stress pathways. Stress. 2011;14:348–356. doi: 10.3109/10253890.2011.586447. [DOI] [PubMed] [Google Scholar]
- 108.Jensen Pena C, Monk C, Champagne FA. Epigenetic effects of prenatal stress on 11beta-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS One. 2012;7:e39791. doi: 10.1371/journal.pone.0039791. [DOI] [PMC free article] [PubMed] [Google Scholar]