

Abstract
Reaction of Cd(OAc)2·2H2O and selenourea in primary-amine/secondary-amine cosolvent mixtures affords crystalline CdSe quantum platelets at room temperature. Their crystallinity is established by X-ray diffraction analysis (XRD), high-resolution transmission electron microscopy (TEM), and their sharp extinction and photoluminescence spectra. Reaction monitoring establishes the magic-size nanocluster (CdSe)34 to be a key intermediate in the growth process, which converts to CdSe quantum platelets by first-order kinetics with no induction period. The results are interpreted to indicate that the critical crystal-nucleus size for CdSe under these conditions is in the range of (CdSe)34 to (CdSe)68. The nanocluster is obtained in isolated form as [(CdSe)34(n-octylamine)16(di-n-pentylamine)2], which is proposed to function as crystal nuclei that may be stored in a bottle.
Introduction
We provide kinetic evidence that the metastable, magic-size (CdSe)34 nanocluster is near the critical-nucleus size for CdSe and supports the growth of wurtzite CdSe nanocrystal platelets at room temperature (20–25 °C). Typical conditions for the synthesis of CdSe nanocrystals are temperatures above 200 °C.1−5 To our knowledge, the growth of CdSe quantum platelets (QPs) reported here, via the intermediacy of (CdSe)34 nanoclusters, constitutes the lowest temperature at which crystalline CdSe has been produced. We ascribe the low-temperature crystal growth to facile nucleation resulting from (CdSe)34 being near to the critical size, such that the nucleation barrier has largely been surmounted in the formation of this magic-size nanocluster.
A crystal nucleation and growth process that is driven by a chemical reaction requires a proper ordering of nucleation, growth, and reaction barriers (activation energies, Scheme 1). According to the classic crystal-growth model,6−9 nucleation barriers are higher than the activation energies for growth steps, such that conditions resulting in nucleation will also support crystal growth. We argue that the barrier for a monomer-generating chemical reaction must be higher than the nucleation barrier to support nanocrystal growth (Scheme 1, curve a). Here, monomers are defined as small (CdSe)n molecules or clusters. If the nucleation barrier exceeds the reaction barrier (Scheme 1, curve b), then monomer is produced by the reaction under conditions that preclude crystal formation, and thus, amorphous aggregates and precipitates are formed instead.
Scheme 1. Reaction-Coordinate Diagrams for Alternate Ordering of the Monomer-Generating-Reaction and Crystal-Nucleation Barriers.
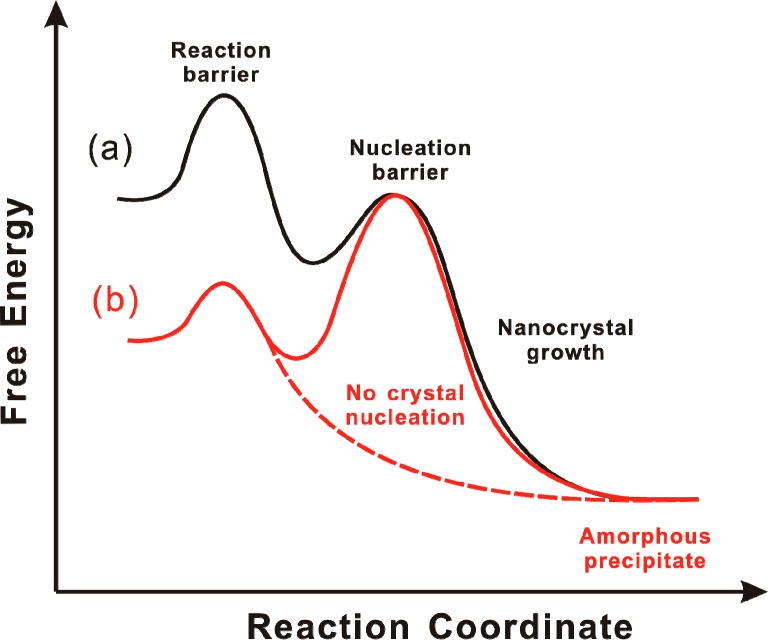
(a) The reaction barrier is higher than the nucleation barrier, and nanocrystal growth occurs on the black free-energy curve. (b) The reaction barrier is lower than the nucleation barrier, precluding nucleation and crystal growth. Instead, amorphous aggregates are formed on the red dashed curve.
We suspect that the high temperatures typically employed in semiconductor-nanocrystal syntheses reflect high nucleation barriers for assembling the critical-size nucleus, such that high-barrier chemical reactions are also required. However, if a critical-size nucleus could be assembled under milder conditions, then in principle semiconductor-nanocrystal nucleation and growth could be achieved at lower temperatures. We propose that (CdSe)34 is near the critical size, such that its binary combination exceeds the critical size. If so, then the critical-nucleus size for CdSe under our conditions is in the range of (CdSe)34–(CdSe)68.
We previously reported syntheses of CdSe quantum belts (nanoribbons) in lamellar, n-octylamine-bilayer templates at the comparatively mild temperatures of 70–80 °C.10 Similar syntheses were also reported by Hyeon and co-workers.11−14 We determined that the magic-size nanocluster (CdSe)13 was an intermediate in the formation of the quantum belts and later isolated and characterized a series of [(CdSe)13(primary amine)13] adducts.15,16
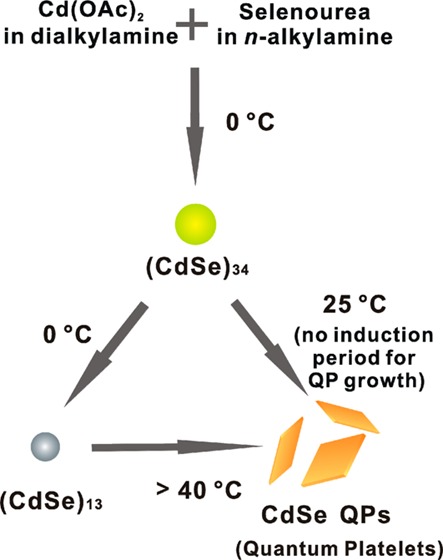
We now report that crystalline, wurtzite, CdSe quantum platelets (QPs; also known as nanoplatelets17−20 or quantum disks21) are formed at room temperature by employing di-n-alkylamine cosolvents or by varying the primary-amine solvent. Reaction monitoring and mechanistic analyses indicate that (CdSe)34 is the magic-size nanocluster intermediate under these conditions, which converts to CdSe QPs at room temperature by first-order kinetics with no detectable induction period. Alternatively, at 0 °C, (CdSe)34 converts to (CdSe)13, which then requires temperatures above 40 °C to form CdSe QPs. Our interpretation of these results is that (CdSe)34 is nearer to the CdSe critical-nucleus size.
A ligated derivative of (CdSe)34 is obtained as a slushy solid that is stable indefinitely at 0 °C. We suggest that this derivative, [(CdSe)34(n-octylamine)16(di-n-pentylamine)2], may effectively function as CdSe crystal nuclei that may be stored in a bottle. Its use in templates of varying geometries may afford low-temperature routes to CdSe nanocrystals having other controlled morphologies.
Experimental Section
Materials and General Procedures
Di-n-octylamine (+98%), di-n-pentylamine (99%), di-n-propylamine (99%), diethylamine (>99.5%), phenethylamine (≥99%), n-dodecylamine (≥99%), n-octylamine (+99%), n-pentylamine (+99%), n-propylamine (+98%), Cd(OAc)2·2H2O (>98%), trin-octylphosphine (TOP) (97%), and oleylamine (or cis-9-octadecenylamine, technical grade, 70%) were obtained from Sigma-Aldrich. Selenourea (99.9%, metal basis) was obtained from Alpha Aesar. All were used as received and stored under N2. Toluene was obtained from Sigma-Aldrich (CHROMASOLV for HPLC, ≥99.9%). Transmission electron microscopy (TEM) sample grids (Cu with holey carbon film) were obtained from Ted Pella, Inc.
All synthetic procedures were conducted under dry N2, except the final washing steps, which were conducted in the ambient atmosphere. The reaction mixtures were not stirred. The synthetic products were generally stored as reaction mixtures, after addition of TOP (see below).
Direct Synthesis of 1.8 nm Thick CdSe QPs
In a typical procedure, Cd(OAc)2·2H2O (65 mg, 0.24 mmol) was dissolved in di-n-octylamine (5.7 g, 24 mmol) in a septum-capped Schlenk tube and placed in a benchtop sonicating bath (10 min) to achieve dissolution. In a glovebox, selenourea (50 mg, 0.41 mmol) was dissolved in n-octylamine (1.2 g, 9.3 mmol) in a septum-capped amber vial. The vial was removed from the glovebox and placed in a benchtop sonicating bath (10 min) to achieve dissolution.
The selenourea solution was injected into the [Cd(OAc)2·2H2O] solution at room temperature (20–25 °C). The colorless reaction mixture became cloudy within 10 s, viscous and light green within 5 min, cloudy and yellow green within 60 min, and cloudy and light yellow at longer reaction times. After 2 h, the mixture was nearly clear and colorless with a light-yellow precipitate. After 2 days, the yellow precipitate remained in the presence of a light-red supernatant, the color of which was due to a Se side product from (CdSe)34 formation. TOP (0.25–0.50 mL) was injected to scavenge the Se side product through the formation of colorless (n-octyl)3P=Se. The light-yellow precipitate of bundled CdSe QPs was then stored at room temperature in the reaction mixture under N2 for further analyses.
Direct Synthesis of 2.2 nm Thick CdSe QPs
The procedure was conducted in the same manner as that for the 1.8 nm thick CdSe QPs (see above), except for the amount of n-octylamine used (2.4 g, 18 mmol) and the reaction temperature, which was raised to 70 °C. The color changes occurred more rapidly, from colorless (0 min) to viscous and yellow (10 s), cloudy and orange (1 min), and cloudy and orange-red (>120 min). After the reaction mixture stood for 2 days at 70 °C, the CdSe QPs were deposited as an orange-red precipitate in the presence of a red supernatant. TOP (0.25–0.50 mL) was injected to scavenge the Se side product from the (CdSe)34 formation responsible for the red coloration of the supernatant, which became colorless. The dispersion of bundled CdSe QPs was then stored at room temperature in the reaction mixture under N2 for further analyses.
Direct Synthesis of 1.4 nm Thick CdSe QPs
In a typical procedure, Cd(OAc)2·2H2O (65 mg, 0.24 mmol) was dissolved in phenethylamine (5.74 g, 47 mmol) in a septum-capped Schlenk tube and heated in a 70 °C oil bath (1 h) to dissolve the cadmium precursor. In a glovebox, selenourea (50 mg, 0.41 mmol) was dissolved in phenethylamine (1.2 g, 9.9 mmol) in a septum-capped amber vial. The vial was removed from the glovebox and placed in a benchtop sonicating bath (10 min) to achieve dissolution of the selenourea.
The selenourea solution was injected into the [Cd(OAc)2·2H2O] solution at 40 °C. The clear, colorless reaction mixture became clear and light yellow within 1 h, viscous and white (60–90 min), and then cloudy and white-yellow (>90 min). After 2 h, the solution became nearly clear and colorless upon formation of a white-yellow precipitate. After 2 days, CdSe QPs were deposited as a white-yellow precipitate in the presence of a light-red supernatant. TOP (0.25–0.50 mL) was injected to scavenge the Se side product from (CdSe)34 formation responsible for the red coloration of the supernatant, which became colorless. The white-yellow precipitate of bundled CdSe QPs was then stored at room temperature in the reaction mixture under N2 for further analyses.
Preparation of [(CdSe)34(n-octylamine)16(di-n-pentylamine)2]
In a typical procedure, Cd(OAc)2·2H2O (65 mg, 0.24 mmol) was dissolved in di-n-pentylamine (5.74 g, 36 mmol) in a septum-capped Schlenk tube and then was stored in an ice bath (0 °C) placed inside a refrigerator. In a glovebox, selenourea (50 mg, 0.41 mmol) was added to n-octylamine (1.2 g, 9.3 mmol) in a septum-capped amber vial. The vial was removed from the glovebox and placed in a benchtop sonicating bath (10 min) to achieve dissolution of the selenourea.
The selenourea solution was injected into the Cd(OAc)2·2H2O solution at 0 °C. The clear, colorless reaction mixture became viscous and light yellow within 6 h, cloudy and yellow within 8 h, and cloudy and green-yellow at longer times (0 °C). After 18 h at 0 °C, (CdSe)34 was formed as a green-yellow precipitate mixed with colorless supernatant. TOP (0.25–0.50 mL) was injected to scavenge excess selenourea.
The greenish-yellow precipitate was separated using a benchtop centrifuge (700g, 30 s) at room temperature, and the colorless supernatant was discarded. The remaining greenish-yellow slush was redispersed into 3–5 mL of toluene. This purification process was repeated, for a total of two such cycles, yielding [(CdSe)34(n-octylamine)16(di-n-pentylamine)2] as a slushy, greenish-yellow solid after drying in vacuo for 12 h (0.061 g, 95.7%). UV–visible (toluene) λmax, nm: 360, 390, 418 (Figure 9a). MS m/z (relative area, assignment): 6508.2572 (100%, (CdSe)34), 6319.5733 (42.6%, (CdSe)33), 3651.5768 (17.0%, (CdSe)19), 2502.4747 (52.4%, (CdSe)13) (Figure 11). Anal. Calcd for [(CdSe)34(n-octylamine)16(di-n-pentylamine)2]: C, 20.00; H, 3.94; N, 2.84. Found, C, 19.94; H, 3.95; N, 2.93. All values are given as percentages.
Figure 9.
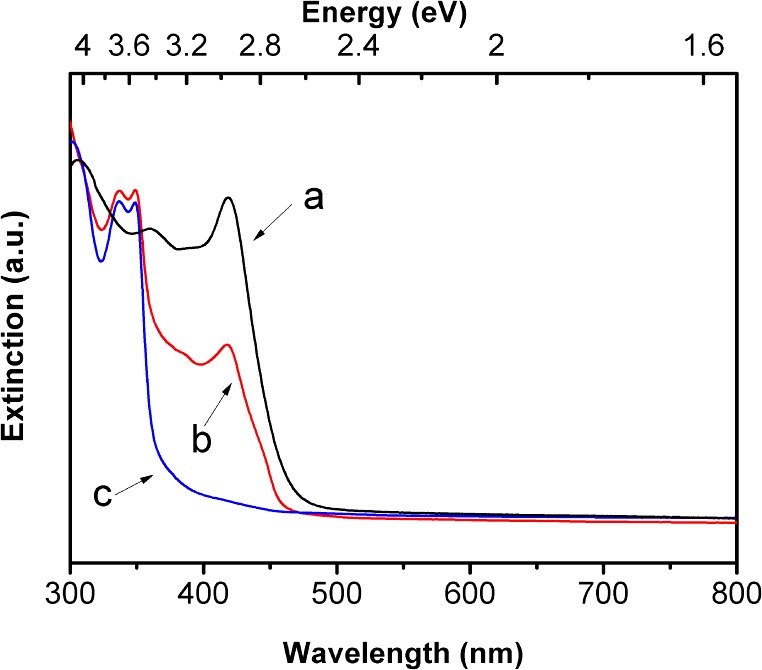
Spectral evolution upon transformation of (CdSe)34 to (CdSe)13 in an n-octylamine/di-n-pentylamine cosolvent at 0 °C. UV–visible extinction spectra of (a) (CdSe)34 after 12 h (black curve), (b) a mixture of (CdSe)34 and (CdSe)13 after 14 days, and (c) (CdSe)13 after 1 month.
Figure 11.

An LDI mass spectrum of [(CdSe)34(n-octylamine)16(di-n-pentylamine)2]. The peaks for the magic-size CdSe nanoclusters are labeled.
[(CdSe)34(n-octylamine)16(di-n-pentylamine)2] was generally used immediately for analyses or further reactions. The compound was stable at room temperature for at least 24 h under N2 and for longer than one month at 0 °C under N2.
Other di-n-alkylamine derivatives of (CdSe)34 were prepared under the same general conditions, except for the reaction cosolvents employed. Di-n-propylamine or diethylamine were used to replace di-n-pentylamine for the Cd(OAc)2·2H2O solution, while n-octylamine was used for the selenourea solution. The purification procedure was the same as that used above.
Conversion of (CdSe)34 Nanoclusters to CdSe QPs
The preparation of (CdSe)34 was conducted as described above. The reaction mixture was then removed from the ice bath and stored at room temperature (20–25 °C) for an additional 12 h. The reaction mixture was periodically monitored by UV–visible spectroscopy to determine the extent of the conversion, which was found to be complete after 12 h. CdSe QPs were deposited as a light-yellow precipitate in the presence of a light-red supernatant. TOP (0.5–1.0 mL) was injected into the reaction mixture to scavenge the red selenium side product from (CdSe)34 formation, resulting in a colorless supernatant. The conversion of (CdSe)34 to CdSe QPs was accelerated by adding additional di-n-pentylamine to the reaction mixture at room temperature after the formation of (CdSe)34.
Measurement of the (CdSe)34-to-CdSe-QPs Conversion Kinetics
A (CdSe)34 sample was prepared as described above. An aliquot (26 mg) taken from the reaction mixture was diluted into a di-n-pentylamine (2.5 g) and n-octylamine (0.07 g) mixture in a quartz cuvette at room temperature (20–25 °C). UV–visible spectra were collected in the wavelength range of 400–500 nm at 1 h intervals. During data collection, the cuvette was stirred by a small stirring bar.
The 418 nm absorption of (CdSe)34 and the 423 and 448 nm absorptions of CdSe QPs were extracted from the spectra by nonlinear least-squares fitting using Origin software (http://originlab.com/). The initial (t = 0 h) spectrum was fit by a single Lorentzian function, yielding the center position of the 418 nm absorption. The final (t = 12 h) was fit with three Lorentzian functions, the first centered at 418 nm, and a background-scattering function (A/λ4, where A was an adjustable parameter), yielding the center positions of the 423 and 448 nm QP absorptions. All of the intermediate spectra were fit with three Lorentzian and the one background-scattering functions, with the Lorentzians initially centered at 418, 423, and 448 nm. These peak centers were refined only in the final stages of fitting.
The peak areas determined from the nonlinear least-squares fits were used for the kinetic analyses. All three absorptions gave first-order plots of absorption peak area vs time over three half-lives. The error in the slope of the plots was determined by conducting three kinetic trials and observing the range in the integrated peak areas in the final (t = 12 h) spectra. The range of slopes that accommodated these final values was assigned as the error in the slopes. The kinetic parameters kobs and t1/2 were extracted from the slopes, and their errors determined by propagation in the normal manner. These values are reported in the Results section.
Conversion of (CdSe)34 to (CdSe)13 Nanoclusters
The preparation of (CdSe)34 was conducted as described above. The reaction temperature equilibrated near 0 °C in the refrigerator, even after the ice in the bath melted. The reaction mixture was periodically monitored by UV–visible spectroscopy to determine the extent of the conversion. When di-n-pentylamine was used as the cosolvent, the complete conversion required longer than 1 month, during which the greenish-yellow precipitate gradually changed to white with formation of a small amount of black precipitate, which was a selenium side product. TOP (0.5–1.0 mL) was injected into the reaction mixture, whereupon the black solid disappeared, leaving (CdSe)13 as a white precipitate. When diethylamine or di-n-propylamine was used as the cosolvent, the conversion of (CdSe)34 to (CdSe)13 was more rapid and completed in 2–3 weeks. The conversion was also accelerated by adding additional n-octylamine to the reaction mixture after the formation of (CdSe)34.
Results
Room-Temperature Growth of CdSe QPs
We previously reported that reaction of Cd(OAc)2·2H2O and selenourea in n-octylamine solvent at room temperature selectively produced magic-size (CdSe)13 nanoclusters entrained within a spontaneously formed, double-lamellar, n-octylamine-bilayer template (eq 1, Scheme 2).10 These intratemplate (CdSe)13 nanoclusters were subsequently converted to crystalline, CdSe quantum belts (QBs) at relatively mild temperatures (70–80 °C, eq 1). The lengths, widths, and thicknesses of the quantum belts were determined by the dimensions within the spontaneously formed, double-lamellar templates (Scheme 2).10 We sought to purposefully vary these dimensions by varying the nature of the amine solvent, and those efforts led to experiments using di-n-alkylamine cosolvents.
 |
1 |
Scheme 2. Low-Temperature Growth of Crystalline CdSe QPs within a Double-Lamellar, Primary-Amine Bilayer Template.
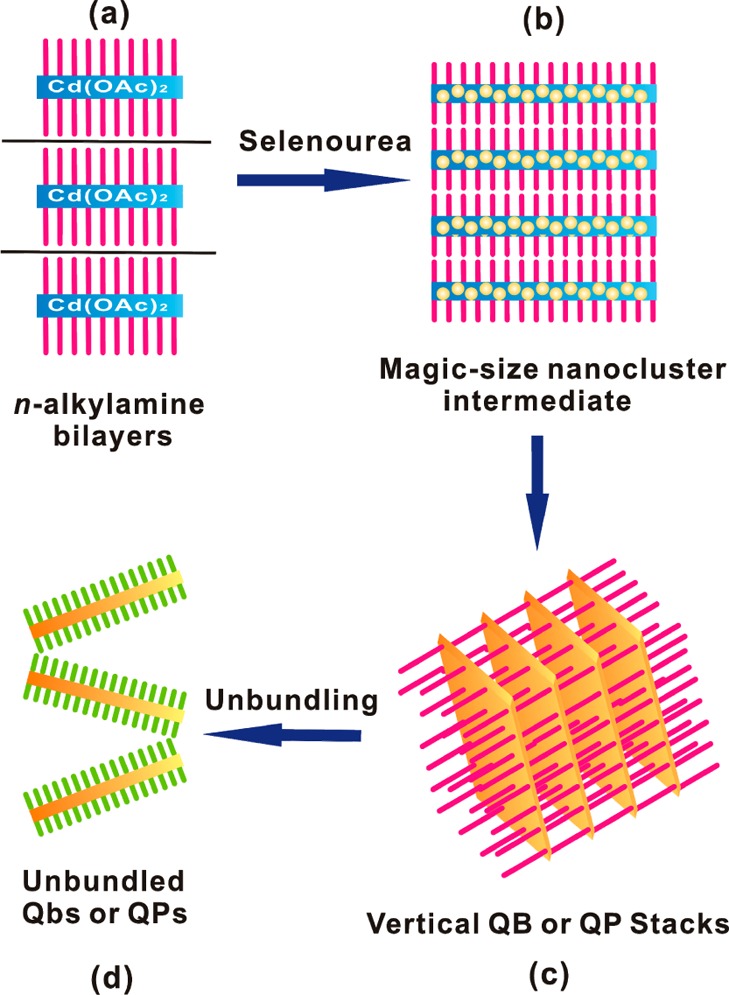
(a) Cd(OAc)2 and the primary-amine solvent forms a lamellar, amine-bilayer mesophase (blue and purple). (b) Magic-size (CdSe)34 clusters are initially formed within the template when primary- and secondary- amine co-solvents are employed (yellow dots, blue and purple). (c) (CdSe)34 clusters are converted to bundled QPs at room temperature in the co-solvent mixtures (orange and purple). (d) Addition of a long-chain primary amine results in the spontaneous exfoliation of the QPs by ligand exchange at room temperature (orange and green).
Reaction of Cd(OAc)2·2H2O and selenourea in an n-octylamine/di-n-octylamine cosolvent mixture at room temperature gave a yellow precipitate, which contrasted with the white (colorless) [(CdSe)13(n-octylamine)13] isolated from the eq 1 reaction.15 The UV–visible spectrum of the yellow precipitate dispersed in toluene (Figure 1) closely matched those previously obtained for CdSe QBs10 and could not be assigned to (CdSe)13 or other magic-size nanoclusters. TEM images (Figure 2) revealed the formation of pseudorectangular CdSe QPs having mean widths and lengths of 7 and 50 nm, respectively. Because the QPs have the electronic properties of quantum wells,17,22 their spectrum depended only on thickness and was effectively indistinguishable from those previously obtained for 1.8 nm thick QBs.22 The sharp PL spectrum (Figure 1) also matched those of the corresponding CdSe QBs.22 Like the QBs, the QPs gave high PL quantum efficiencies (PL QE = 25%). We surmised that, in the cosolvent mixture, the reaction proceeded through magic-size nanocluster intermediates (see below) to CdSe QP nanocrystals at room temperature (eq 2).
 |
2 |
Figure 1.
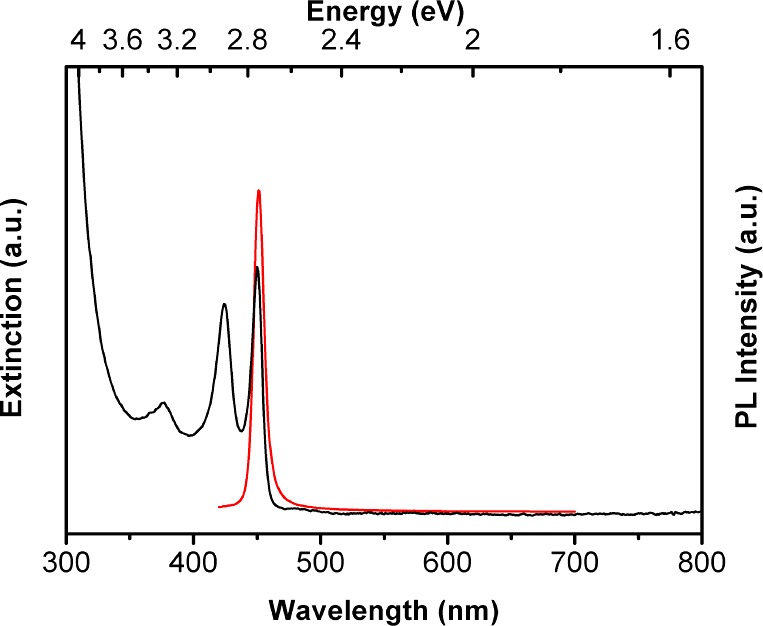
A UV–visible extinction spectrum in a toluene dispersion (black curve) and a photoluminescence spectrum in an oleylamine–toluene solution (12% w/w, red curve) of 1.8 nm thick CdSe QPs.
Figure 2.

TEM images of CdSe QPs synthesized in n-octylamine and various di-n-alkylamine cosolvents. (a, b) di-n-octylamine, (c, d) di-n-pentylamine, (e, f) di-n-propylamine, and (g, h) diethylamine. The right panels are at higher magnification.
We next sought to establish the crystallinity of the QPs grown by the room-temperature synthesis. An X-ray diffraction (XRD) pattern of the as-prepared material (Figure 3) matched those previously obtained for wurtzite CdSe QBs.22 Like those QBs, the QPs exhibited a lattice contraction associated with the surface tension of the thin nanocrystals.11 The lattice parameters extracted from the XRD data (a = 4.07 ± 0.02 Å, c = 6.82 ± 0.03 Å) were smaller than the bulk values (a = 4.30 Å, c = 7.02 Å) by nearly the same amounts as those of the QBs.22
Figure 3.
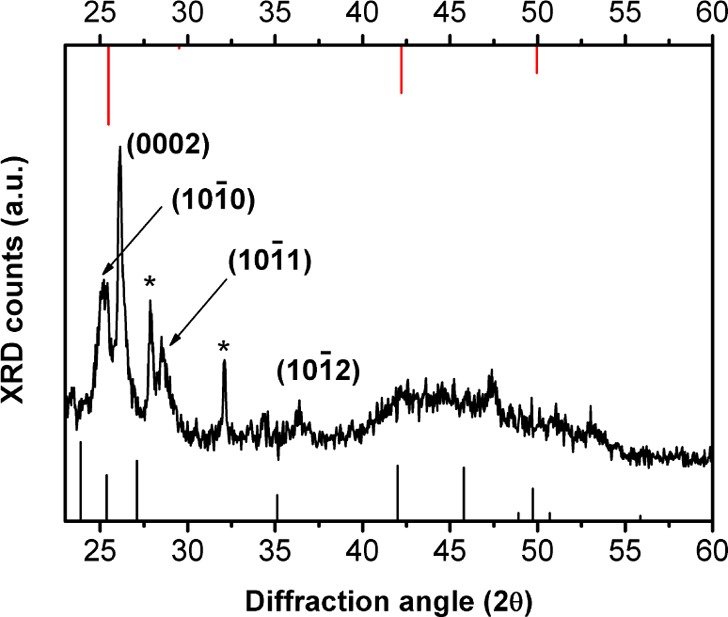
An XRD pattern of 1.8 nm thick CdSe QPs. The black sticks are the peak positions for bulk CdSe in the wurtzite structure, and the red sticks are the peak positions for bulk CdSe in the zinc-blende structure. The indexed reflections for the wurzite QPs are shifted to a higher angle than in the bulk pattern because of the lattice contraction (see the text and ref (22)). The asterisks identify unassigned peaks.
High-resolution TEM images were also consistent with crystalline CdSe QPs. Figure 4a views a stack of bundled QPs parallel to the QP edges (individual QPs are identified by arrows). The 0002 lattice spacings appearing as parallel fringes were clearly evident and provided another measure of the lattice parameter c = 6.86 ± 0.04 Å. A Fourier transform of the HRTEM image of the face of a QP was consistent with the (112̅0) plane of wurtzite (Figure 4b inset), as with the previously reported QBs.22 The lattice parameter a = 4.04 ± 0.08 Å was extracted from the fringe pattern in the image of the face (Figure 4b).
Figure 4.

HRTEM images of the 1.8 nm thick CdSe QPs. (a) Edge view of bundled QPs. (b) Face view of a single QP. Fast Fourier transforms of the images are inset. White arrows in (a) indicate the length dimension of the bundled QPs.
Although amorphous nanoparticles may be crystallized under the electron beam in the TEM, we did not observe such a process; the QPs were crystalline from the outset of TEM observations. Thus, the sharp extinction and PL spectra (Figure 1), the comparatively sharp XRD pattern that clearly indexed to wurtzite (Figure 3), and the high-resolution TEM data (Figure 4) all indicated that the CdSe QPs obtained from the room-temperature synthesis were crystalline as formed.
The QP synthesis was repeated using other combinations of primary and secondary amines. Experiments were conducted using n-octylamine and various di-n-alkylamine cosolvents. As summarized in Table 1, the secondary amine influenced the mean lengths of the QPs, without strongly affecting widths or thicknesses. Interestingly, the mean QP lengths were inversely proportional to the lengths of the alkyl groups on the di-n-alkylamine cosolvent (Figure 5). We do not understand the origin of this effect.
Table 1. Dependence of QP Dimensions on the Di-n-alkylamine Co-solvent.
| R2NH, R = | thickness (nm) | width range (nm) | mean length (nm) |
|---|---|---|---|
| n-octyl | 1.8 | 5–10 | 50.3 ± 3.4 |
| n-pentyl | 1.8 | 5–10 | 63.2 ± 5.6 |
| n-propyl | 1.8 | 5–10 | 99.1 ± 6.5 |
| ethyl | 1.8 | 5–10 | 151 ± 6.8 |
Figure 5.

A plot of the QP mean length vs the inverse of the carbon number of the di-n-alkylamine cosolvent alkyl chain. The carbon number is the number of carbon atoms in the alkyl chain.
Another set of experiments was conducted in which the primary amine was varied and the secondary amine was held constant. In contrast to the above, systematic dependences of the QP sizes or morphologies on the primary amine were not observed. However, the results established, as described below, that the top and bottom QP facets were predominantly passivated by the primary amine. The QPs were produced in bundled stacks that were derived from the lamellar, amine-bilayer templates in which they grew (see Scheme 2 and Figures 2 and 4a).10 Consequently, the inter-QP spacing (d spacing) provided a measure of the amine-bilayer thickness. Low-angle XRD patterns of QPs obtained from various primary-amine and diethylamine cosolvents are given in Figure S1 (Supporting Information). The d spacing was dependent on the primary amine and consistent with the lengths of the alkyl chains. The experiments described above in n-octylamine and di-n-alkylamine cosolvents gave d spacings consistent with n-octylamine, with no influence by the di-n-alkylamine (Figure S2, Supporting Information). These results required that the primary amine was responsible for lamellar, amine-bilayer template formation, and consequently, the large QP facets inherited primary-amine passivation from the growth template.
Many combinations of primary and secondary amines were investigated as cosolvents (See Table S1, Supporting Information). The best results were achieved when the length of the alkyl chain on the primary amine (CH3(CH2)nNH2) was equal to or longer than the length of the alkyl chains on the secondary amine [CH3(CH2)m]2NH (n ≥ m). When this empirical rule was violated, the UV–visible spectra of the resulting QPs were broadened and in some cases contained absorptions for platelets of other discrete thicknesses (see below; Figure S3, Supporting Information).
Each of the syntheses conducted at room temperature and as described above gave QPs with a discrete thickness of 1.8 nm. The 2.2 nm thick QPs were obtained with an n-octylamine/di-n-octylamine cosolvent mixture when the synthesis was conducted at 70 °C. Low-resolution TEM images of the QPs (Figure 6a,b) showed widths of 10–20 nm and a mean length of 50 nm. A discrete QP thickness of 2.2 nm was established by high-resolution TEM (Figure 6c). As expected,10,18,19 the three characteristic QP absorptions were red-shifted from those of the 1.8 nm thick QPs (Figure 7).
Figure 6.

TEM and HRTEM images of bundles of the 2.2 nm thick CdSe QPs. (a, b) TEM image and (c) HRTEM image of the QPs viewed from the edge. Inset is the fast Fourier transform of the image.
Figure 7.
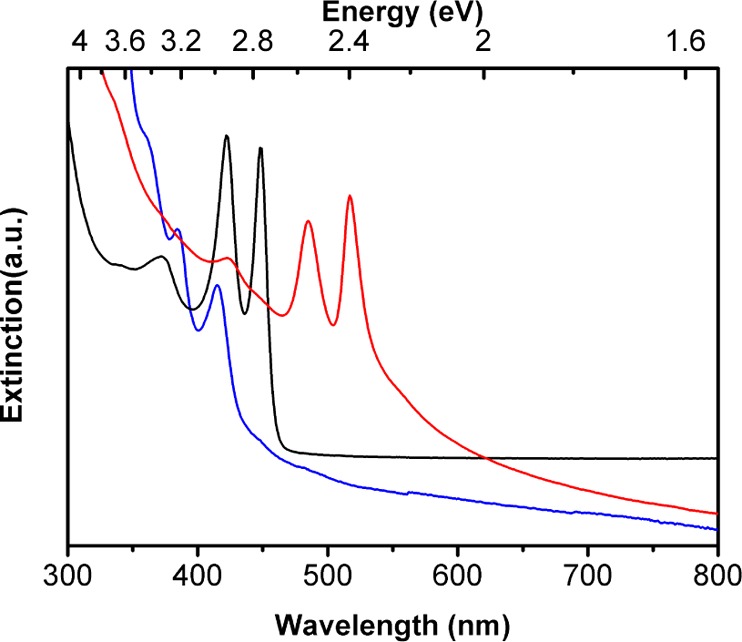
UV–visible extinction spectra in toluene dispersions of bundled 1.4 nm thick CdSe QPs (blue curve), 1.8 nm thick CdSe QPs (black curve), and 2.2 nm thick CdSe QPs (red curve).
1.4 nm thick QPs were obtained in the solvent 2-phenethylamine at 40 °C with no secondary-amine cosolvent. Low-resolution TEM images of the QPs (Figure 8a,b) showed widths of 2–4 nm and a mean length of 700 nm. We note that these lengths are closer to QBs along the QP–QB length spectrum than the cases discussed above. A discrete QP thickness of 1.4 nm was established by high-resolution TEM (Figure 8c). In this case, the three characteristic QP absorptions were blue-shifted from those of the 1.8 nm thick QPs (Figure 7). Thus, we have prepared CdSe QPs of three discrete thicknesses (1.4, 1.8, and 2.2 nm). By comparison, Dubertret and co-workers have prepared CdSe QPs of four discrete thicknesses.18
Figure 8.

TEM and HRTEM images of bundles of 1.4 nm thick CdSe QPs (1.4 nm thickness). (a, b) TEM image and (c) HRTEM image. Inset is the fast Fourier transform of the image.
Spectroscopic Monitoring of (CdSe)34 Formation and Conversion
Our prior study established magic-size CdSe nanoclusters to be intermediates in the formation of CdSe QBs.10 However, the eq 1 reaction in primary-amine solvents at room temperature gave (CdSe)13,16 whereas the eq 2 reaction in primary-amine/secondary-amine cosolvents at room temperature gave CdSe QPs. Thus, we monitored the eq 2 reaction by UV–visible spectroscopy to determine if magic-size nanocluster intermediates participated in the reaction.
The eq 2 reaction was conducted in an n-octylamine/di-n-octylamine cosolvent mixture at room temperature as described above. An aliquot removed from the reaction mixture after 1 h gave the spectrum in Figure S4a, Supporting Information, which has been previously assigned to the magic-size nanocluster (CdSe)34.14,23 In our prior study,10 we mistakenly assigned one of these spectroscopic features to (CdSe)66, but the results reported here (see below) demonstrate that the spectrum does indeed correspond to (CdSe)34. A second aliquot was removed from the reaction mixture after 2 days, which gave the spectrum in Figure S4b, Supporting Information, clearly assignable to CdSe QPs. Thus, spectroscopic monitoring suggested that (CdSe)34 was an intermediate in the formation of the QPs; other magic-size nanoclusters were not observed.
We then combined the eq 2 reactants in an n-octylamine/di-n-pentylamine cosolvent mixture at the lower temperature of 0 °C, to determine if other nanocluster intermediates would be detected. (The reaction was conducted in a different cosolvent mixture because n-octylamine/di-n-octylamine mixtures are solid at 0 °C.) The spectrum of an aliquot taken after 12 h at 0 °C corresponded exclusively to (CdSe)34 (Figure 9a). The mixture was then monitored for over 1 month at 0 °C. The spectrum after 14 days corresponded to a mixture of (CdSe)34 and (CdSe)13 (Figure 9b). After 1 month, the (CdSe)34 was completely converted to (CdSe)13 (Figure 9c). For a similar reaction conducted at 0 °C in a n-propylamine/di-n-ethylamine cosolvent mixture, the conversion of (CdSe)34 to (CdSe)13 was complete in about 1 week (see Figure S5, Supporting Information). The results established that, under these conditions, (CdSe)13 was more thermodynamically stable than (CdSe)34, a conclusion supported by another observation (see below).
We next sought to determine if the secondary-amine cosolvent was merely an inert diluent of the primary-amine component (an inert cosolvent) or was an active participant in the initial, selective formation of (CdSe)34. Consequently, the room-temperature synthesis described above was conducted using the inert cosolvent 1-octadecene in place of the secondary-amine cosolvent. Reaction monitoring after 5 min revealed the (unselective) formation of a mixture of (CdSe)13 and (CdSe)34, from which the (CdSe)34 was gradually converted to a mixture of (CdSe)13 and CdSe QPs. The results indicated that (CdSe)34 is a kinetic product, and its conversion to the thermodynamically more stable (CdSe)13 is actively hindered in the presence of a secondary amine.
The conversion kinetics of (CdSe)34 to CdSe QPs were determined by UV–visible spectroscopy. Figure S6, Supporting Information, shows the spectrum of (CdSe)34 prepared in an n-octylamine/di-n-pentylamine cosolvent mixture at 0 °C, as described above, having a prominent absorption feature at 418 nm (black curve). Over the course of several hours at room temperature, a sharp absorption feature emerged at 448 nm corresponding to the lowest-energy transition in the spectrum of CdSe QPs (red and blue curves). A second QP feature grew in at 423 nm, only slightly shifted from the 418 nm absorption of (CdSe)34. The blue curve in Figure S6, Supporting Information, corresponds to the fully transformed sample. The kinetics of the appearance of CdSe QPs and the disappearance of (CdSe)34 were monitored by curve fitting of the 418, 423, and 448 nm absorptions (Figure S7, Supporting Information).
For kinetic analysis, (CdSe)34 was diluted into a cosolvent mixture having a lower n-octylamine/di-n-pentylamine ratio (which increased the conversion rate). The appearance of CdSe QPs at room temperature was followed by the integrated area of the QP absorption at 448 nm derived from the curve fitting. As shown in Figure 10, the log of the integrated absorption vs time was linear over three half-lives (kobs = (4.94 ± 0.47) × 10–5 s–1; t1/2 = 233 ± 22.2 min), establishing a first-order process. The inverse of the integrated absorption vs time was nonlinear, ruling out second-order kinetics (Figure 10). Significantly, no induction (nucleation) period was observed; first-order QP growth began immediately upon warming the (CdSe)34 solution to room temperature.
Figure 10.
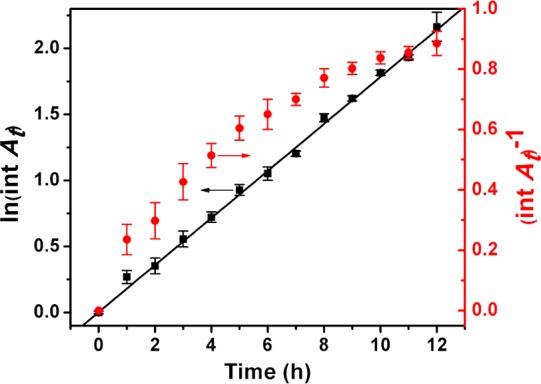
Kinetic data for the conversion of (CdSe)34 to CdSe QPs at room temperature. The black squares in the first-order plot (left axis) were obtained from the integrated area of the 448 nm QP absorption (see the text). The black line is the linear least-squares fit. The data are also plotted for second-order kinetics (red points), which are nonlinear.
The kinetics were also analyzed by the disappearance of the fitted 418 nm (CdSe)34 feature and the appearance of the fitted 423 nm CdSe QP feature. These data also gave linear first-order plots over three half-lives (Figures S8 and S9, Supporting Information). The kinetic parameters for the disappearance of (CdSe)34 were determined to be kobs = −(4.69 ± 0.81) × 10–5 s–1; t1/2 = 247 ± 42.6 min. This rate constant is, within experimental error, the opposite of that for the appearance of CdSe QPs (see above), establishing that the conversion of (CdSe)34 to CdSe QPs occurs without the accumulation of an intermediate. The appearance of CdSe QPs analyzed using the 423 nm feature gave kobs = (5.14 ± 1.03) × 10–5 s–1; t1/2 = 225 ± 50.5 min, in good agreement with the more-precise 448 nm data. Thus, the conversion was demonstrated to be a first-order process, with no induction period.
The room-temperature conversion rates of (CdSe)34 were influenced by the alkyl-chain lengths on the primary and secondary amine cosolvents and on the primary/secondary amine ratio. Shorter-chain lengths on both the primary and secondary amines increased the rates of room-temperature conversion of (CdSe)34 to CdSe QPs, presumably by enhancing diffusion. Lower primary/secondary amine ratios increased the room-temperature conversion rates of (CdSe)34 to CdSe QPs, perhaps by the increased lability of secondary-amine ligands on (CdSe)34. Higher primary/secondary amine ratios decreased the room-temperature conversion rates of (CdSe)34 to CdSe QPs, by facilitating the conversion of (CdSe)34 to (CdSe)13. The cluster (CdSe)13 seems particularly stabilized by primary-amine ligation.
Characterization of [(CdSe)34(n-octylamine)16(di-n-pentylamine)2]
A ligated derivative of (CdSe)34 as a slushy, greenish-yellow solid was obtained from preparations conducted in n-octylamine and di-n-pentylamine cosolvents. The UV–visible spectrum (Figure S10, Supporting Information) matched those of (CdSe)34 in Figures 9a and S6, Supporting Information. Although no features assignable to other magic-size nanoclusters were detected, the presence of (CdSe)13, (CdSe)19, or (CdSe)33 in small amounts was not ruled out, because their absorptions appear at shorter wavelengths and may have been obscured by the absorptions of (CdSe)34.
The isolated (CdSe)34 specimen was further characterized by laser-desorption-ionization (LDI) mass spectrometry (see Figure 11). The spectrum contained a prominent ion centered at m/z 6508 corresponding to the bare (CdSe)34 nanocluster, indicating ligand desorption had occurred during the experiment. Peaks were also present for each fragment nanocluster (CdSe)x, over the range of x = 33–13. The (CdSe)19 (m/z 3652) and (CdSe)33 (m/z 6320) ions were slightly more abundant, and the (CdSe)13 (m/z 2502) ion was significantly more abundant, than those of the other fragment ions. Significantly, the Figure 11 LDI mass spectrum differed markedly from the one that we previously reported for isolated [(CdSe)13(n-octylamine)13]15 and was consistent with a simple fragmentation process from the (CdSe)34 parent. However, the data did not confirm that (CdSe)34 had been isolated in a pure form, because of the presence of the fragment ions corresponding to the other magic sizes (CdSe)13, (CdSe)19, and (CdSe)33. The results did establish that the sample was at least highly enriched in (CdSe)34.
The composition of the ligand shell was determined by calibrated mass spectrometry (see the Supporting Information). Analysis of the isolated (CdSe)34 derivative gave an n-octylamine/di-n-pentylamine ligand ratio of 8.1 ± 0.5. That ratio was used to fit the C, H, and N analyses, providing an excellent fit to the formula [(CdSe)34(n-octylamine)16(di-n-pentylamine)2]. On the basis of that formula, the (CdSe)34 derivative was isolated in a 96% yield.
Nanoclusters of (CdSe)34 dispersed in a di-n-alkylamine solvent were stable for over one month at room temperature. Isolated [(CdSe)34(n-octylamine)16(di-n-pentylamine)2] was stable at room temperature for over one week and stable for longer periods when stored at 0 °C. A color change to reddish orange was observed when the greenish yellow [(CdSe)34(n-octylamine)16(di-n-pentylamine)2] was subjected to a vacuum (0.1 Torr) for longer than 12 h. However, after redispersion of such samples in toluene, analysis by UV–visible spectroscopy showed that the (CdSe)34 nanocluster remained intact, with no evidence for other species.
Discussion
Syntheses of flat CdSe nanocrystals may be categorized in two general types. In the first type, long-chain cadmium carboxylate precursors and high reaction temperatures (≥170 °C) are employed, yielding QPs having zinc-blende structures.17−19,21 The second type, used here, employs simple cadmium salts and amine solvents at comparatively low temperatures (25–100 °C), producing CdSe QPs and QBs having wurtzite structures.10,11 The spectroscopic properties of the two types of flat CdSe nanocrystals are closely related and produce comparable quantum-well absorption and emission spectra.17,22
The preparation of CdSe QPs having three discrete thicknesses, 1.4, 1.8, and 2.2 nm, is described here. These discrete thicknesses correspond to integer numbers of CdSe monolayers. Because the wurtzite QPs exhibit a [112̅0] orientation, the monolayer thickness is a/2 = 0.20 nm, which is half of the basal unit-cell face diagonal. Note that both a and c are compressed in the QPs relative to the bulk values, so that a/2 here is smaller than the bulk value (see the Results). Therefore, the three discrete QP thicknesses we have obtained are 7, 9, and 11 monolayers. In comparison, Dubertret and co-workers18 have prepared CdSe QPs having 4, 5, 6, and 7 monolayers, corresponding to discrete thicknesses of 1.2, 1.5, 1.8, and 2.1 nm. Because of the zinc-blende structure and [100] orientation of Dubertret’s QPs, the monolayer thickness is a/2 = 0.30 nm, explaining the apparent discrepancy between the monolayer and actual thicknesses of the two sets of nanocrystals.
We also report here the isolation of a ligated form of (CdSe)34 having the empirical formula [(CdSe)34(n-octylamine)16(di-n-pentylamine)2]. Our results may be compared to the room-temperature synthesis and isolation of n-octylamine-ligated (CdSe)34 recently described by Sardar and co-workers.24 Their nanocluster specimen is a bright yellow solid having a strong, narrow lowest-energy absorption feature at 418 nm, where we observed it in [(CdSe)34(n-octylamine)16(di-n-pentylamine)2] (Figure 9a). The Sardar absorption spectrum also contains the two higher-energy features we also observed for our amine-ligated (CdSe)34 (Figure 9a). Moreover, Sardar and co-workers reported LDI mass spectra in which the (CdSe)34 parent ion was the (most-intense) base peak. By recording LDI mass spectra at varying laser powers, they demonstrated that the lower-mass peaks present in the spectra were fragment ions of (CdSe)34. The evidence strongly supported the isolation of a purified, ligated form of (CdSe)34, which compares very closely to the [(CdSe)34(n-octylamine)16(di-n-pentylamine)2] isolated in this study.
Prominent, reasonably narrow absorption features having λmax values in the range of 410–420 nm have been frequently observed in studies of small CdSe nanocrystals or nanoclusters. In some cases, these absorptions have been assigned to magic-size nanocrystals or nanoclusters of unspecified stoichiometry.4,25−28 More recently, such features have been assigned to (CdSe)33, (CdSe)34 mixtures,23,29−31 or exclusively to (CdSe)34,24 as in the present study. Cossairt and Owen isolated a nanocluster having a Cd35Se28 stoichiometry (with additional charge-balancing ligation), which also gave a prominent absorption at 418 nm.32 Whether a variety of small CdSe nanoclusters has absorption features in this range or whether they equilibrate to the same absorbing species is presently unknown.
The ligand-to-cluster stoichiometry of [(CdSe)34(n-octylamine)16(di-n-pentylamine)2] is not readily reconciled with the theoretically proposed structures of the bare (CdSe)34 nanocluster. Both cage33 and core-cage23 structures have been proposed for (CdSe)34, the latter of which has a structure (CdSe)6@28, in which 6 formula units of CdSe are in the core of an outer cage structure.23 If one presumes amine binding to each surface Cd atom, the expected ligand/cluster ratio would be 34 or 28, respectively. That we have measured a ligand/cluster ratio of 18 suggests either that not all surface Cd atoms are ligated or that the cluster structure has only 18 surface Cd atoms.
In our previously reported synthesis of (CdSe)13 at room temperature in primary-amine solvents, an initial mixture of (CdSe)13, (CdSe)19, (CdSe)33, and (CdSe)34, was observed to equilibrate exclusively to (CdSe)13.15,16 The results here show that the same synthesis conducted in a primary-amine/secondary-amine cosolvent mixture initially produces (CdSe)34 as the only detectable nanocluster product, at both room temperature and at 0 °C (Scheme 3). At the lower temperature, (CdSe)34 eventually converts to (CdSe)13, establishing that (CdSe)13 is more thermodynamically stable under these conditions and that (CdSe)34 is a kinetic product (Scheme 3). The secondary-amine cosolvent slows the conversion of (CdSe)34 to (CdSe)13. The (CdSe)13 generated from (CdSe)34 in this manner requires temperatures above 40 °C for conversion to CdSe QPs, whereas this conversion occurs readily at room temperature from (CdSe)34 (Scheme 3). Thus, (CdSe)34 is a more potent nanocrystal precursor than is (CdSe)13, because we propose (CdSe)34 is much closer to the critical crystal-nucleus size.
Scheme 3. Reaction Scheme Summarizing the Formation and Interconversion of CdSe Nanoclusters and QPs in Primary-Amine/Secondary-Amine Co-Solvent Mixtures.
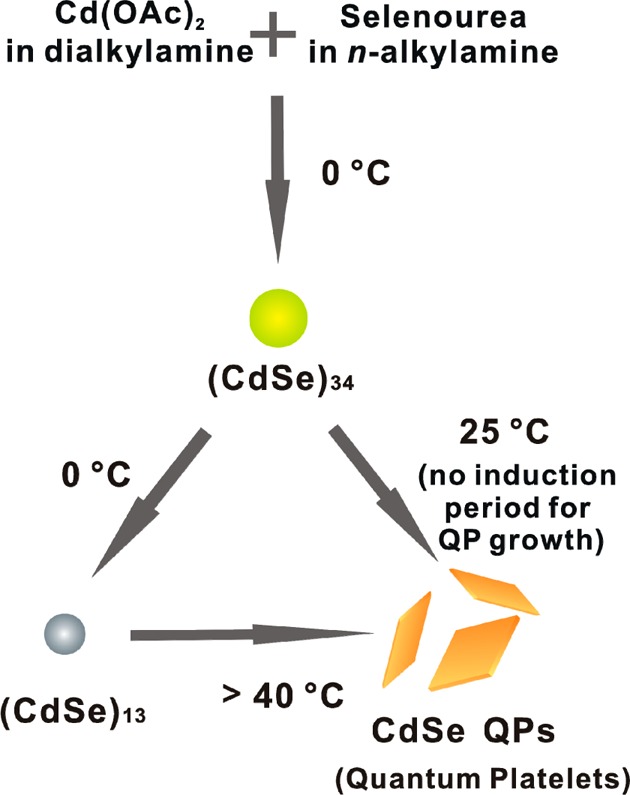
The nanocluster (CdSe)34 (yellow-green dot) is the kinetic product at 0 °C, which slowly converts to the thermodynamic product (CdSe)13 (gray dot) at 0 °C or to crystalline, wurtzite CdSe QPs (orange platelets) at 25 °C. Temperatures of >40 °C are required to convert the (CdSe)13 generated by the scheme to CdSe QPs.
The room-temperature conversion of (CdSe)34 to CdSe QPs occurs by first-order kinetics, with no induction period. The result requires that crystal nucleation is spontaneous under these conditions. The first-order nature of the conversion suggests that an activated, partially ligated form of (CdSe)34 is generated by a ligand dissociation in the rate-determining step, which itself either functions as a critical-size nucleus or coalesces with a fully ligated (CdSe)34 nanocluster in a subsequent fast bimolecular collision to exceed the critical-nucleus size. If correct, then the critical-nucleus size (CdSe)x is in the range of x = 34–68. Other experimental determinations of the CdSe critical-nucleus are in the diameter range of 1.2–1.6 nm.1,34 For comparison, (CdSe)34 has a theoretical diameter of 1.45 nm,23 and thus, the critical-size range we elucidate here is consistent with the prior measurements.
The very low temperature (25 °C) at which crystalline CdSe is produced here is surprising. The early syntheses of CdSe colloids conducted at room temperature within the water pools of inverse micelles gave materials of low crystallinity.35,36 The crystalline coherence lengths in such colloids were shown to be much smaller than the particle sizes.37 Consequently, most syntheses of CdSe nanocrystals are conducted at temperatures well above 200 °C.1−3,38,39 For example, the now-classical CdSe quantum-dot synthesis in TOPO solvent reported by Murray, Norris, and Bawendi employed nanocrystal-growth temperatures of 230–260 °C.38 Crystalline CdSe nanosheets12,14 and quantum belts10 have been grown at the low temperatures of 100 °C and 45–80 °C, respectively, using n-octylamine as the solvent and via magic-size nanocluster intermediates. Crystalline CdSe quantum dots have been obtained under aqueous conditions at 55 °C.40 To our knowledge, the synthesis of CdSe quantum platelets reported here, via the intermediacy of (CdSe)34 nanoclusters, constitutes the lowest temperature at which crystalline CdSe has been obtained.
In the Introduction, we argue that the monomer-generating reaction and crystal nucleation must be the two highest-barrier processes participating in semiconductor-nanocrystal growth. Therefore, we surmise that the high temperatures typically employed in nanocrystal synthesis reflect either high reaction barriers or high nucleation barriers with the use of conventional precursors and conditions. The very mild conditions for CdSe QP growth found here suggest that the nucleation barrier has nearly been surmounted in (CdSe)34. Magic-size nanoclusters should be ideal precursors to support semiconductor-nanocrystal growth and have been observed as reaction intermediates in nanocrystal synthesis2,5,41 since the early observations of Henglein and co-workers.42
The critical sizes and stoichiometries of crystal nuclei are likely precursor and condition dependent. To our knowledge, a stoichiometry for the critical-size crystal nucleus has not been previously determined for CdSe but has been reported to be (ZnO)25±4 for ZnO and (ZnSe)181±109 for ZnSe by Gamelin and co-workers.43,44 Our phenomenological determination of (CdSe)34–68 for CdSe is nicely consistent with these stoichiometries.
Conclusion
The magic-size nanocluster (CdSe)34 has been shown to be a potent, room-temperature precursor for crystalline CdSe QPs. The first-order conversion kinetics suggest that the critical nucleus is achieved in a deligated form of (CdSe)34 or in its combination with a second (CdSe)34, which supports room-temperature crystal growth. The nanocluster is obtained in isolable form as [(CdSe)34(n-octylamine)16(di-n-pentylamine)2], which functions as critical crystal nuclei that may be stored in a bottle.
The results suggest a strategy for making low-temperature nanocrystal synthesis more generally achievable. Magic-size nanoclusters like (CdSe)34 of other compositions should be near to the critical size and function as potent nucleating agents. Incorporating these into other mesophase-template geometries may provide low-temperature routes to well-passivated nanocrystals having a range of compositions and morphologies.
Acknowledgments
This work was supported by the NSF under grants CHE-1306507 (to W.E.B.) and DMR-0906966 (to R.A.L.) and grants from the National Institute of General Medical Sciences (8 P41 GM103422-35) from the National Institutes of Health.
Supporting Information Available
Additional experimental methods, XRD patterns, UV–visible spectra, and kinetic data (12 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bullen C. R.; Mulvaney P. Nano Lett. 2004, 4, 2303. [Google Scholar]
- Cumberland S. L.; Hanif K. M.; Javier A.; Khitrov G. A.; Strouse G. F.; Woessner S. M.; Yun C. S. Chem. Mater. 2002, 14, 1576. [Google Scholar]
- Lovingood D. D.; Oyler R. E.; Strouse G. F. J. Am. Chem. Soc. 2008, 130, 17004. [DOI] [PubMed] [Google Scholar]
- Murray C. B.; Norris D. J.; Bawendi M. G. J. Am. Chem. Soc. 1993, 115, 8706. [Google Scholar]
- Peng Z. A.; Peng X. G. J. Am. Chem. Soc. 2002, 124, 3343. [DOI] [PubMed] [Google Scholar]
- LaMer V. K.; Dinegar R. H. J. Am. Chem. Soc. 1950, 72, 4847. [Google Scholar]
- LaMer V. K. Ind. Eng. Chem. 1952, 44, 1270. [Google Scholar]
- Reiss H. J. J. Chem. Phys. 1951, 19, 482. [Google Scholar]
- Sugimoto T. Adv. Colloid Interface Sci. 1987, 28, 65. [Google Scholar]
- Liu Y. H.; Wang F. D.; Wang Y. Y.; Gibbons P. C.; Buhro W. E. J. Am. Chem. Soc. 2011, 133, 17005. [DOI] [PubMed] [Google Scholar]
- Joo J.; Son J. S.; Kwon S. G.; Yu J. H.; Hyeon T. J. Am. Chem. Soc. 2006, 128, 5632. [DOI] [PubMed] [Google Scholar]
- Son J. S.; Wen X. D.; Joo J.; Chae J.; Baek S.; Park K.; Kim J. H.; An K.; Yu J. H.; Kwon S. G.; Choi S. H.; Wang Z.; Kim Y. W.; Kuk Y.; Hoffmann R.; Hyeon T. Angew. Chem., Int. Ed. 2009, 48, 6861. [DOI] [PubMed] [Google Scholar]
- Son J. S.; Park K.; Kwon S. G.; Yang J.; Choi M. K.; Kim J.; Yu J. H.; Joo J.; Hyeon T. Small 2012, 8, 2394. [DOI] [PubMed] [Google Scholar]
- Yu J. H.; Liu X.; Kweon K. E.; Joo J.; Park J.; Ko K. T.; Lee D. W.; Shen S. P.; Tivakornsasithorn K.; Son J. S.; Park J. H.; Kim Y. W.; Hwang G. S.; Dobrowolska M.; Furdyna J. K.; Hyeon T. Nat. Mater. 2009, 9, 47. [DOI] [PubMed] [Google Scholar]
- Wang Y. Y.; Liu Y. H.; Zhang Y.; Wang F. D.; Kowalski P. J.; Rohrs H. W.; Loomis R. A.; Gross M. L.; Buhro W. E. Angew. Chem., Int. Ed. 2012, 51, 6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. Y.; Liu Y. H.; Zhang Y.; Kowalski P. J.; Rohrs H. W.; Buhro W. E. Inorg. Chem. 2013, 52, 2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ithurria S.; Dubertret B. J. Am. Chem. Soc. 2008, 130, 16504. [DOI] [PubMed] [Google Scholar]
- Ithurria S.; Tessier M. D.; Mahler B.; Lobo R. P. S. M.; Dubertret B.; Efros A. L. Nat. Mater. 2011, 10, 936. [DOI] [PubMed] [Google Scholar]
- Ithurria S.; Bousquet G.; Dubertret B. J. Am. Chem. Soc. 2011, 133, 3070. [DOI] [PubMed] [Google Scholar]
- Mahler B.; Nadal B.; Bouet C.; Patriarche G.; Dubertret B. J. Am. Chem. Soc. 2012, 134, 18591. [DOI] [PubMed] [Google Scholar]
- Li Z.; Peng X. J. Am. Chem. Soc. 2011, 133, 6578. [DOI] [PubMed] [Google Scholar]
- Liu Y. H.; Wayman V. L.; Gibbons P. C.; Loomis R. A.; Buhro W. E. Nano Lett. 2010, 10, 352. [DOI] [PubMed] [Google Scholar]
- Kasuya A.; Sivamohan R.; Barnakov Y. A.; Dmitruk I. M.; Nirasawa T.; Romanyuk V. R.; Kumar V.; Mamykin S. V.; Tohji K.; Jeyadevan B.; Shinoda K.; Kudo T.; Terasaki O.; Liu Z.; Belosludov R. V.; Sundararajan V.; Kawazoe Y. Nat. Mater. 2004, 3, 99. [DOI] [PubMed] [Google Scholar]
- Dolai S.; Nimmala P. R.; Mandal M.; Muhoberac B. B.; Dria K.; Dass A.; Sardar R. Chem. Mater. 2014, 26, 1278. [Google Scholar]
- Bowers M. J. II; McBride J. R.; Rosenthal S. J. J. Am. Chem. Soc. 2005, 127, 15378. [DOI] [PubMed] [Google Scholar]
- Bowers M. J. II; McBride J. R.; Garrett M. D.; Sammons J. A.; Duckes A. D. III; Schreuder M. A.; Watt T. L.; Lupini A. R.; Pennycook S. J.; Rosenthal S. J. J. Am. Chem. Soc. 2009, 131, 5730. [DOI] [PubMed] [Google Scholar]
- Lands C.; El-Sayed M. A. J. Phys. Chem. A 2002, 106, 7621. [Google Scholar]
- Yu K.; Hu M. Z.; Wang R.; Piolet M. L.; Frotey M.; Zaman M. B.; Wu X.; Leek D. M.; Tao T.; Wilkinson D.; Li C. J. Phys. Chem. C 2010, 114, 3329. [Google Scholar]
- Duckes A. D. III; McBride J. R.; Rosenthal S. J. Chem. Mater. 2010, 22, 6402. [Google Scholar]
- Harrell S. M.; McBride J. R.; Rosenthal S. J. Chem. Mater. 2013, 25, 1199. [Google Scholar]
- Kasuya A.; Noda Y.; Dmitruk I.; Romanyuk V.; Barnakov Y.; Tohji K.; Kumar V.; Belosludov R.; Kawazoe Y.; Ohuchi N. Eur. Phys. J. D 2005, 34, 39. [Google Scholar]
- Cossairt M.; Owen J. S. Chem. Mater. 2011, 23, 3114. [Google Scholar]
- Nguyen K. A.; Day P. N.; Pachter R. J. Phys. Chem. C 2010, 114, 16197. [Google Scholar]
- Xie C.; Hao H. X.; Chen W.; Wang J. K. J. Cryst. Growth 2008, 310, 3504. [Google Scholar]
- Ptatschek V.; Schmidt T.; Lerch M.; Muller G.; Spanhel L.; Emmerling A.; Fricke J.; Foitzik A. H.; Langer E. Ber. Bunsenges. Phys. Chem. 1998, 102, 85. [Google Scholar]
- Steigerwald M. L.; Alivisatos A. P.; Gibson J. M.; Harris T. D.; Kortan R.; Muller A. J.; Thayer A. M.; Duncan T. M.; Douglass D. C.; Brus L. E. J. Am. Chem. Soc. 1988, 110, 3046. [Google Scholar]
- Bawendi M. G.; Kortan A. R.; Steigerwald M. L.; Brus L. E. J. Chem. Phys. 1989, 91, 7282. [Google Scholar]
- Murray C. B.; Norris D. J.; Bawendi M. G. J. Am. Chem. Soc. 1993, 115, 8706. [Google Scholar]
- Peng Z. A.; Peng X. G. J. Am. Chem. Soc. 2002, 124, 3343. [DOI] [PubMed] [Google Scholar]
- Al-Amri A. M.; Yaghmour S. J.; Mahoud W. E. J. Cryst. Growth 2011, 334, 76. [Google Scholar]
- Soloviev V. N.; Eichhofer A.; Fenske D.; Banin U. J. Am. Chem. Soc. 2001, 123, 2354. [DOI] [PubMed] [Google Scholar]
- Fojtik A.; Weller H.; Koch U.; Henglein A. Ber. Bunsenges. Phys. Chem. 1984, 88, 969. [Google Scholar]
- Bryan D. J.; Schwartz D. A.; Gamelin D. R. J. Nanosci. Nanotechnol. 2005, 5, 1472. [DOI] [PubMed] [Google Scholar]
- Norberg N. S.; Parks G. L.; Salley G. M.; Gamelin D. R. J. Am. Chem. Soc. 2006, 128, 13195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.