

Abstract
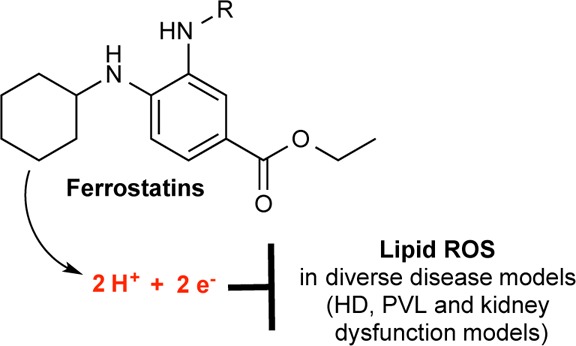
Ferrostatin-1 (Fer-1) inhibits ferroptosis, a form of regulated, oxidative, nonapoptotic cell death. We found that Fer-1 inhibited cell death in cellular models of Huntington’s disease (HD), periventricular leukomalacia (PVL), and kidney dysfunction; Fer-1 inhibited lipid peroxidation, but not mitochondrial reactive oxygen species formation or lysosomal membrane permeability. We developed a mechanistic model to explain the activity of Fer-1, which guided the development of ferrostatins with improved properties. These studies suggest numerous therapeutic uses for ferrostatins, and that lipid peroxidation mediates diverse disease phenotypes.
Introduction
Reactive oxygen species (ROS) are required for a wide range of normal biological processes,1 but also have been linked to tissue degeneration2 and cell death.3 Thus, there is a clear rationale for the development of small molecule antioxidants with high potency and specificity for damage-inducing or death-promoting ROS.4 Ideally, such compounds would be readily synthesized and selectively prevent the formation or accumulation of ROS that are damaging or lethal to cells, without impinging upon the production of normal homeostatic ROS. We recently identified ferrostatin-1 (Fer-1) as a potent and selective inhibitor of ferroptosis, a form of nonapoptotic, iron-dependent, oxidative cell death.4 Ferroptosis can be triggered in cancer cells by several small molecules, including erastin, sulfasalazine, and RSL3, that perturb redox homeostasis, allowing the iron-dependent accumulation of lethal ROS.4−7 In human HT-1080 fibrosarcoma cells, erastin treatment blocks the uptake of cystine, the oxidized form of cysteine,4 which is the rate-limiting precursor for the production of the antioxidant tripeptide glutathione. Erastin treatment leads after several hours to the accumulation of lipid ROS, as detected using the oxidizable lipid ROS probe C11-BODIPY, and, ultimately, cell death.4 Both C11-BODIPY oxidation and cell death are blocked by Fer-1.4 The role of iron in ferroptosis is unclear, and it may act to catalyze free radical formation directly in the cytosol (e.g., via Fenton chemistry) and/or as a cofactor for an enzyme essential for the ROS production that precedes cell death.8
Here, we provide evidence that Fer-1 (Figure 1a) is a drug-like, easily synthesized, potent inhibitor of ferroptosis that acts via a reductive mechanism to prevent damage to membrane lipids and thereby inhibit cell death.
Figure 1.

The effects of ferrostatins in Huntington’s disease (HD), periventricular leukomalacia (PVL), and kidney proximal tubules cell death models. (a) Chemical structure of ferrostatins. (b) Effect of ferrostatins on cell survival in an HD brain-slice model. YFP = yellow fluorescent protein transfection control. httN90Q73 is mutant huntingtin (N-terminal 90aa with a Q73 repeat). KW + SP is a combination used as a positive control for protection. (c) Dose–response test of the effect of ferrostatins in a model of PVL. Cys = Cystine supplementation, a positive control for cell death rescue. (d) Effect of ferrostatins, at various concentrations, in a primary kidney renal tubule damage model.
Fer-1 is an arylalkylamine. It is established that diarylamines and hindered dialkylamines are antioxidants (also known as scavengers or reducing agents) in the food industry to prevent spoilage,9 and in the materials industry to slow the oxidative degradation of polymers, plastics, and fine chemicals.10 In the chemical industry, these antioxidants retard autoxidation of substrates, such as rubbers and plastics. The autoxidation is caused primarily by radical chain reactions between oxygen and the substrates. To prevent autoxidation form occurring, hindered amines such as 2,2,6,6-tetramethyl-piperidine, for example, have been used in the synthesis of highly stable polyvinylchlorides,11 plastics, and rubbers.12 In an academic setting, Sheltons,13−15 and Ingold16−18 were among the first to report in the 1960s studies on the inhibition of free radical chain mechanisms of autoxidation of organic materials. Since then, there has been a tremendous interest in investigating the effect of phenols, diphenylamines, and hindered alkylamines as potential antioxidants in medically relevant systems in the search for therapeutics.19,20 Recently, Michelle Coote and co-workers reported21 ab initio calculations on a new mode of the inhibition of hindered amine light stabilizers (HALSs). These amines are based on radical-trapping antioxidants (RTAs). Secondary amines, in particular diarylamines, are regarded as important scavengers that prevent radical chain reactions and that can serve as antioxidants, since their reducing activity allow them to act as radical scavengers.22,23 Most studies of amine antioxidants have been focused either on diarylamines or hindered dialkylamines. In this study, we report a third family of this type of antioxidant, the arylalkyl amines, exemplified by Fer-1, and its therapeutic relevance.
Results and Discussion
We previously showed that Fer-1 attenuates oxidative, iron-dependent cell death in cancer cells treated with small molecules that block cystine import and glutathione production, as well as cell death in rat organotypic hippocampal brain slices treated with toxic concentrations of glutamate,4 which also blocks cystine import.24,25 We hypothesized that Fer-1 would be effective at preventing other forms of cell death involving iron-dependent oxidative stress and/or glutamate toxicity. Huntington’s disease (HD) is reported to involve perturbation of glutamate, glutathione, and iron levels;26 the accumulation of lipid oxidation breakdown products;27 and nonapoptotic neuronal cell death.28 We therefore tested whether Fer-1 could prevent, in rat corticostriatal brain slices, cell death induced by the expression, via biolistic transfection, of a huntingtin (htt) exon 1 fragment with a pathogenic repeat (73Q) (mN90Q73), along with yellow fluorescent protein (YFP) to mark transfected neurons29 (Figure 1b). Slices were treated with DMSO (vehicle control), a positive control death inhibitor combination of the adenosine A2A receptor antagonist KW-6002 (KW, 50 μM) and the JNK inhibitor SP600125 (SP, 30 μM), or ferrostatins (Fer-1 or SRS11-92) at increasing concentrations (1 nM to 1 μM). Four days later, the number of healthy medium spiny neurons (MSNs) was quantified, as previously described.29 A significant increase in the number of healthy MSNs was observed upon Fer-1 and SRS11-92 treatment at 10 nM, 100 nM, and 1 μM. Moreover, with 1 μM treatment of Fer-1, the number of healthy MSNs was statistically indistinguishable (P > 0.05) from both the YFP (no htt) control and the control inhibitor combination (KW + SP) (Figure 1b).
We then tested the effect of Fer-1 in an in vitro model of periventricular leukomalacia (PVL), a syndrome afflicting infants born prematurely that is caused primarily by the death of developing oligodendrocytes (OLs).30 It has been suggested that OL death is iron-dependent; in autopsy studies, elevated levels of lipid ROS biomarkers are observed,31−34 potentially suggesting the involvement of ferroptosis. To trigger death, cultured OLs were exposed to cystine-free conditions, which ultimately deplete glutathione. Fer-1, SRS11-92 (Figure 1c), and Fer-1 analogues (Supporting Information Figure S1) fully protected OLs from cystine deprivation when tested at 100 nM (Figure 1c).
Finally, we tested the effects of Fer-1, SRS11-92, and selected analogues in a model of iron-induced cell death in freshly isolated mouse kidney proximal tubules that simulates major elements of rhabdomyolysis-induced acute kidney injury.35 Cell death in this model is known to involve oxidative lipid damage;35 we therefore suspected that Fer-1 might be protective. Hydroxyquinoline and ferrous ammonium sulfate (HQ + Fe; 10 μM each) were used to induce cell death, which was quantified by monitoring LDH release after 60 min. In this model, Fer-1, SRS11-92, and analogues prevented lethality (Figure 1d, Supporting Information Figure S2). Together, the results of these assays demonstrated that Fer-1, SRS11-92, and analogues are potent inhibitors of a variety of clinically relevant oxidative cell death phenotypes. By implication, cell death in these diverse models may involve, at least in part, the induction of ferroptosis.
Using the general lipid ROS probe C11-BODIPY (581/591),36 we previously suggested that erastin-induced ferroptosis involves Fer-1-sensitive oxidative lipid modification.4 We confirmed this result using an independent assay. HT-1080 cells were incubated with an alkyne containing form of the oxidizable fatty acid linoleic acid (linoleamide alkyne, LAA) and treated with erastin (10 μM) +/– Fer-1 or SRS11-92 (both at 1 μM). After 7.5 h, cells were washed, fixed, and permeabilized, and a copper-catalyzed cycloaddition reaction (“click” reaction) was performed with fluorescein azide. When viewed under a microscope, cells treated with erastin displayed accumulation of fluorescein-positive puncta, indicative of the formation of oxidative breakdown products of LAA, while co-treatment with Fer-1 or SRS11-92 prevented the accumulation of this marker (Supporting Information Figure S4).
The above results suggested that the oxidative destruction of specific fatty acids was likely to occur during ferroptosis. To search in an unbiased manner for Fer-1-sensitive effects on specific membrane lipids or other metabolites, we characterized changes in the metabolome of HT-1080 cells treated with erastin (10 μM) +/– Fer-1 (1 μM) (Figure 2a). Cells from four independent biological replicates were harvested following 6 h of compound treatment, before death became apparent, and subjected to GC/MS and LC/MS/MS analysis. From a set of 328 metabolites consistently identified in all conditions, 78 were altered significantly (either depleted or enriched) in erastin versus DMSO treatment conditions (P < 0.05, Figure 2a, Supporting Information Table S1). Consistent with our proposed mechanism of action for erastin as an inhibitor of cystine import via the system xc– transporter,4 we detected significant erastin-induced depletion of cysteine and cysteinylglycine (indicated in Figure 2a). We likewise observed substantial depletion of glutathione, suggesting that the synthesis of this tripeptide is compromised in these cells when cystine import is inhibited, as expected. Furthermore, the most highly enriched metabolite was the glutathione analogue ophthalmate (12-fold increase in erastin versus DMSO-treated control), which has a methyl group in place of the thiol found in glutathione. Ophthalmate accumulation has previously been observed under conditions where glutathione, and presumably cysteine, are depleted.37 Under these conditions, the enzymes of the glutathione synthesis pathway use 2-aminobutyrate in place of cysteine to form ophthalmate. While this compound is redox inactive, it may serve to inhibit GSH-consuming enzymes, thereby conserving intracellular GSH stores.37 Ophthalmate is thus a potential metabolic biomarker for cysteine-depleted cells that could be used to assess the efficacy of treatments that target cystine import or cysteine metabolism.37 The finding that the levels of ophthalmate are elevated following erastin treatment is consistent with our model of erastin’s mechanism of action.
Figure 2.

Fer-1 inhibits the oxidative destruction of unsaturated fatty acids. (a) Significant (P < 0.05) changes in metabolite levels in HT-1080 cells treated with erastin (10 μM, 6 h) versus DMSO (left) or with erastin + Fer-1 (1 μM) versus erastin alone (right). 2-LG, 2-linoleoylglycerol; 1-LG, 1-linoleoylglycerol; 2-AG, 2-arachidonoyl glycerol. (b) Spot dilutions of Saccharomyces cerevisiaecoq3Δ cells treated with linolenic acid (LA, 500 μM) ± trolox (50 μM, a positive control antioxidant) or ferrostatin-1 (Fer-1, 10 μM).
Among the metabolites affected by erastin treatment, the levels of small subset (17/78 = 22%) were observed to be significantly (P < 0.05) closer to baseline (i.e., maintained at normal levels) when cells were co-treated with Fer-1. While Fer-1 co-treatment did not significantly change the observed levels of cysteine, glutathione, or ophthalmate (as expected), it did preserve the normal levels of polyunsaturated fatty acids (PUFAs) and PUFA-derivatives, including eicosapentaenoate (20:5n3), linoleate (18:2n6), linolenate (18:3n3/6), and docosahexaenoate (22:6n3). PUFAs and PUFA-containing lipids are highly susceptible to oxidation, via both enzymatic (e.g., lipoxygenase-enzyme-mediated) and non-enzymatic (e.g. iron-dependent Fenton chemistry-mediated) processes.38 A parsimonious explanation of these results is that under conditions that favor ferroptosis (e.g., cystine and glutathione depletion) specific membrane PUFAs are oxidized and subsequently fragmented, resulting in the net depletion of these species, as detected in our MS analyses. Fer-1 is able to prevent the depletion of these species by inhibiting their oxidative destruction. Mechanistically, we hypothesize that Fer-1 acts either to reduce a lipid peroxide to an alcohol (R-OOH → R-OH) and/or to intercept and scavenge a lipid radical through hydrogen atom transfer or direct reduction (R-O· → R-OH). Both mechanisms would explain the sparing of PUFAs by Fer-1 under ferroptosis-inducing conditions. While these results cannot conclusively rule out the possibility that the observed Fer-1-sensitive changes in lipid metabolites are merely a correlate of rescue from erastin-induced death by another means, we speculate that the oxidative destruction of these lipids contributes directly to cell death, either through the physical disruption of the plasma membrane permeability barrier or through the formation of PUFA breakdown products (e.g., reactive aldehydes) that are toxic to cells.
On the basis of these results, we hypothesized that the Fer-1-sensitive oxidative destruction of PUFAs is sufficient for ferroptotic cell death. This hypothesis is difficult to test in mammalian cells, given the compositional complexity of the plasma membrane. Thus, we employed a yeast (S. cerevisiae) model of cell death, where it is possible to isolate the lethal effects of individual PUFAs.39 PUFAs are not normally present in the plasma membrane of S. cerevisiae, but these cells normally tolerate growth in the presence of high concentrations of PUFAs (which are susceptible to auto-oxidation), in part, through a moonlighting antioxidant function of the mitochondrial electron carrier coenzyme Q.40S. cerevisiae cells in which coenzyme Q biosynthesis has been eliminated by deletion of the gene COQ3 (coq3Δ) are hypersensitive to incubation in PUFAs, providing a model of PUFA-oxidation-induced death. Thus, coq3Δ cells were incubated with 500 μM linolenic acid (LA, 18:3) for 6 h +/– Fer-1 (10 μM) or a positive control lipophilic antioxidant (trolox, 50 μM), then spotted in a dilution series on agar to examine clonogenic growth. Like trolox, Fer-1 restored the clonogenic growth of LA-treated coq3Δ cells. Thus, Fer-1 can inhibit the loss of clonogenic growth potential induced by PUFA auto-oxidation, entirely consistent with a model where Fer-1 prevents cell death by inhibiting the oxidative destruction of membrane lipid PUFAs.
To obtain further insight into the Fer-1 protective mechanism, we designed a three-step synthesis based on the previously reported synthetic route of Fer-1.4 A nucleophilic aromatic substitution reaction (SNAr) between the commercially available ethyl 4-chloro-3-nitrobenzoate and cyclohexylamine followed by catalytic hydrogenolysis of the nitro group provided the desired Fer-1 derivatives.40 The aniline of the latter compounds was reacted through reductive amination in the presence of sodium triacetoxyborohydride or alkylation conditions in the presence of either arylaldehydes41 or 1-(bromomethyl)aryl compounds, respectively (Figure 4a). We used this general route to synthesize 67 analogues. We established a structure–activity relationship (SAR) for the Fer-1 scaffold (Figure 4a, Tables S2–S8 and Schemes S1–S5). We sought to test the hypothesis that Fer-1 acts as an antioxidant.4 Examination of the Fer-1 structure suggested a possible release of two protons and two electrons, through the tautomer intermediate compound A, resulting in the formation of a redox-stable compound B (Figure 4b). This mechanism could form the basis for the antioxidant activity of Fer-1.
Figure 4.

SAR study of Fer-1. (a) General scheme for the synthesis of ferrostatins. (b) Fer-1 as a reducing agent: release of 2 protons and 2 electrons results in a formation of ethyl 4-(cyclohexylimino)-3-iminocyclohexa-1,5-dienecarboxylate intermediate B.
Figure 3.
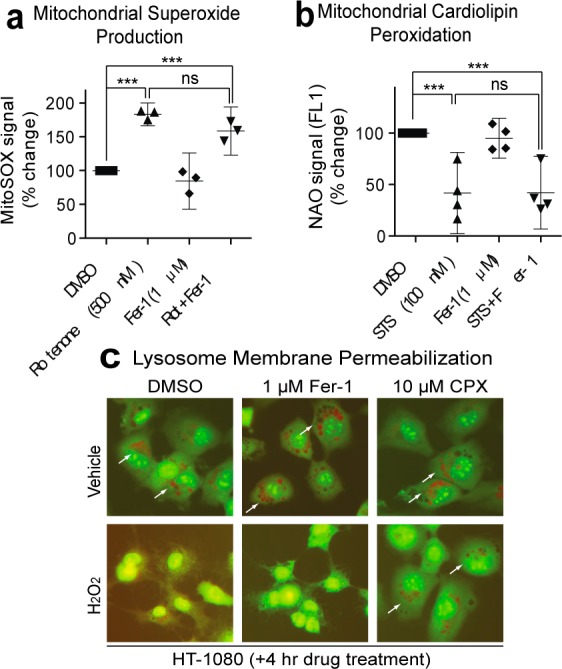
Fer-1 does not inhibit all forms of ROS production or ROS-induced death. (a) Mitochondrial ROS production in response to rotenone (Rot, 250 nM, 3 h) ± Fer-1 (1 μM) was detected using MitoSOX. (b) Cardiolipin peroxidation in response to staurosporine (STS,100 nM, 3 h) detected using 10-nonyl acridine orange (NAO). Data in (a) and (b) were analyzed by one-way ANOVA ***P < 0.001, ns = not significant; (c) Lysosomal membrane permeabilization detected in response to H2O2 using acridine orange (AO) relocalization. An iron chelator, ciclopirox olamine (CPX), protects from lysosomal rupture.
To test this hypothesis, we evaluated the 67 analogues for their ability to prevent erastin-(10 μM)-induced cell death4−6 (see Supplementary Synthesis Section in Supporting Information). Cell death inhibition (EC50) values were determined by treating HT-1080 fibrosarcoma cells with a lethal concentration of erastin (10 μM) in the presence of each ferrostatin analogue in a 10-point, 2-fold dilution series starting at 4 μg/mL, or a 20-point, 2-fold dilution starting at 2 μg/mL, for subsequently identified more potent compounds. These results are presented in Tables S2–S8 in the Supporting Information.
To assess the importance of the primary aromatic amine in the activity of ferrostatins, we designed mono- and disubstituted amines (Supporting Information Tables S5–S7). We hypothesized that if the above mechanism were correct; these modifications should decrease or abolish the ability of the compounds to prevent erastin-induced ferroptosis. In these studies, the primary aromatic amine of the ferrostatin compounds was used as a starting material for a reductive amination or alkylation reaction using arylaldehydes or 1-(bromomethyl)aryl compounds, respectively (Supporting Information Tables S5–S7). In addition, other electrophiles, such as acylchlorides, alkylchloroformates, or benzylchloroformates, were also used to generate ferrostatin derivatives efficiently (Supporting Information Table S5).
We observed that, first, all analogues with a nitro group in place of the amine were inactive (Supporting Information Table S8, entry 1 vs entry 2), confirming the concept that this amine is essential for suppression of ferroptosis. In addition, analogues lacking a hydrophobic cyclohexylamine moiety were not active (Supporting Information Table S8, entry 1 vs entries 3 and 4). Second, introduction of heteroatoms into the N-cyclohexyl moiety resulted in consistent reductions in potency, consistent with the hypothesis that the hydrophobicity of this portion of the scaffold is crucial for anchoring within lipophilic membrane environments. Third, modifications of the ethyl ester4 (Supporting Information Table S4 and Scheme S1), involving substantial extensions, were generally well-tolerated, and may serve as substrates for future probes. Fourth, both amines are essential for full activity; while there was no difference in potency between the nonalkylated and monomethylated analogues (Supporting Information Table S5, entry 6 vs entry 7), the N,N-dimethyl substituted derivative was 43-fold less potent than the mono-N-methyl substituted derivative (Supporting Information Table S5, entry 7 vs entry 8). Thus, it appears that the ability to oxidize both amines is crucial for the potency of ferrostatins (Figure 4b).
In addition, compounds with electron withdrawing groups, such as methyl-, benzyl-, or tert-butylcarbamates, as well as amides, block tautomerization, which results in the inhibition of the release of 2 protons and 2 electrons (Figure 4b). This mechanism was supported by the loss of the activity of these analogues (Supporting Information Table S5, entries 2–5). Indeed, a tertiary amine (dioctyl) derivative was 190-fold less active than a secondary amine (monooctyl) (Supporting Information Table S5, entry 9 vs entry 10). These data support our hypothesis that the activity of ferrostatins requires both secondary amines (Figure 4b). Fifth, we extended the reductive amination or the selective monoalkylation reaction of Fer-1 to aryl aldehydes and aryl halides, respectively (Supporting Information Tables S6 and S7). These aryl aldehydes and aryl halides were ortho, meta, or para mono- or disubstituted with electron withdrawing or electron donating groups (−Cl, −Br, −F, −CN, −CF3, −NO2, esters, or −OCH3). In addition, we also incorporated a nitrogen atom in the phenyl ring in the design of the pyridine and pyrimidine analogues of Fer-1 (Supporting Information Table S7). Overall, the introduction of these variations to the mono-alkylated analogues did not affect potency, suggesting that these modifications did not inhibit the electron delocalization and release of reducing equivalents into the cell. These analogues further support our hypothesized model of Fer-1’s antioxidant activity. Sixth and finally, we modified the aniline ring of the parent compound (Fer-1) and created additional Fer-1 analogues with (1) a nitrogen incorporated in the aniline ring (pyridine) (Supporting Information Scheme S2); and (2) with various substitutions (F, Cl, CH3) in the aniline ring (Supporting Information Schemes S3–S5). Overall, we observed a loss of potency with the pyridine moiety analogues (Supporting Information Scheme S2), while the aniline-substituted analogues have equivalent or better potency than the Fer-1 (Supporting Information Schemes S3–S5). Overall, this series of Fer-1 analogues provided insight into the Fer-1 mechanism of action and also allowed us to identify analogues with greater potency and improved properties. Indeed, we discovered a number of compounds (SRS11-92, SRS12-45, SRS13-35, and SRS13-37) that were more potent than Fer-1 and may be suitable for further translational studies (Figure 5a). For example, SRS11-92 was 15-fold more potent than the parent Fer-1, with an EC50 = 6 nM (Figure 5a, entry 3). An X-ray structure of SRS11-92 analogue was obtained (Figure 5b) to confirm its purity and the orientation of both secondary amines.
Figure 5.
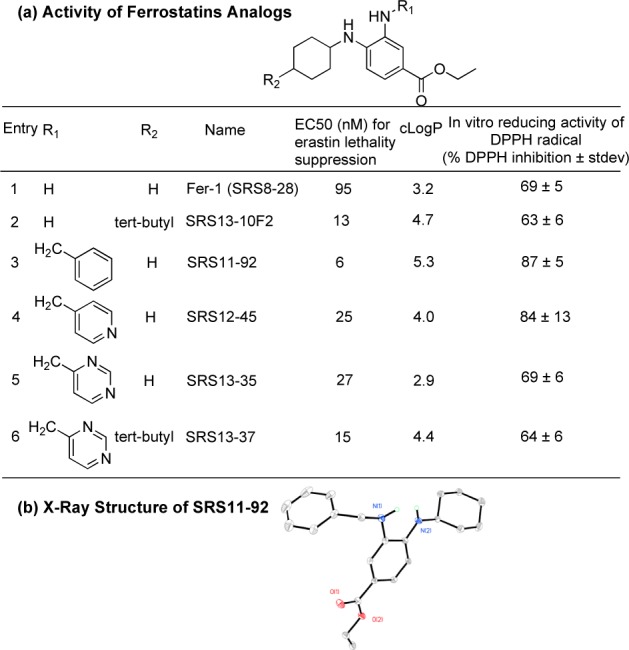
SAR study of Fer-1. (a) EC50, cLogP, and % DPPH inhibition of selected potent ferrostatins. DPPH: 2,2-diphenyl-1-picrylhydrazyl radical. (b) Xray structure of the most potent Fer-1 analogue (SRS11-92).
In parallel with the above cell-based assays, we used a 2,2- diphenyl-1-picrylhydrazyl (DPPH) reduction assay4,42−44 to examine the ability of each analogue to reduce this free radical in an in vitro test of intrinsic reducing (and hence antioxidant) capacity. The reduction of the DPPH radical by ferrostatins results in a decrease in absorbance, indicating the radical quenching capacity of the ferrostatins. Antioxidant active analogues scavenged the DPPH radical by 60–90% within 30 min (Figure 5a). Consistent with the notion that the lipophilic character of the N-cyclohexyl ring is essential for cell death inhibition in cells, but not intrinsic antioxidant activity,4 Fer-1 analogues bearing heteroatoms substitutions such as O- or N-methyl in this moiety retained antioxidant activity in the DPPH assay (68% and 86% reduction of DPPH, respectively, Supporting Information Figure S3), despite resulting in much lower potency in the HT-1080 death-suppression assay. Conversely, analogues with electron-withdrawing functional groups on the primary amine did not scavenge the DPPH radical (0–10%, Supporting Information Figure S3) and did not prevent cell death, suggesting that these groups act to prevent the delocalization and oxidation necessary for radical scavenging (Figure 4b).
Conclusion
In conclusion, the testing of ferrostatins, generated via an efficient three-step synthesis, has provided insights into the ferroptotic phenotype, confirming that it involves the depletion of specific membrane lipids, most likely due to oxidative destruction. As Fer-1 and analogues are protective against cell death in a brain slice model of HD, an oligodendrocyte model of PVL, and in isolated kidney proximal tubules that model kidney dysfunction, these results imply that these processes may converge upon or require oxidative destruction of specific PUFA-containing membrane lipids and might represent examples of ferroptotic cell death that occur during pathological situations. The ferrostatins represents useful probes with which to dissect ferroptosis in a variety of contexts and could form the basis of future drugs to combat lipid-peroxidation-mediated tissue injury in diverse diseases.
Acknowledgments
This work was funded by grants from the New York State Stem Cell Science (NYSTEM, Contract No. C026715 for the CPS Facility) and the US National Institute of Health (NIH R01CA097061, R01GM085081, R01 CA161061, HD18655, NS066019, and DK34275), the Whitehall Foundation, the William Randolph Hearst Foundation, Alzheimer's Drug Discovery Foundation, and the Baby Alex Foundation. B.R.S. is an Early Career Scientist of the Howard Hughes Medical Institute. S.J.D. was supported by a K99 Award from the National Cancer Institute (1K99CA166517-01). We acknowledge the assistance of J. Decatur, Y. Itagaki, G. Parkin, and J. Palmer, and NSF grants CHE 0840451 and CHE-06196, and NIH grant 1S10RR025431-01A1.
Supporting Information Available
Additional synthetic and characterization details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sena L. A.; Chandel N. S. Mol. Cell 2012, 48, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kell D. B. BMC Med. Genomics 2009, 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xun Z.; Xun Z.; Rivera-Sánchez S.; Ayala-Peña S.; Lim J.; Budworth H.; Skoda E. M.; Robbins P. D.; Niedernhofer L. J.; Wipf P.; McMurray C. T. Cell Rep. 2012, 2, 1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon S. J.; Lemberg K. M.; Lamprecht M. R.; Skouta R.; Zaitsev E. M.; Gleason C. E.; Patel D. N.; Bauer A. J.; Cantley A. M.; Yang W. S.; Morrison B.; Stockwell B. R. Cell 2012, 149, 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouta R.; Hayano M.; Shimada K.; Stockwell B. R. Bioorg. Med. Chem. Lett. 2012, 22, 5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. S.; Stockwell B. R. Chem. Biol. 2008, 15, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolma S.; Lessnick S. L.; Hahn W. C.; Stockwell B. R. Cancer Cell 2003, 3, 285. [DOI] [PubMed] [Google Scholar]
- Dixon S. J.; Stockwell B. R. Nat. Chem. Biol. 2014, 10, 9. [DOI] [PubMed] [Google Scholar]
- Finley J. W.; Kong A. N.; Hintze K. J.; Jeffery E. H.; Ji L. L.; Lei X. G. J. Agric. Food. Chem. 2011, 59, 6837. [DOI] [PubMed] [Google Scholar]
- Klemchuk P. P.Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar]
- Adeniyi J. B.; Al-Malika S.; Scott G. J. Appl. Polym. Sci. 1986, 32, 6063. [Google Scholar]
- Lye P. H.; Toh H. K. J. Polym. Sci., Polym. Lett. Ed. 1984, 22, 327. [Google Scholar]
- Shelton J. R. Rubber Rev. Rubber Chem. Technol. 1957, 30, 1270. [Google Scholar]
- Shelton J. R.; Cox W. L. Ind. Eng. Chem. 1953, 45, 392. [Google Scholar]
- Shelton J. R.; Vincent D. N. J. Am. Chem. Soc. 1963, 85, 2433. [Google Scholar]
- Ingold K. U. Chem. Rev. 1961, 61, 563. [Google Scholar]
- Brownlie I. T.; Ingold K. U. Can. J. Chem. 1966, 44, 861. [Google Scholar]
- Adamic K.; Ingold K. U. Can. J. Chem. 1969, 47, 295. [Google Scholar]
- Behl C. Prog. Neurobiol. 1999, 57, 301. [DOI] [PubMed] [Google Scholar]
- Behl C.; Moosmann B. Free Radical Biol. Med. 2002, 33, 182. [DOI] [PubMed] [Google Scholar]
- Gryn’ova G.; Ingold K. U.; Coote M. L. J. Am. Chem. Soc. 2012, 134, 12979. [DOI] [PubMed] [Google Scholar]
- Esteves M. A.; Narender N.; Marcelo-Curto M. J.; Gigante B. J. Nat. Prod. 2001, 64, 761. [DOI] [PubMed] [Google Scholar]
- Scott G. Bull. Chem. Soc. Jpn. 1988, 61, 165. [Google Scholar]
- Bannai S. J. Biol. Chem. 1986, 261, 2256. [PubMed] [Google Scholar]
- Murphy T. H.; Miyamoto M.; Sastre A.; Schnaar R. L.; Coyle J. T. Neuron 1989, 2, 1547. [DOI] [PubMed] [Google Scholar]
- Johnson W. M.; Wilson-Delfosse A. L.; Mieyal J. J. Nutrients 2012, 4, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Kosaras B.; Del Signore S. J.; Cormier K.; McKee A.; Ratan R. R.; Kowall N. W.; Ryu H. Acta Neuropathol. 2011, 121, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmaine M.; Raza A.; Mahal A.; Mangiarini L.; Bates G. P.; Davies S. W. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffstrom B. G.; Kaplan A.; Letso R.; Schmid R. S.; Turmel G. J.; Lo D. C.; Stockwell B. R. Nat. Chem. Biol. 2010, 6, 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe J. J.; Kinney H. C.; Jensen F. E.; Rosenberg P. A. Int. J. Dev. Neurosci. 2011, 29, 565. [DOI] [PubMed] [Google Scholar]
- Back S. A.; Luo N. L.; Mallinson R. A.; O’Malley J. P.; Wallen L. D.; Frei B.; Morrow J. D.; Petito C. K.; Roberts C. T. Jr.; Murdoch G. H.; Montine T. J. Ann. Neurol. 2005, 58, 108. [DOI] [PubMed] [Google Scholar]
- Inder T.; Mocatta T.; Darlow B.; Spencer C.; Volpe J. J.; Winterbourn C. Pediatr. Res. 2002, 52, 213. [DOI] [PubMed] [Google Scholar]
- Brault S.; Martinez-Bermudez A. K.; Roberts J. 2nd; Cui Q. L.; Fragoso G.; Hemdan S.; Liu H. N.; Gobeil F. Jr.; Quiniou C.; Kermorvant-Duchemin E.; Lachance C.; Almazan G.; Varma D. R.; Chemtob S. Free Radical Biol. Med. 2004, 37, 358. [DOI] [PubMed] [Google Scholar]
- Welin A. K.; Svedin P.; Lapatto R.; Sultan B.; Hagberg H.; Gressens P.; Kjellmer I.; Mallard C. Pediatr. Res. 2007, 61, 153. [DOI] [PubMed] [Google Scholar]
- Sogabe K.; Roeser N. F.; Venkatachalam M. A.; Weinberg J. M. Kidney Int. 1996, 50, 845. [DOI] [PubMed] [Google Scholar]
- Drummen G. P.; Gadella B. M.; Post J. A.; Brouwers J. F. Free Radical Biol. Med. 2004, 36, 1635. [DOI] [PubMed] [Google Scholar]
- Soga T.; Baran R.; Suematsu M.; Ueno Y.; Ikeda S.; Sakurakawa T.; Kakazu Y.; Ishikawa T.; Robert M.; Nishioka T.; Tomita M. J. Biol. Chem. 2006, 28, 16768. [DOI] [PubMed] [Google Scholar]
- Loscalzo J. Cell Metab. 2008, 8, 182. [DOI] [PubMed] [Google Scholar]
- Do T. Q.; Schultz J. R.; Clarke C. F. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu P. L.; Hache B.; Von Moos E. Synthesis 2003, 11, 1683. [Google Scholar]
- Abdel-Magid A. F.; Carson K. G.; Harris B. D.; Maryanoff C. A.; Shah R. D. J. Org. Chem. 1996, 61, 3849. [DOI] [PubMed] [Google Scholar]
- Lehuédé J.; Fauconneau B.; Barrier L.; Ourakow M.; Piriou A.; Vierfond J.-M. Eur. J. Med. Chem. 1999, 34, 991. [DOI] [PubMed] [Google Scholar]
- Okawa M.; Kinjo J.; Nohara T.; Ono M. Biol. Pharm. Bul. 2001, 24, 1202. [DOI] [PubMed] [Google Scholar]
- Huang D.; Ou B.; Prior R. L. J. Agric. Food. Chem. 2005, 53, 1841. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.