

Abstract
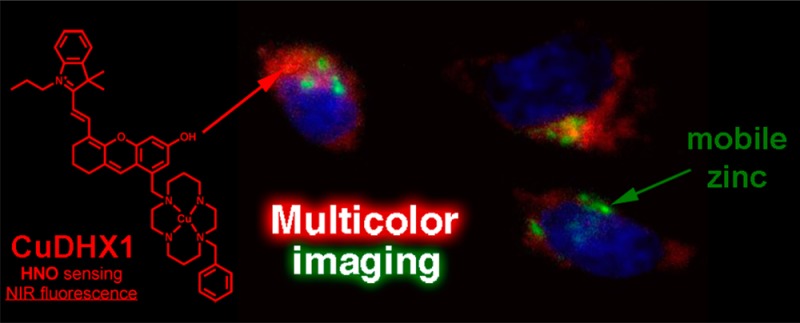
The first near-infrared fluorescent turn-on sensor for the detection of nitroxyl (HNO), the one-electron reduced form of nitric oxide (NO), is reported. The new copper-based probe, CuDHX1, contains a dihydroxanthene (DHX) fluorophore and a cyclam derivative as a Cu(II) binding site. Upon reaction with HNO, CuDHX1 displays a five-fold fluorescence turn-on in cuvettes and is selective for HNO over thiols and reactive nitrogen and oxygen species. CuDHX1 can detect exogenously applied HNO in live mammalian cells and in conjunction with the zinc-specific, green-fluorescent sensor ZP1 can perform multicolor/multianalyte microscopic imaging. These studies reveal that HNO treatment elicits an increase in the concentration of intracellular mobile zinc.
Introduction
Nitric oxide (NO) is an important biological signaling molecule present in the cardiovascular,1 immune,2 and nervous systems.3 NO is released by macrophages during the immune response,2 controls smooth muscle relaxation,4 and regulates neurotransmission.3,5 Dysregulation of NO homeostasis is associated with numerous diseases, including diabetes,6 stroke,7 Alzheimer’s disease,8 and cancer.9 HNO, the one-electron reduced and protonated derivative of NO, displays distinctive chemistry and biochemistry from that of NO.10 Exogenously applied HNO confers vasoprotective effects,11 increases heart muscle contractility,12 and inhibits platelet aggregation.13 HNO also exacerbates ischemia-related injury14 and induces neurotoxicity.15 Under biological conditions, HNO is both an electrophile that oxidizes thiols and a nucleophile that can coordinate and reduce metal ions.16 Thiolates that bind zinc are a potential biological target of HNO. Nitrosation of thiolates induces the release of mobile zinc,17 which may trigger a variety of signaling pathways.18 Understanding these and other downstream effects of exogenously applied HNO is essential if nitroxyl donors are to be used as therapeutic agents.19
Endogenous generation of HNO has not been directly observed in live organisms. Some proposed biosynthetic pathways are shown in Scheme 1. HNO can be produced by nitric oxide synthase (NOS). Under normal physiological conditions, NOS catalyzes the conversion of arginine to citrulline and NO in the presence of the cofactor tetrahydrobiopterin (THB).20,21 In the absence of THB, isolated neuronal NOS produces HNO instead of NO.21 Additionally, HNO can be generated by oxidation of hydroxylamine (NH2OH) with heme-containing proteins.14 NO and HNO interconvert in the presence of superoxide dismutase (SOD),22 and cytochrome c can reduce NO to HNO.23 Recently, HNO production was detected in the reaction between HSNO and H2S24 and in the heme-iron-catalyzed reduction of nitrite with H2S.25 These results suggest that HNO can be produced under biological conditions and might be associated with particular physiological or pathological states. Improved methods for detecting HNO selectively, with well-defined spatiotemporal resolution in live cells, tissues, and animals, are needed to fully understand the biology of nitroxyl.
Scheme 1. Possible Biosynthetic Pathways to HNO.

l-NHA = N-hydroxy-l-arginine; nNOS = neuronal nitric oxide synthase; SOD = superoxide dismutase; THB = tetrahydrobiopterin.
A number of fluorescent sensors that detect HNO in live cells are available (Chart 1). One approach to HNO sensing relies on the use of fluorophores functionalized with amines that bind Cu(II). The fluorescence of these sensors is quenched upon binding of the paramagnetic cupric ion. Upon exposure to HNO, Cu(II) reduces to Cu(I), which renders the complex diamagnetic and restores the fluorescence of the probe. Sensors based on this principle include BODIPY-based complex CuBOT1 (Chart 1),26,27 coumarin-based derivative CuCOT1 (Chart 1),28 and the CuBRNO series of benzoresorufin-based sensors (Chart 1) that detect both HNO and NO.29 Recently, the metal-free probe P-Rhod (Chart 1) was reported.30 This sensor exploits the reactivity of HNO with triphenylphosphine to produce an azaylide, which undergoes an intramolecular nucleophilic attack that releases rhodol, a bright green fluorophore. Despite their value, these probes have shortcomings associated with their lack of selectivity or short emission wavelengths. It would be valuable to have a sensor that not only detects HNO with high selectivity but would also emit in the near-infrared (NIR) region, desired for in vivo imaging because of deeper tissue penetration.31 Moreover, sensors with narrow emission profiles at long wavelengths can be used together with visible-light probes in multicolor microscopy experiments.32,33 In the present article we describe the design, synthesis, and implementation of CuDHX1 (Chart 1), a novel sensor that satisfies all of these requirements.
Chart 1. Chemical Structures and Photophysical Properties of Some Previously Reported and Newly Developed (CuDHX1) Probes for HNO26−30.

The structure of CuDHX1 comprises a cyclam binding site for Cu(II) and a dihydroxanthene (DHX) NIR fluorophore. We chose cyclam because Cu(II) complexes of this ligand react very slowly with the interfering species H2S and glutathione (GSH) under physiological conditions.34 The DHX fluorophore was selected because it is a bright NIR emitter with excellent biocompatibility.35 Here we report the results of our studies using CuDHX1 to image HNO in live cells as well as multicolor imaging experiments to investigate the ability of Angeli’s salt, an HNO donor, to affect the levels of mobile zinc in live cells.
Experimental Section
General Methods
All reactions were performed under a nitrogen atmosphere unless otherwise specified. ZP1 was prepared as previously described.36 Reagents were purchased from commercial sources and used as received. Solvents were purified and degassed by standard procedures. Nitric oxide was passed through an Ascarite column and a 6 ft coil containing silica gel at −78 °C to remove impurities and then collected and stored under nitrogen in a gas storage bulb. NMR spectra were acquired on a Varian Inova-500 or a Varian Mercury-300 instrument. 1H NMR chemical shifts are reported in ppm relative to SiMe4 (δ = 0) and were referenced internally with respect to residual protons in the solvent (δ = 3.31 for CD3OD or δ = 2.50 for DMSO-d6). Coupling constants are reported in Hz. 13C NMR chemical shifts are reported in ppm relative to SiMe4 (δ = 0) and were referenced internally with respect to solvent signal (δ = 39.51 for DMSO-d6). 19F NMR chemical shifts are reported in ppm relative to CFCl3 (δ = 0) and were referenced internally with respect to 2,2,2-trifluoroethanol (δ = −77.03). Low-resolution mass spectra (LRMS) were acquired on an Agilent 1100 Series LC/MSD Trap spectrometer (LCMS), using electrospray ionization (ESI). High-resolution mass spectrometry (HR-ESI-MS) was conducted by staff at the MIT Department of Chemistry Instrumentation Facility on a Bruker Daltonics APEXIV 4.7 T FT-ICR-MS instrument. Silica gel 60 321 (0.015–0.040 mm) was used for flash column chromatography. Semipreparative HPLC separations were carried out on an Agilent 1200 HPLC instrument with a multiwavelength detector and automated fraction collector using a C18 reverse stationary phase (Zorbax-SB C18, 5 μm, 9.5 × 250 mm) and a mobile phase composed of two solvents (A: 0.1% (v/v) trifluoroacetic acid (TFA) in H2O; B: 0.1% (v/v) TFA in CH3CN). Specific purification protocols are described below for each compound. IUPAC names of all compounds are provided and were determined using CS ChemBioDrawUltra 12.0.
Synthesis of (E)-2-(2-(6-hydroxymethyl)-2,3-dihydro-1H-xanthen-4-yl)vinyl)-3,3-dimethyl-1-propyl-3H-indol-1-ium iodide (1)
A solution of IR780 (300 mg, 0.45 mmol) in dry DMF (5 mL) was added to 5-(hydroxymethyl)benzene-1,3-diol (63 mg, 0.45 mmol). Triethylamine (0.6 mL, 4.49 mmol) was added, and the mixture was stirred at 110 °C for 30 min. The solvent was evaporated under reduced pressure and the solid was purified by column chromatography (SiO2; CH2Cl2/CH3OH 9:1) to give the product as a dark blue solid (195 mg, yield 76%). 1H NMR (300 MHz, CD3OD): 1.04 (t, 3J = 7.4 Hz, 3H), 1.80 (s, 6H), 1.89–1.96 (m, 4H), 2.69–2.82 (m, 4H), 4.26 (t, 3J = 7.2 Hz), 4.79 (s, 2H), 6.40 (d, 3J = 14.7 Hz, 1H), 6.74 (d, 4J = 2 Hz, 1H), 6.91 (d, 4J = 2 Hz, 1H), 7.36–7.41 (m, 1H), 7.45–7.49 (m, 2H), 7.60–7.63 (m, 1H), 7.70 (s, 1H), 8.69 (d, 3J = 14.5 Hz, 1H). LRMS (ESI). Calcd for [C29H32NO3]+: 442.2, found 442.2.
Synthesis of (E)-2-(2-(8-(chloromethyl)-6-hydroxy-2,3-dihydro-1H-xanthen-4-yl)vinyl)-3,3-dimethyl-1-propyl-3H-indol-ium iodide (2)
Thionyl chloride (75 μL, 1.03 mmol) and dry pyridine (83 μL, 1.03 mmol) were dissolved in dry CH2Cl2 (1 mL) and cooled to 0 °C. Compound 1 (195 mg, 0.34 mmol) was dissolved in dry CH2Cl2 (1 mL) and dry DMF (0.1 mL) and added slowly to the mixture of pyridine and thionyl chloride. After 30 min, H2O (0.1 mL) was added, and the mixture was dried with Na2SO4. The solvent was evaporated under reduced pressure. The dark blue solid was dissolved in CH2Cl2/CH3OH (9:1) and filtered through a plug of SiO2 with CH2Cl2/CH3OH (9:1, 200 mL). The solvent was evaporated, and the crude product was used immediately. LRMS (ESI). Calcd for [C29H31ClNO2]+: 460.2, found 460.1.
Synthesis of (E)-2-(2-(8-((1,4,8,11-tetraazacyclotetradecan-1-yl)methyl)-6-hydroxy-2,3-dihydro-1H-xanthen-4-yl)vinyl)-3,3-dimethyl-1-propyl-3H-indol-1-ium trifluoroacetate (3)
A solution of crude compound 2 in dry CH3CN (3 mL) was added to 1,4,8,11-tetraazacyclotetradecane (cyclam, 136 mg, 0.68 mmol). Diisopropylethylamine (0.12 mL, 0.68 mmol) was added, and the mixture was heated to reflux. After 30 min, the solvent was evaporated, and the residue was purified by RP-HPLC according to the following protocol: constant flow rate 3 mL min–1; isocratic flow 2% B, 0–5 min; gradient, 35–95% B, 10–25 min. The product was collected between 16.5–16.8 min. All equivalent fractions recovered from independent runs were combined and lyophilized to dryness to yield the TFA salt of compound 3 (46 mg, 18% over two steps). Mp: 110–120 °C. 1H NMR (500 MHz, DMSO-d6): 0.96 (t, 3J = 7.4 Hz, 3H), 1.72 (s, 7H), 1.81 (m, 7H), 2.67 (m, 10H), 3.10 (m, 10H), 3.80 (s, 2H), 4.35 (t, 3J = 7.1 Hz, 2H), 6.53 (d, 3J = 15 Hz, 1H), 6.92 (m, 1H), 7.42, (t, 3J = 7.7 Hz, 1H), 7.51 (t, 3J = 8.3 Hz, 1H), 7.66 (d, 3J = 8.0 Hz, 1H), 7.72 (d, 3J = 7.7 Hz, 2H), 8.55 (d, 3J = 15 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): 11.08, 20.10, 20.92, 23.60, 27.59, 28.55, 40.44, 45.95, 49.56, 50.24, 101.59, 103.73, 113.12, 113.38, 113.71, 115.75, 115.96, 118.11, 120.48, 122.76, 125.85, 126.85, 127.96, 128.97, 136.75, 141.65, 141.88, 144.50, 154.73, 158.13, 158.42, 158.65, 158.90, 160.62, 161.12, 177.01. 19F NMR (282 MHz, CD3OD): −75.31. HR-ESI-MS. Calcd for [C39H54N5O2]+: 624.4273, found: 624.4259.
Synthesis of (E)-2-(2-(8-((8-benzyl-1,4,8,11-tetraazacyclotetradecan-1-yl)methyl)-6-hydroxy-2,3-dihydro-1H-xanthen-4-yl)vinyl)-3,3-dimethyl-1-propyl-3H-indol-1-ium trifluoroacetate (DHX1)
A solution of compound 3 (46 mg, 0.06 mmol) in CH3CN (3 mL) and (bromomethyl)benzene (4 μL, 0.03 mmol) were combined. Diisopropylethylamine (22 μL, 0.12 mmol) was added, and the reaction mixture stirred at room temperature for 2 h. The solvent was evaporated, and the product was purified by RP-HPLC according to the following protocol: constant flow rate 3 mL min–1; isocratic flow 2% B, 0–5 min; gradient, 35–95% B, 10–25 min. The product was collected between 18.1–18.5 min. All equivalent fractions recovered from independent runs were combined and lyophilized to dryness to yield the TFA salt of compound DHX1 (14.2 mg, 28%). Mp: 115–120 °C. 1H NMR (500 MHz, DMSO-d6): 0.97 (t, 3J = 7 Hz, 3H), 1.72 (s, 6H), 1.80 (m, 6H), 2.06 (m, 2H), 2.66 (m, 3H), 2.72 (m, 5H), 3.14 (m, 8H), 3.83 (s, 3H), 4.35 (t, 3J = 7 Hz, 2H), 6.52 (d, 3J = 15 Hz, 1H), 6.91 (m, 1H), 7.00 (m, 1H), 7.40 (m, 6H), 7.50 (t, 3J = 7, 1H), 7.66 (d, 3J = 5 Hz, 1H), 7.72 (m, 2H), 8.54 (d, 3J = 15 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): 11.09, 20.09, 20.89, 23.60, 27.57, 27.62, 28.50, 40.43, 45.91, 50.20, 50.24, 53.71, 57.10, 101.27, 103.55, 113.07, 113.32, 113.52, 113.69, 113.85, 115.69, 116.11, 118.06, 120.42, 122.76, 125.63, 126.78, 128.29, 128.44, 128.93, 130.06, 130.37, 131.40, 141.64, 141.85, 144.41, 154.66, 158.02, 158.04, 158.51, 158.81, 160.80, 161.29, 176.87. 19F NMR (282 MHz, CD3OD): −75.18. HR-ESI-MS. Calcd for [C46H60N5O2]+: 714.4742, found: 714.4758.
Spectroscopic Methods
All aqueous solutions were prepared using deionized water with resistivity 18.2 mΩ cm–1, obtained using a Milli-Q water purification system. All solvents were supplied by Aldrich and used as received. Piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) and 99.999% KCl were purchased from Calbiochem. Stock solutions of compound 3 and ligand DHX1 in DMSO were prepared in the 1–8 mM range and stored at −20 °C in 1 mL aliquots and thawed immediately before each experiment. The copper complexes were prepared by dissolving the ligand in 1 mL of CH3OH, adding 1 equiv of CuCl2 to each solution, and stirring overnight. The solvent was evaporated, and the resulting sensor was redissolved in 1 mL DMSO and stored at −20 °C. All spectroscopic measurements were conducted in aqueous buffer containing 50 mM PIPES (pH 7.0) and 100 mM KCl, with the exception of those performed in CH3OH and at varying pH values. UV–vis spectra were acquired using a Cary 50 spectrometer using quartz cuvettes from Starna (1 cm path length). Fluorescence spectra were acquired on a Photon Technology International fluorimeter. All measurements were conducted on solutions maintained at 25 °C by circulating water baths. Extinction coefficients of the ligands and sensors were determined by using 1–3 μM solutions in aqueous buffer. Fluorescence quantum yields were determined using the same solutions, λex = 650 nm. Fluorescence emission spectra were integrated from 660 to 900 nm. Quantum yields were referenced to IR780, which has a reported quantum yield of 0.076 in CH3OH, λex = 725 nm.37
Anaerobic Sample Preparation
Degassed aliquots of the stock solutions of each sensor were brought into an anaerobic chamber under a nitrogen atmosphere (O2 < 1 ppm) dedicated to work with aqueous solutions (hereafter called the “wet box”). Solutions containing 5 μM Cu-3 and 2 μM CuDHX1 were prepared using 2 mL of either degassed aqueous buffer or CH3OH in gastight cuvettes. Angeli’s salt (Na2N2O3, Cayman Chemical) was used as the source of HNO, because it decomposes rapidly (t1/2 = 3 min) at pH = 7 to produce HNO and NaNO2.38 Solutions of Angeli’s salt (4 mM) were prepared in the wet box in degassed 10 mM NaOH (2 mL) and brought out of the wet box in a gastight syringe. NO gas was removed from the wet box in a gastight syringe and injected into the headspace of each gastight cuvette before measuring fluorescence.
Cyclic Voltammetry
Cyclic voltammograms were measured in a three-electrode cell with a 2.0 mm diameter glassy carbon working electrode, a platinum auxiliary electrode, and Ag/Ag+ pseudoreference electrode in acetonitrile. The solvent contained 0.1 M n-Bu4NPF6 as the supporting electrolyte. The measurements were performed at room temperature with a VersaSTAT3 potentiostat from Princeton Applied Research operated with V3 studio software. Measurements were carried out at a scan rate of 200 mV s–1 on quiescent solutions that had been sparged with N2 for 5 min. All data were referenced to the Fc/Fc+ couple as an internal standard.
Electron Paramagnetic Resonance (EPR) Spectroscopy
Low-temperature X-band EPR spectra (77 K, 9 GHz) were collected with a Bruker EMS spectrometer equipped with an ER 4199HS cavity and a Gunn diode microwave source. EPR samples were prepared anaerobically. Solid DHX1 and 3 were brought into a glovebox. 0.8 equiv of Cu(MeCN)4BF4 were added to 400 μM DHX1 or 3 in 350 μL CH3OH, stirred overnight, and brought out of the glovebox in sealed EPR tubes. Angeli’s salt (100 equiv) was prepared in 10 mM NaOH anaerobically and brought out of the wet box in a gastight syringe. When CuCl2 was used to prepare CuDHX1 for the HNO reactivity EPR studies, different results were observed (Figure S21). For the NO reactivity test, degassed aliquots from the stock solution of CuDHX1 were brought into a wet box. Solutions containing 400 μM CuDHX1 were prepared in 350 μL of degassed CH3OH and brought out of the wet box in sealed EPR tubes. NO gas was taken out of the wet box in a gastight syringe and injected into the headspace of the EPR tube. Spectra were simulated in Matlab using the solid-state/frozen-solution functionality (‘pepper’) implemented in EasySpin.39
Analyte Selectivity Studies
Selectivity of the fluorescence turn-on toward biologically relevant analytes was determined by comparing the fluorescence emission spectra of a 2 μM solution of CuDHX1 in aqueous buffer at pH = 7, before and after treatment with 100 equiv of CaCl2, MgCl2, NaCl, ZnCl2, KNO3, NaNO2, KO2, H2O2, NaClO, sodium ascorbate, NaONOO, l-(+)-cysteine hydrochloride, gluthathione, methionine, Na2S, or Angeli’s salt. NO gas, 5000 equiv, and 100 μL of 10 mM NaOH (solvent of Angeli’s salt solutions) were also tested. In each case, the response was quantified by integrating the emission intensity from 660 to 900 nm and normalized to that of 2 μM CuDHX1 in aqueous buffer. For the NO and HNO selectivity studies, the samples were prepared anaerobically. For the NO, HNO, and Na2S studies, fluorescence spectra were acquired every 1 min for 10 min. For the remaining analytes, the fluorescence spectra were recorded at 0, 5, and 10 min. In addition, the fluorescence turn-on of 2 μM CuDHX1 in the presence of 100 equiv of KO2 and Angeli’s salt was measured in CH3CN. To determine the effect of pH on the fluorescence emission of CuDHX1, 2 μM solutions of CuDHX1 were prepared anaerobically in aqueous buffer (either 50 mM MES, 100 mM KCl; pH 4 and 5 or 50 mM PIPES, 100 mM KCl; pH 6, 7, and 8), and fluorescence spectra were recorded before and after addition of 100 equiv of Angeli’s salt.
Cell Culture and Staining Procedures
HeLa cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Cellgro, MediaTec, Inc.), supplemented with 10% fetal bovine serum (FBS; HyClone), 1% penicillin-streptomycin, 1% sodium pyruvate, and 1% l-glutamine. The cells were grown to 90% confluence at 37 °C with 5% CO2 before being passed and plated onto poly-d-lysine-coated plates 48 h before imaging. All cells were used between passage number 5 and 15. Imaging was conducted when plates reached 50–70% confluence. The growth medium was replaced with phosphate-buffered saline (PBS) containing 5 μM CuDHX1 and 3 μM Hoechst 33528 dye, and the cells were incubated for 15 min. Cells were rinsed with PBS (2 × 2 mL) followed by addition of fresh PBS (2 mL) and mounted on the microscope. All cell imaging experiments involving addition of HNO were carried out in PBS because addition of Angeli’s salt to plates containing DMEM did not lead to any turn-on response (Figure S23), presumably because of reaction of Angeli’s salt with cysteine or other components of DMEM. For cell imaging experiments with ZP1, the growth medium was replaced with dye-free DMEM containing 5 μM ZP1, 3 μM Hoechst 33528 dye, and the cells were incubated for 1 h. Cells were rinsed with PBS (2 × 2 mL) before addition of fresh PBS containing 5 μM CuDHX1 and incubated for 15 min. Cells were rinsed with PBS (2 × 2 mL) followed by addition of fresh PBS (2 mL) and mounted on the microscope.
Fluorescence Microscopy
Imaging experiments were performed using a Zeiss Axiovert 200 M inverted epifluorescence microscope equipped with an EM-CCD digital camera (Hamamatsu) and a MS200 XY Piezo Z stage (Applied Scientific Instruments). The light source was an X-Cite 120 metal-halide lamp (EXFO), and the fluorescence images were obtained using an oil-immersion objective at 63× or 100× magnification. The filters sets used are defined as blue: excitation G 365 nm, beamsplitter FT 395 nm, emission BP 445/50 nm; green: excitation BP 470/40 nm, beamsplitter FT 495 nm, emission 525/50 nm; NIR: excitation HQ 650/45 nm, beamsplitter Q 680 nm, emission HQ 710/50 nm. The microscope was operated using Volocity software (Perkin-Elmer).
The exposure times for acquisition of fluorescence images were kept constant for each series of images at each channel. To measure analyte-induced fluorescence changes, a solution of Angeli’s salt, Na2S, or S-nitrosoglutathione was added to the plate on the microscope stage to reach a concentration of 1.5 mM, and images were taken immediately after addition and after 5 min. Mobilization of intracellular zinc was induced by treating the cells with 3 mM Angeli’s salt and the released Zn2+ was chelated by bathing the cells in a solution of fresh PBS containing 50 μM N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN). Quantification of fluorescence intensity was performed using ImageJ (version 1.45, NIH). The whole cell was selected as the region of interest, and the integrated fluorescence from the background region was subtracted from the cell body region.
Results and Discussion
Synthesis and Photophysical Properties
Scheme 2 summarizes the synthesis of ligand DHX1. 5-(Hydroxymethyl)benzene-1,2-diol was allowed to react with IR780, a commercially available tricarbocyanine dye, to produce compound 1.35 Alcohol 1 was transformed into chloride 2 with thionyl chloride. Compound 2 was used immediately in a nucleophilic substitution reaction with 1,4,8,11-tetraazacyclotetradecane (cyclam) to give compound 3. This product was obtained after purification by RP-HPLC using a mobile phase containing 0.1% TFA and isolated as the trifluoroacetate salt, as ascertained by 13C and 19F NMR spectroscopy. Compound 3 was alkylated with benzyl bromide to produce DHX1, which was purified by RP-HPLC and isolated as the trifluoroacetate salt of the trans (1,8-substituted cyclam) isomer. Small amounts of other isomers were recovered after HPLC but not further characterized. The purity of both 3 and DHX1 was determined to be >95% by analytical HPLC analysis (Figure S6).
Scheme 2. Synthesis of 3 and DHX1.

Reagents and conditions: (a) Et3N, DMF, 110 °C, 20 min, 78%; (b) SOCl2, pyridine, CH2Cl2, DMF, 0 °C, 30 min; (c) Cyclam, DIPEA, CH3CN, reflux, 30 min, 18% (over steps b and c); (d) Benzyl bromide, DIPEA, CH3CN, 25 °C, 2 h, 28%. Counterions are omitted for clarity. DIPEA = diisopropylethylamine.
Compounds 3 and DHX1 were treated with CuCl2 to form their Cu(II) complexes. ESI-MS analysis revealed that under these conditions the complexes Cu-3 and CuDHX1 were formed with a coordinated trifluoroacetate ligand with m/z = 799.33 and 889.38, respectively.
The photophysical properties of 3 (Figure S12) and DHX1, as well as their Cu(II) complexes, as measured in buffered aqueous solution (50 mM PIPES, 100 mM KCl, pH = 7) are summarized in Table 1. The absorption spectrum of DHX1 is broad with a maximum at 693 nm (Figure 1). The absorbance of the Cu(II) complex is also broad and, in addition to a maximum at 693 nm, contains a peak at 650 nm. Excitation at 650 nm gives a fluorescence spectrum with a maximum at 715 nm that extends across the NIR region (Figure 1). The brightness (εϕ) of DHX1 is 1.1 × 103 M–1 cm–1. This value is comparable to that of Indocyanine Green (1.1 × 103 M–1 cm–1),40 a benchmark NIR fluorophore for in vivo imaging,41 indicating that the photophysical properties of DHX1 are suitable for in vivo applications. The quantum yield of this compound is decreased by more than 1 order of magnitude upon binding of Cu(II), a consequence of paramagnetic quenching induced by the transition metal.
Table 1. Summary of Photophysical Properties of Compounds 3 and DHX1.
| absorption: λmax (nm); ε (cm–1 M–1) |
emission: λmax (nm); φ |
|||
|---|---|---|---|---|
| ligand | Cu complex | ligand | Cu complex | |
| 3 | 693; 2.7(1) × 104 | 693; 3.3(2) × 104 | 715; 0.059(2) | 715; 0.0050(4) |
| DHX1 | 693; 2.3(1) × 104 | 693; 2.9(1) × 104 | 715; 0.048(3) | 715; 0.0027(1) |
Figure 1.
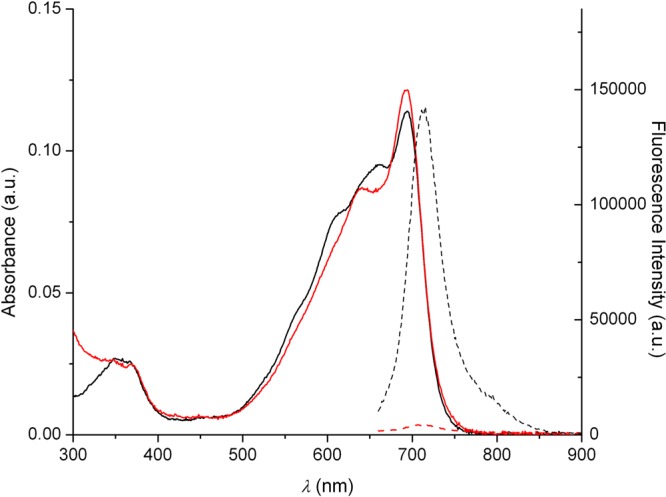
Fluorescence (dotted lines) and absorbance (solid lines) spectra of DHX1 (black lines) and CuDHX1 (red lines) in aqueous buffer (50 mM PIPES, 100 mM KCl, pH = 7).
Analyte Selectivity Studies
The ability of a molecule to sense a particular analyte in a complex biological sample depends on the specificity of its chemical reactions. We tested the reactivity of Cu-3 and CuDHX1 toward HNO, the analyte of interest, and other species that are present in live cells and can potentially interfere with nitroxyl sensing. All reactivity tests were performed in aqueous buffer (50 mM PIPES, 100 mM KCl, pH = 7). Samples of Cu-3 and CuDHX1 were prepared anaerobically to ensure that the sensors react directly with HNO or NO and not with an oxidation product. Cu-3 displayed no change in fluorescence intensity upon addition of NO gas and showed only a very small fluorescence increase upon addition of Angeli’s salt, an HNO donor (Figure S11). CuDHX1, however, reacted rapidly and selectively with HNO. Upon addition of 100 equiv of Angeli’s salt, CuDHX1 underwent a ∼5-fold increase in fluorescence intensity (Figure 2) after only 2 min (Figure S13). We determined 50 equiv to be the minimum amount of Angeli’s salt needed to induce turn-on of 2 μM CuDHX1 in aqueous buffer at pH = 7. It is difficult to estimate the effective concentration of HNO in a solution of Angeli’s salt because of the high reactivity of nitroxyl.42 We therefore can only conclude that the detection limit of HNO by CuDHX1 is ≤50 equiv.
Figure 2.
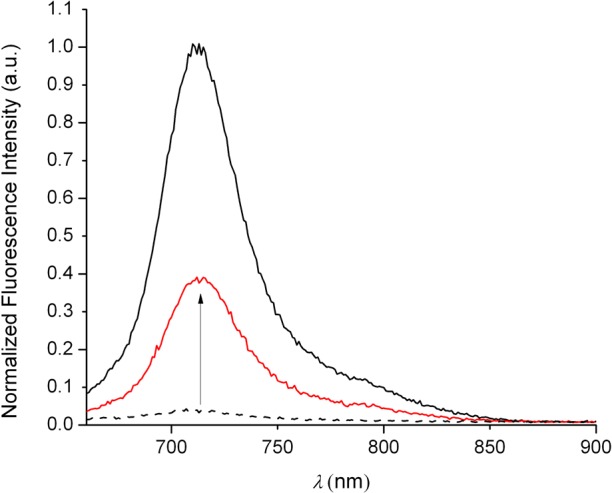
Fluorescence spectra of 2 μM CuDHX1 (black dashed line) in aqueous buffer (50 mM PIPES, 100 mM KCl, pH = 7) and 2 min after the addition of 100 equiv of Angeli’s salt (red solid line). The black solid line is the spectrum of ligand DHX1. λex: 650 nm.
Addition of 5000 equiv of NO resulted in no detectable change in fluorescence intensity, demonstrating that CuDHX1 detects HNO selectively over NO (Figure 3). The reaction of CuDHX1 with HNO did not restore the fluorescence to that of the free ligand (Figure 2). Moreover, after 2 min, the fluorescence intensity decreased slowly (Figure S13). Upon reaction of ligand DHX1 with Angeli’s salt, there was a decrease in fluorescence intensity (Figure S14), indicating that one of the products of the decomposition of the salt reacts with DHX1 to diminish its emission in buffered solution.
Figure 3.

Normalized integrated fluorescence intensity (660–900 nm) of 2 μM CuDHX1 in aqueous buffer (50 mM PIPES, 100 mM KCl, pH = 7) and 10 min after addition of 100 equiv of the analyte or 2 min after addition of 100 equiv of HNO. λex: 650 nm.
Angeli’s salt, which decomposes readily (t1/2 = 3 min)38 at pH = 7 to give HNO and NaNO2, is usually handled as an aqueous solution at pH = 12. To verify that HNO is responsible for the observed fluorescence increase, the reactivity of CuDHX1 toward NaNO2 (100 equiv) and 10 mM NaOH (100 μL) was evaluated. No increase in fluorescence intensity was observed upon reaction of CuDHX1 with either 100 equiv of NaNO2 or 10 mM NaOH in aqueous buffer (Figure 3). Additionally, no turn-on was induced when 100 equiv of CaCl2, MgCl2, NaCl, ZnCl2, KNO3, H2O2, NaClO, sodium ascorbate, NaONOO, l-(+)-cysteine, GSH, or methionine were added to a solution of CuDHX1 (Figure 3). CuDHX1 is selective for HNO over superoxide (KO2) both in buffer (Figure 3) and in CH3CN (Figure S16). The only analyte that promoted some detectable fluorescence enhancement after 10 min was H2S (Figure 3), which elicited a much slower turn-on compared to that induced by HNO. This selectivity is remarkable in view of the susceptibility of some Cu-based sensors toward reducing thiols26−28 and makes CuDHX1 a much more valuable sensor for use in biological samples where thiols are abundant. In addition, the fluorescence of CuDHX1 was measured at different pH values. A blue shift and decrease in intensity was observed at pH < 6, consistent with protonation of the hydroxyl group on the fluorophore (pKa ∼5.6).35 At low pH values, HNO-induced turn-on was considerably smaller than at neutral pH (Figure S17).
Mechanistic Investigation
Cyclic voltammetric studies of CuDHX1 and Cu-3 revealed quasi-reversible Cu(II)/Cu(I) reductions at 370 and 325 mV (vs Fc/Fc+), respectively (Figures S18 and S19). The reduction of NO to give HNO at pH = 7 occurs at −0.11 V (vs NHE,)43 and the reduction potential of the Cu(II)/Cu(I) couple in CuDHX1 is 1.01 V (vs NHE). These values indicate that HNO is thermodynamically able to reduce CuDHX1. Reduction of the nitrosonium cation (NO+) to NO occurs at 1.52 V (vs NHE),44 and consequently NO cannot reduce Cu(II) in CuDHX1. These data explain the selectivity of CuDHX1 for HNO over NO.
ESI-MS studies in buffer (50 mM PIPES, 100 mM KCl, pH = 7) revealed that both CuDHX1 and Cu-3 exist as mixtures of complexes with Cu(II) bound to trifluoroacetate or chloride as fifth ligand. Addition of 100 equiv of Angeli’s salt to CuDHX1 or Cu-3 resulted in loss of the copper ion to give primarily the m/z of ligands DHX1 and 3, respectively (Figures S9 and S10). In the case of Cu-3, the m/z of the copper complex without the fifth ligand was also detected (Figure S10).
The X-band EPR spectrum of CuDHX1, measured at 77 K in CH3OH, revealed a rhombic signal (Figure 4), which disappeared upon treatment with 100 equiv of Angeli’s salt, as expected for reduction of Cu(II) to Cu(I). In contrast, addition of 5000 equiv of NO gas to a solution of CuDHX1 in CH3OH did not evoke a noticeable change in the EPR spectrum (Figure 4). The reduced form of CuDHX1 could be reoxidized by allowing air into the EPR tube, giving a more axially symmetric signal; simulated traces are provided in Figure S20. Notably, we could not observe an EPR-silent species after reaction of Cu-3 with Angeli’s salt (Figure S22). This observation suggests that, if Cu(I)-3 is formed, it converts immediately to Cu(II), even under anaerobic conditions. The nature of the species responsible for this reoxidized copper material was not further investigated.
Figure 4.
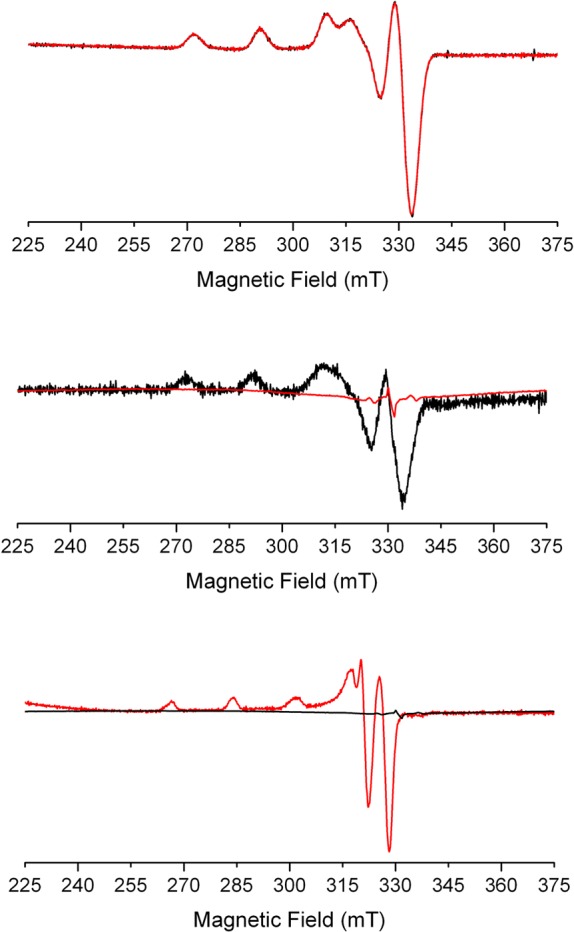
X-band EPR spectra of 400 μM CuDHX1 in CH3OH. Top: CuDHX1 before (black line) and after addition of 5000 equiv of NO (red line). Middle: CuDHX1 before (black line) and after (red line) addition of 100 equiv of Angeli’s salt. Bottom: CuDHX1 after reduction by HNO (black line) and after reoxidation by air (red line). Collection parameters: temperature, 77 K; modulation amplitude, 20 G; microwave power, 0.2 mW at 9.23 GHz.
Live Cell Imaging
We assessed the ability of CuDHX1 to detect HNO in live cells. Human cervical cancer (HeLa) cells were incubated with 5 μM CuDHX1 and 3 μM Hoechst 33528 nuclear stain in PBS for 15 min at 37 °C. Fluorescence microscopy images revealed only faint fluorescence in the NIR channel (Figure 5C), consistent with the low brightness of CuDHX1. Treatment with 1.5 mM Angeli’s salt resulted in a ∼3-fold increase in fluorescence intensity after only 5 min to restore a bright signal in the NIR channel (Figure 5D).
Figure 5.
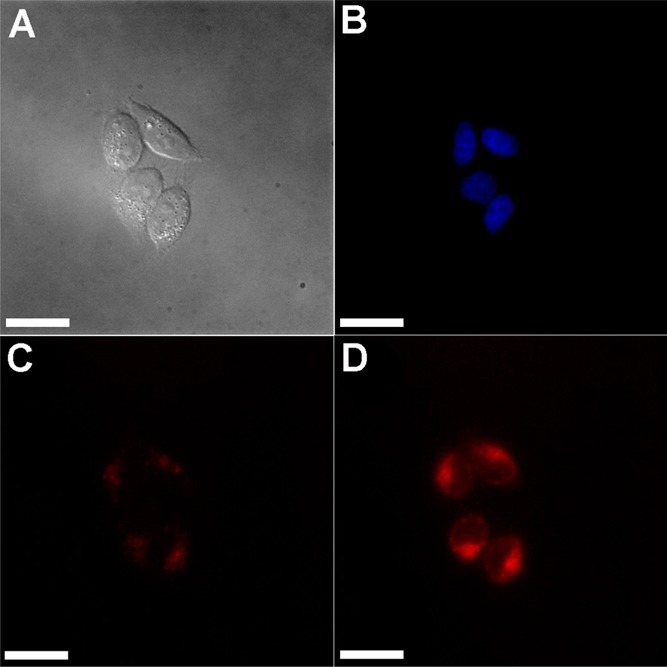
Fluorescence microscopy images of cells incubated with CuDHX1 in PBS before and after addition of Angeli’s salt: (A) Differential interference contrast (DIC) image; (B) blue channel showing nuclei; (C) NIR channel before addition of Angeli’s salt; and (D) NIR channel 5 min after treatment with 1.5 mM Angeli’s salt. Scale bar =25 μm.
Time-lapsed microscopy experiments indicated that, in the absence of HNO, CuDHX1 displays only a slight turn-on over time in live cells (Figure S24), significantly less than that induced by HNO. Treatment of HeLa cells with GSNO, an NO donor, did not elicit any detectable fluorescence turn-on (Figure S25), demonstrating that the sensor is selective for HNO over NO and S-nitrosothiols in live cells. In addition, incubation of live cells with Na2S resulted in only a very small increase in fluorescence, and subsequent treatment of the same cells with Angeli’s salt still induced a strong fluorescence turn-on (Figure S26). This experiment proves that the presence of H2S does not interfere with intracellular HNO sensing by CuDHX1.
An advantage of NIR probes is that they can be used in combination with visible-color sensors to image two or more analytes simultaneously. In this kind of experiment, it is possible to investigate in real time the interplay between signaling molecules in live organisms. An example of such a relationship is the release of mobile zinc upon nitrosation of metallothionein.45,46 Mobilization of endogenous zinc induced by NO donors can be detected in live cells by fluorescent sensors,47 but NO is unable to nitrosate thiolates directly.48 HNO, however, reacts directly with thiols16 and may be capable of releasing chelatable zinc. To investigate this possibility, we incubated HeLa cells with CuDHX1 and the green-fluorescent, zinc-selective, sensor ZP1.36 Before any treatment, the cells showed only very faint fluorescence in both the NIR and green channels (Figure 6D,G). Treatment of these cells with 3 mM Angeli’s salt led to an increase in fluorescence in both the NIR channel and the green channel (Figure 6C,E,H). Addition of 50 μM TPEN reduced the fluorescence only in the green channel (Figure 6C,F), demonstrating that the observed turn-on corresponded to an increase in the intracellular levels of mobile zinc. Addition of TPEN did not significantly change the fluorescence of CuDHX1 in the NIR channel (Figure 6C,I).
Figure 6.
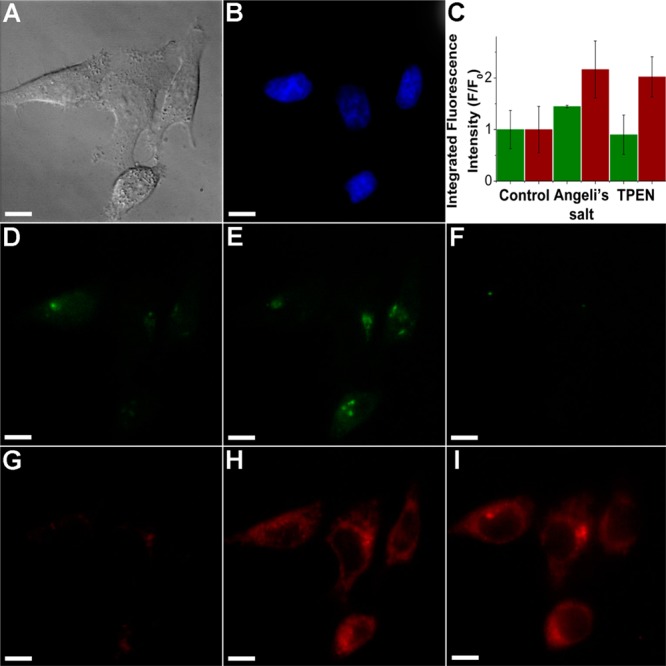
Multicolor/multianalyte imaging of HNO and mobile zinc. (A) DIC image; (B) blue channel showing nuclei; (C) quantification of the fluorescence intensity in the green and NIR channels; (D) green channel before addition of Angeli’s salt; (E) green channel after addition of Angeli’s salt; (F) green channel after addition of TPEN; (G) NIR channel before addition of Angeli’s salt; (H) NIR channel after addition of Angeli’s salt; and (I) NIR channel after addition of TPEN. Scale bar =10 μm.
As discussed above, Angeli’s salt was handled as a solution in aqueous 10 mM NaOH, which produces HNO and NaNO2. To prove that the mobilization of Zn2+ was induced by HNO, HeLa cells were incubated with 3 mM NaNO2 in aqueous 10 mM NaOH. No significant change in fluorescence intensity was detected in the green channel under these conditions (Figure S27).
These experiments reveal the value of CuDHX1 as a versatile sensor for multicolor imaging, allowing for simultaneous observation of HNO and mobile Zn2+. They also prove that release of mobile Zn2+ is a downstream effect of addition of HNO. To the best of our knowledge, our results provide the first application of a multicolor microscopy experiment to study the cellular chemistry of HNO.
Summary and Conclusions
CuDHX1 provides a fast, selective, NIR fluorescent sensor for the detection of HNO. CuDHX1 is selective against a variety of analytes present in live cells, including thiols and H2S, which is an advantage over previous Cu(II)-based HNO sensors. EPR spectroscopic and ESI-MS studies showed that the sensing mechanism relies on reduction of Cu(II) by HNO, with concomitant dissociation of Cu(I) from the cyclam-based ligand. The sensor detects HNO in live cells, even in the presence of H2S. The narrow emission of CuDHX1 in the NIR region makes it useful for multicolor imaging experiments, exemplified here by its use in combination with ZP1 to track the increase in intracellular levels of mobile zinc elicited by exogenously applied Angeli’s salt. These results represent the first direct observation of a relationship between HNO and mobile zinc in a biological environment. We anticipate that CuDHX1 will be useful for investigating the biology of HNO and its interplay with other signaling molecules by multicolor fluorescence microscopy in the NIR.
Acknowledgments
This work was supported by the National Science Foundation (Grant CHE-1265770 to S.J.L.). Instrumentation in the MIT DCIF is maintained with funding from NIH Grant 1S10RR13886-01. A.T.W. acknowledges support from the Paul E. Gray (1954) Endowed Fund for UROP. A.D.L. thanks the NIH for support under Interdepartmental Biotechnology Training Grant T32 GM008334. P. R.-F. thanks the Swiss National Science Foundation for a postdoctoral fellowship. We thank Dr. Robert Radford for providing a sample of ZP1 and Dr. Ulf-Peter Apfel for insightful discussions.
Supporting Information Available
NMR spectroscopic data, analytical HPLC traces, high-resolution ESI-MS, fluorescence and absorbance spectra of 3, time-dependent fluorescence experiments, cyclic voltammograms, simulated EPR spectra, time-lapsed microscopy measurements, and control imaging experiments. This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cannon R. O. III Clin. Chem. 1998, 44, 1809–1819. [PubMed] [Google Scholar]
- Bogdan C. Nat. Immunol. 2001, 2, 907–916. [DOI] [PubMed] [Google Scholar]
- Bredt D. S.; Hwang P. M.; Snyder S. H. Nature 1990, 347, 768–770. [DOI] [PubMed] [Google Scholar]
- Palmer R. M. J.; Ferrige A. G.; Moncada S. Nature 1987, 327, 524–526. [DOI] [PubMed] [Google Scholar]
- Bult H.; Boeckxstaens G. E.; Pelckmans P. A.; Jordaens F. H.; Van Maercke Y. M.; Herman A. G. Nature 1990, 345, 346–347. [DOI] [PubMed] [Google Scholar]
- Pieper G. M. Hypertension 1998, 31, 1047–1060. [DOI] [PubMed] [Google Scholar]
- Wei G.; Dawson V. L.; Zweier J. L. Biochim. Biophys. Acta, Mol. Basis Dis. 1999, 1455, 23–34. [DOI] [PubMed] [Google Scholar]
- Heales S. J. R.; Bolaños J. P.; Stewart V. C.; Brookes P. S.; Land J. M.; Clark J. B. Biochim. Biophys. Acta, Bioenerg. 1999, 1410, 215–228. [DOI] [PubMed] [Google Scholar]
- Hussain S. P.; Hofseth L. J.; Harris C. C. Nat. Rev. Cancer 2003, 3, 276–285. [DOI] [PubMed] [Google Scholar]
- Fukuto J. M.; Cisneros C. J.; Kinkade R. L. J. Inorg. Biochem. 2013, 118, 201–208. [DOI] [PubMed] [Google Scholar]
- Bullen M. L.; Miller A. A.; Andrews K. L.; Irvine J. C.; Ritchie R. H.; Sobey C. G.; Kemp-Harper B. K. Antioxid. Redox Signal. 2011, 14, 1675–1686. [DOI] [PubMed] [Google Scholar]
- Paolocci N.; Saavedra W. F.; Miranda K. M.; Martignani C.; Isoda T.; Hare J. M.; Espey M. G.; Fukuto J. M.; Feelisch M.; Wink D. A.; Kass D. A. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 10463–10468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautov R. F.; Ngo D. T. M.; Licari G.; Liu S.; Sverdlov A. L.; Ritchie R. H.; Kemp-Harper B. K.; Horowitz J. D.; Chirkov Y. Y. Nitric Oxide 2013, 35, 72–78. [DOI] [PubMed] [Google Scholar]
- Choe C.-u.; Lewerenz J.; Fischer G.; Uliasz T. F.; Espey M. G.; Hummel F. C.; King S. B.; Schwedhelm E.; Böger R. H.; Gerloff C.; Hewett S. J.; Magnus T.; Donzelli S. J. Neurochem. 2009, 110, 1766–1773. [DOI] [PubMed] [Google Scholar]
- Choe C.-U.; Lewerenz J.; Gerloff C.; Magnus T.; Donzelli S. Antioxid. Redox Signal. 2011, 14, 1699–1711. [DOI] [PubMed] [Google Scholar]
- Fukuto J. M.; Carrington S. J. Antioxid. Redox Signal. 2011, 14, 1649–1657. [DOI] [PubMed] [Google Scholar]
- Kröncke K. D.; Fehsel K.; Schmidt T.; Zenke F. T.; Dasting I.; Wesener J. R.; Bettermann H.; Breunig K. D.; Kolb-Bachofen V. Biochem. Biophys. Res. Commun. 1994, 200, 1105–1110. [DOI] [PubMed] [Google Scholar]
- Maret W. Adv. Nutr. 2013, 4, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switzer C. H.; Flores-Santana W.; Mancardi D.; Donzelli S.; Basudhar D.; Ridnour L. A.; Miranda K. M.; Fukuto J. M.; Paolocci N.; Wink D. A. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N.; Kaufman S.; Milstien S. Mol. Pharmacol. 1993, 43, 6–10. [PubMed] [Google Scholar]
- Adak S.; Wang Q.; Stuehr D. J. J. Biol. Chem. 2000, 275, 33554–33561. [DOI] [PubMed] [Google Scholar]
- Murphy M. E.; Sies H. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 10860–10864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe M. A.; Cooper C. E. Biochem. J. 1998, 332, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovic M. R.; Miljkovic J. L.; Nauser T.; Royzen M.; Klos K.; Shubina T.; Koppenol W. H.; Lippard S. J.; Ivanović-Burmazović I. J. Am. Chem. Soc. 2012, 134, 12016–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljkovic J. L.; Kenkel I.; Ivanović-Burmazović I.; Filipovic M. R. Angew. Chem., Int. Ed. 2013, 52, 12061–12064. [DOI] [PubMed] [Google Scholar]
- Rosenthal J.; Lippard S. J. J. Am. Chem. Soc. 2010, 132, 5536–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royzen M.; Wilson J. J.; Lippard S. J. J. Inorg. Biochem. 2013, 118, 162–170. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Liu K.; Li J.-Y.; Fang Y.; Zhao T.-C.; Yao C. Org. Lett. 2011, 13, 1290–1293. [DOI] [PubMed] [Google Scholar]
- Apfel U.-P.; Buccella D.; Wilson J. J.; Lippard S. J. Inorg. Chem. 2013, 52, 3285–3294. [DOI] [PubMed] [Google Scholar]
- Kawai K.; Ieda N.; Aizawa K.; Suzuki T.; Miyata N.; Nakagawa H. J. Am. Chem. Soc. 2013, 135, 12690–12696. [DOI] [PubMed] [Google Scholar]
- Hilderbrand S. A.; Weissleder R. Curr. Opin. Chem. Biol. 2010, 14, 71–79. [DOI] [PubMed] [Google Scholar]
- Egawa T.; Hirabayashi K.; Koide Y.; Kobayashi C.; Takahashi N.; Mineno T.; Terai T.; Ueno T.; Komatsu T.; Ikegaya Y.; Matsuki N.; Nagano T.; Hanaoka K. Angew. Chem., Int. Ed. 2013, 52, 3874–3877. [DOI] [PubMed] [Google Scholar]
- Kobayashi H.; Koyama Y.; Barrett T.; Hama Y.; Regino C. A. S.; Shin I. S.; Jang B.-S.; Le N.; Paik C. H.; Choyke P. L.; Urano Y. ACS Nano 2007, 1, 258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasakura K.; Hanaoka K.; Shibuya N.; Mikami Y.; Kimura Y.; Komatsu T.; Ueno T.; Terai T.; Kimura H.; Nagano T. J. Am. Chem. Soc. 2011, 133, 18003–18005. [DOI] [PubMed] [Google Scholar]
- Yuan L.; Lin W.; Zhao S.; Gao W.; Chen B.; He L.; Zhu S. J. Am. Chem. Soc. 2012, 134, 13510–13523. [DOI] [PubMed] [Google Scholar]
- Burdette S. C.; Walkup G. K.; Spingler B.; Tsien R. Y.; Lippard S. J. J. Am. Chem. Soc. 2001, 123, 7831–7841. [DOI] [PubMed] [Google Scholar]
- Chapman G.; Henary M.; Patonay G. Anal. Chem. Insights 2011, 6, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda K. M.; Nagasawa H. T.; Toscano J. P. Curr. Top. Med. Chem. 2005, 5, 649–664. [DOI] [PubMed] [Google Scholar]
- Stoll S.; Schweiger A. J. Magn. Reson. 2006, 178, 42–55. [DOI] [PubMed] [Google Scholar]
- Philip R.; Penzkofer A.; Bäumler W.; Szeimies R. M.; Abels C. J. Photochem. Photobiol. A 1996, 96, 137–148. [Google Scholar]
- Alander J. T.; Kaartinen I.; Laakso A.; Pätilä T.; Spillmann T.; Tuchin V. V.; Venermo M.; Välisuo P. Int. J. Biomed. Imaging 2012, 2012, 940585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda K. M.; Dutton A. S.; Ridnour L. A.; Foreman C. A.; Ford E.; Paolocci N.; Katori T.; Tocchetti C. G.; Mancardi D.; Thomas D. D.; Espey M. G.; Houk K. N.; Fukuto J. M.; Wink D. A. J. Am. Chem. Soc. 2005, 127, 722–731. [DOI] [PubMed] [Google Scholar]
- Armstrong D. A.; Huie R. E.; Lymar S.; Koppenol W. H.; Merényi G.; Neta P.; Stanbury D. M.; Steenken S.; Wardman P. BioInorg. React. Mech. 2014, 9, 59–61. [Google Scholar]
- Lee K. Y.; Kuchynka D. J.; Kochi J. K. Inorg. Chem. 1990, 29, 4196–4204. [Google Scholar]
- Berendji D.; Kolb-Bachofen V.; Meyer K. L.; Grapenthin O.; Weber H.; Wahn V.; Kröncke K.-D. FEBS Lett. 1997, 405, 37–41. [DOI] [PubMed] [Google Scholar]
- Stamler J. S.; Simon D. I.; Osborne J. A.; Mullins M. E.; Jaraki O.; Michel T.; Singel D. J.; Loscalzo J. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahl D. U.; Berendji-Grün D.; Suschek C. V.; Kolb-Bachofen V.; Kröncke K.-D. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13952–13957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozhukh J.; Lippard S. J. Inorg. Chem. 2012, 51, 7346–7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.