

Abstract
Biofilms are surface-attached communities of bacteria enclosed in a polysaccharide matrix. Bacteria in a biofilm are extremely resistant to antibiotics. Several recent reports have linked the signaling molecule nitric oxide (NO) with biofilm dispersal. We have previously reported that an H-NOX (heme-nitric oxide/oxygen binding) protein in the biofilm-dwelling bacterium Shewanella woodyi mediates NO-induced biofilm dispersal. In S. woodyi, H-NOX (SwH-NOX) is cocistronic with a gene encoding a dual-functioning diguanylate cyclase/phosphodiesterase enzyme, designated here as HaCE (H-NOX-associated cyclic-di-GMP processing enzyme). Enzymes such as these are responsible for regulating the intracellular concentrations of cyclic-di-GMP, a secondary signaling molecule essential to biofilm formation in bacteria. We have demonstrated that NO-bound SwH-NOX regulates both enzymatic activities of SwHaCE, resulting in decreased cellular cyclic-di-GMP levels and disruption of biofilm formation. Thus, H-NOX/HaCE represents a potential drug target for regulating biofilm formation. In this work, the SwH-NOX surface residues critical for the formation of a protein complex with SwHaCE are identified using nuclear magnetic resonance, fluorescence quenching, and cosedimentation. Enzyme assays confirm this protein–protein interface and its importance for H-NOX/HaCE function.
Biofilm formation is an important phenotype in bacteria, where cells switch from a motile, free-swimming, single-cell growth mode to a surface-attached, sessile, community of cells encapsulated in a matrix consisting of exopolysaccharide, protein, and DNA.1,2 Bacteria within biofilms are extremely resistant to antibiotics and are thought to be responsible for several drug-resistant infectious diseases. Biofilm development is poorly understood in general, but the intracellular concentration of bis-(3′–5′)-cyclic dimeric guanosine monophosphate (cyclic-di-GMP or c-di-GMP) has been shown to be important for the regulation of motility and biofilm formation,3,4 in addition to many other important signaling processes, including cell division, transcriptional regulation, and virulence.5−8
In bacteria, two classes of enzymes maintain the intracellular pool of cyclic-di-GMP. Cyclic-di-GMP is synthesized from two molecules of GTP by GGDEF domain-containing diguanylate cyclases (DGCs).9−11 Cyclic-di-GMP is hydrolyzed by ExL domain-containing phosphodiesterases (PDEs)12−14 to form the linear metabolite 5′-phosphoguanylyl-(3′→5′)-guanosine (pGpG). Because cyclic-di-GMP signaling and bacterial biofilms are central to many infectious diseases, the pathways that contribute to the regulation of intracellular cyclic-di-GMP concentrations are potentially important antibiotic drug targets.
Nitric oxide (NO) is a well-established and important signaling molecule in mammals that regulates a variety of physiological processes, including vasodilation and neurotransmission.15 The enzyme soluble guanylate cyclase (sGC) acts as an NO sensor via its N-terminal heme-nitric oxide/oxygen binding domain.16 Interestingly, several different groups have recently observed that low concentrations of NO induce biofilm dispersal in a variety of bacterial species.17−19 In bacteria, H-NOX domains typically exist as stand-alone proteins that regulate cocistronic histidine kinase [H-NOX-associated histidine kinase (HahK)] or c-di-GMP synthase and/or phosphodiesterase [H-NOX-associated cyclic-di-GMP processing enzyme (HaCE)] proteins.20,21 In general, the biophysical characterization of these H-NOX/enzyme complexes is lacking.
H-NOX signaling in Legionella pneumophila and Shewanella woodyi, both of which encode a diguanylate cyclase/phosphodiesterase protein in the same operon as an H-NOX domain, has been characterized.22 It is reported that the H-NOX-associated protein Lpg1057 (LpHaCE) from L. pneumophila has in vitro diguanylate cyclase activity that is inhibited by NO-bound H-NOX.23 The phosphodiesterase domain was found to be inactive. In S. woodyi, H-NOX regulates the DGC and PDE activities of SwHaCE (Swoo_2750), previously termed SwDGC.24 In the absence of NO, SwH-NOX stimulates diguanylate cyclase activity while the phosphodiesterase activity remains at basal levels. In the presence of NO-bound H-NOX, however, this pattern is reversed. SwHaCE displays basal level cyclase activity, whereas the phosphodiesterase activity is upregulated. These results underlie the additional finding that nanomolar NO leads to less biofilm and a decreased level of cyclic-di-GMP in S. woodyi. These data are consistent with H-NOX acting as an NO sensor that regulates biofilm formation by modulating the intracellular level of cyclic-di-GMP in bacteria.
Therefore, the H-NOX/HaCE protein–protein interaction is important for regulating the biofilm formation pathway in some bacteria. Here we use a combination of techniques to study how HaCE proteins are regulated by H-NOX. We employ NMR chemical shift perturbation experiments to identify the residues of SwH-NOX that interact with SwHaCE. The role of these residues was confirmed by enzyme activity assays as well as fluorescence binding studies with mutant proteins. Sedimentation equilibrium studies were used to assess the stoichiometry of the SwH-NOX/SwHaCE complex. The data were used to generate a model for H-NOX regulation of HaCE activity.
Experimental Procedures
Materials and General Methods
All reagents were obtained at their highest available purity and used as received. All enzymes were purchased from New England Biolabs.
Protein Expression and Purification
SwH-NOX and SwHaCE were expressed and purified as described previously.22,24 In brief, the plasmid containing the SwH-NOX gene was transformed into Escherichia coli Tuner(DE3)pLysS cells for protein expression. The cells were grown in yeast extract medium containing a final sodium phosphate concentration of 100 mM. The protein expression was induced with 10 μM IPTG (final concentration) at 18 °C overnight. The pellet was harvested and stored at −80 °C until it was purified. For purification, the pellet was thawed on ice and resuspended in H-NOX A buffer [50 mM sodium phosphate and 300 mM sodium chloride (NaCl) (pH 8.0)]. After sonication, the lysate was spun at 18000 rpm (39200g) for 1 h. The cleared supernatant was loaded on a nickel-NTA (GE) column, and the protein was eluted using an imidazole gradient of H-NOX B buffer [50 mM sodium phosphate, 300 mM NaCl, and 250 mM imidazole (pH 8.0)]. The plasmid encoding SwHaCE24 was transformed into E. coli BL21(DE3)pLysS cells for protein expression. The cells were grown in 2XYT medium containing 16 g of tryptone, 10 g of yeast extract, and 5 g of NaCl per liter. Protein expression was induced with 100 μM IPTG (final concentration) at 16 °C overnight. The pellet was resuspended in lysis buffer containing 50 mM Tris base, 10 mM magnesium chloride (MgCl2), 25 mM potassium chloride (KCl), 300 mM NaCl, 10% glycerol, and 2 mM βME (β-mercaptoethanol) (pH 8.0). Phenylmethanesulfonyl fluoride (PMSF) dissolved in methanol was added to a final concentration of 1 mM to the resuspended pellet prior to lysis to inhibit protease activity. After sonication, the lysate was spun at 18500 rpm (41400g) for 1 h. The cleared supernatant was loaded on a nickel-NTA column (GE), and the protein was eluted using an imidazole gradient in lysis buffer. After elution off the Ni column, the protein was loaded on Superdex-200 (GE) equilibrated in lysis buffer without glycerol and PMSF to obtain >95% pure protein.
To produce 13C- and 15N-labeled protein, E. coli Tuner(DE3)pLysS cells transformed with the SwH-NOX plasmid were grown in M9 minimal medium supplemented with 15NH4Cl and 13C-labeled glucose (Cambridge Isotope Laboratories, Inc.) as the sole sources of 15N and 13C, respectively. Protein expression was induced by adding 10 μM IPTG (final concentration) at 25 °C and allowing cultures to grow overnight. Then the protein was purified by nickel-NTA affinity chromatography followed by gel filtration using Superdex 200. The yield of the isotope-labeled protein was ∼10 mg/L. For all protein-based assays described below, the protein concentrations were determined using the Bradford Protein Assay (Thermo Scientific, Pierce).25 The standard curve for the assay was prepared using BSA (Thermo Scientific, Pierce).
Site-Directed Mutagenesis
The Stratagene QuikChange protocol was used to introduce several SwH-NOX point mutations. The following complementary primers were used to make the E16K, F17A, and E20K SwH-NOX mutants: E16K, 5′-GGAG TTG ATT GAA GAT AAA TTC GGA TAT GAA ACC-3′; F17A, 5′-GAG TTG ATT GAA GAT GAG GCT GGA TAT GAA ACC-3′; and E20K, 5′-GAG TTC GGA TAT AAA ACC TTA GAT ACT TTA CTT G-3′ (the underlined portions are the mutated codons). Phusion polymerase was purchased from New England Biolabs. The mutagenesis products were transformed into E. coli Dh5α cells for propagation. The mutations were confirmed using DNA sequencing results obtained from the DNA sequencing facility at Stony Brook University. The expression and purification of SwH-NOX surface mutants were conducted using the same protocols as described above for the wild-type SwH-NOX protein.
NMR and Data Analysis
The SwH-NOX Fe(II)–CO complex was used in the NMR studies to avoid paramagnetic effects resulting from the unpaired electron in the Fe(II)–NO and Fe(II)–unligated SwH-NOX complexes. The Fe(II)–CO complex was prepared in an anaerobic glovebag as previously described.26 The SwH-NOX concentration was 400 μM, dissolved in 50 mM sodium phosphate, 50 mM NaCl, and 10 mM sodium dithionite (pH 7.6). Triple-resonance NMR experiments for the backbone assignments of SwH-NOX (HNCO, HNCACB, CBCACONH, and HNCA) were performed at 25 °C on a Bruker 900 MHz spectrometer equipped with a cryoprobe at the New York Structural Biology Center (New York, NY). Data were processed using NMRPipe27 followed by analysis using Sparky;28 75% of the backbone resonances were assigned.
Transverse relaxation-optimized spectroscopy (TROSY)-edited 15N–1H HSQC experiments were performed on the same spectrometer at 25 °C for the following samples: (1) the 15N-labeled Fe(II)–CO SwH-NOX complex and (2) the 15N-labeled Fe(II)–CO SwH-NOX complex and the nonisotopically labeled binding partner SwHaCE, at a ratio of 1:1.25. The buffer used was the same as that used in the three-dimensional (3D) NMR experiments. Then, the weighted average chemical shift difference (Δ in parts per million) was calculated using the equation
| 1 |
Tryptophan Fluorescence Quenching
Intrinsic fluorescence experiments were conducted on a PTI spectrofluorometer. SwHaCE (1 μM) was placed in a cuvette, and the SwH-NOX Fe(II)–NO complex (WT and mutants E16K, F17A, and E20K, separately) was titrated serially into the protein while being stirred constantly at 25 °C. SwHaCE was diluted to 10% of the starting volume (2000 μL) . Titration was continued until the maximal change in fluorescence had been achieved. The data were fit to a first-order binding equation in Origin:
| 2 |
Sedimentation Velocity and Sedimentation Equilibrium
Experiments were conducted at 4 °C on a Beckman Optima XL-Analytical ultracentrifuge using an An-60 Ti rotor. For sedimentation velocity experiments, SwHaCE was incubated with the SwH-NOX Fe(II)–NO complex (WT and mutant E16K, separately), and the protein complex was centrifuged at 50000 rpm (201240g) for 24 h with 60 scans at 280 nm. The protein concentration was adjusted so that the absorbance was 0.8 at 280 nm. The data were analyzed using SEDFIT.29 The buffer consisted of 50 mM Tris-HCl, 250 mM NaCl, 25 mM KCl, 10 mM MgCl2, and 2 mM βME (pH 8.0).
For the equilibrium experiments with SwHaCE alone, SwHaCE was analyzed at three rotor speeds (9000, 14000, and 18000 rpm, which are equal to 6500g, 15800g, and 26100g, respectively), which were calculated using the molecular mass of SwHaCE (74.5 kDa). Two scans were collected at the end of each speed. The protein absorbance was followed at 280 and 260 nm. The data were analyzed using HeteroAnalysis (University of Connecticut Analytical Ultracentrifugation Facility) to determine the molecular mass for SwHaCE alone in solution. The data were fit globally across samples and speeds to yield the apparent molar mass.
For the equilibrium experiments with the SwH-NOX/SwHaCE complex, SwHaCE, either the WT or the E16K mutant, was incubated with WT SwH-NOX, as either the Fe(II)-unligated or SwH-NOX Fe(II)–NO complex, in 1:1, 1:2, and 1:5 molar ratios, and analyzed at three rotor speeds (9000, 14000, and 18000 rpm, which are equal to 6500g, 15800g, and 26100g, respectively) with two scans at the end of each speed. The scans were collected at an absorbance of 400 nm for experiments with the Fe(II)–NO complex or 430 nm for experiments with the Fe(II)-unligated complex to follow the heme absorbance of SwH-NOX. The protein concentration was adjusted so that the absorbance was 0.5 at 430 nm. For the equilibrium experiments with SwH-NOX alone, SwH-NOX was analyzed at three rotor speeds (17000, 27000, and 34000 rpm, which are equal to 23300g, 58700g, and 93000g, respectively), which were calculated using the molecular mass of SwH-NOX (22.5 kDa).
Enzyme Activity Measurements Using the Malachite Green Assay
The SensoLyte MG phosphate assay kit was purchased from AnaSpec. For the diguanylate cyclase enzyme assays, SwHaCE by itself (1 μM), or in the presence of the SwH-NOX Fe(II)-unligated complex (WT, E16K, F17A, or E20K in a 1:20 concentration ratio), was incubated on ice for 10 min to allow the proteins to form a complex. The protein mixtures (20 μL) were then added to 180 μL a prepared solution containing 200 μM GTP (substrate), 5 mM MgCl2, and 1× buffer [50 mM Tris base and 10 mM MgCl2 (pH 7.5)]. The reaction mixture was incubated for 10 min at 25 °C followed by boiling at 100 °C to quench the reaction. After being cooled, the mixture was spun at 14000 rpm (16900g) for 5 min to remove any precipitate; 70 μL of the supernatant was mixed with 10 μL of 3 units/mL IPP [inorganic pyrophosphatase (NEB)] in a 96-well plate and incubated at 25 °C for 10 min to hydrolyze the inorganic pyrophosphate (PPi) produced during the reaction. Then, 20 μL of Malachite green reagent was added and the solution mixed, and the plate was read at 610 nm using a Perkin-Elmer Viktor X5 microplate reader. The absorbance readings were corrected for protein only and GTP only background. This experiment measured the initial velocity for the diguanylate cyclase activity for SwHaCE and SwHaCE in complex with SwH-NOX. This experiment was repeated in at least three fully independent experiments.
NO Dissociation Kinetics
NO dissociation kinetics of SwH-NOX mutants E16K, F17A, and E20K were conducted as previously described.30
Results
Despite the importance of biofilms in infectious diseases, the mechanisms that regulate biofilm formation and dispersal are poorly understood. In this study, we have characterized the H-NOX/HaCE protein complex from S. woodyi to improve our understanding of how NO causes biofilm dispersal in this bacterium.
Chemical Shift Perturbation Assays Reveal the Binding Interface for SwHaCE
We collected HNCO, HNCACB, CBCACONH, and HNCA spectra to assign the backbone resonances of SwH-NOX. Using standard methods, resonances from 75% of the SwH-NOX residues (∼140 residues) could be assigned, and these assignments were used to probe the binding of SwHaCE (Figure 1). HSQC NMR spectra of the 15N-labeled SwH-NOX Fe(II)–CO complex were compared to the spectra of the 1:1.25 complex of the 15N-labeled SwH-NOX Fe(II)–CO complex in the presence of SwHaCE. In the presence of SwHaCE, there was a significant loss of signal in the SwH-NOX HSQC spectrum because of peak broadening, presumably because the total size of the SwH-NOX/SwHaCE complex is expected to be at least 100 kDa (74.5 kDa for SwHaCE and 22.5 kDa for SwH-NOX). Hence, TROSY spectra were recorded for SwH-NOX alone (Figure 2, red) and in complex with SwHaCE (Figure 2, blue). No line broadening effects were observed, and the TROSY spectrum for the complex showed only one set of peaks, indicating fast exchange on the NMR time scale.
Figure 1.
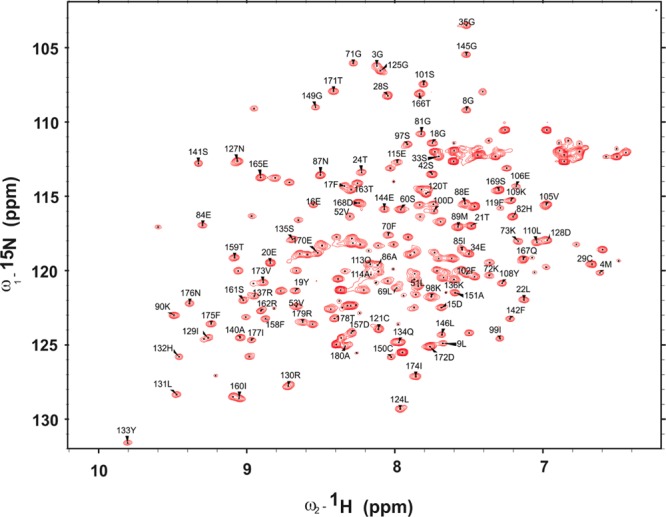
Backbone assignment for SwH-NOX [as the Fe(II)–CO complex]. Approximately 75% of the peaks were assigned using extensive 3D experiments. The x-axis represents the chemical shift in 1H, and the y-axis represents the chemical shift in 15N.
Figure 2.
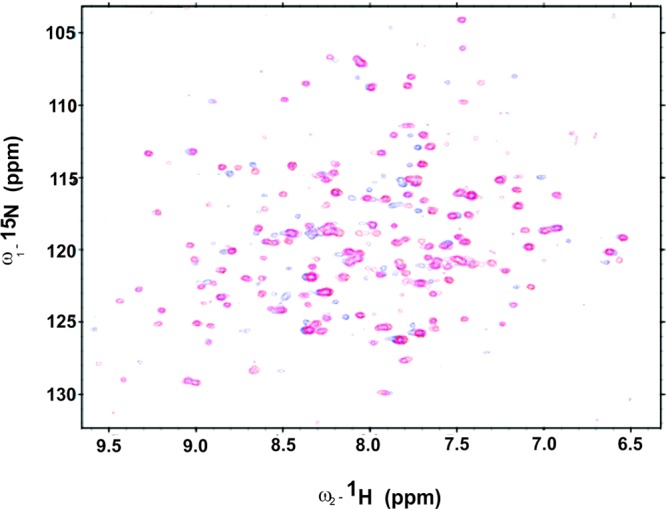
Chemical shift mapping for SwHaCE and SwH-NOX complex formation. The 15N–1H HSQC spectrum (red) was recorded for a sample containing uniformly 15N-labeled 50 μM SwH-NOX [as the Fe(II)–CO complex]. The blue spectrum was recorded for a 1:1.25 ratio of 15N-labeled SwH-NOX [as the Fe(II)–CO complex] to unlabeled SwHaCE. An overlay of these spectra indicates that peaks for some residues are shifted, indicating a change in their chemical environment in the presence of SwHaCE.
The chemical shifts of some residues shifted significantly for the protein/protein complex (Figure 2, blue), compared to the spectrum of SwH-NOX alone (Figure 2, red). To correlate the changes in chemical shift with structure, a homology model of SwH-NOX was generated (Figure 3A). The change in chemical shift for each assigned residue of SwH-NOX (with and without SwHaCE) was plotted (Figure 3B). Assuming a ratio of 0.02 to be a significant change, there are ∼11 SwH-NOX residues that shift upon exposure to SwHaCE.
Figure 3.
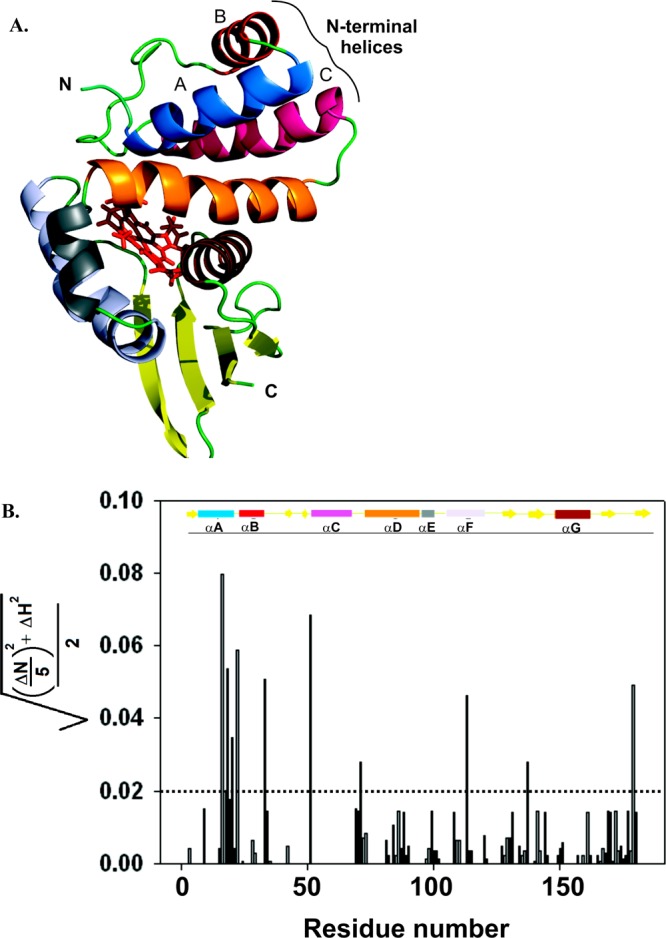
(A) Ribbon structure of the SwH-NOX protein from S. woodyi. This figure is a homology model generated in PyMOL using the solution-state NMR structure of Shewanella oneidensis(26) (So2144, Protein Data Bank entry 2KII, chain A). The heme is shown as red sticks. The N- and C-termini of the protein are denoted. The seven different helices are depicted in different colors: blue for helix αA, red for helix αB, hot pink for helix αC, orange for helix αD, gray for helix αE, purple for helix αF, and chocolate brown for helix αG. (B) Chemical shift changes for SwH-NOX upon addition of SwHaCE. The shifts (SwH-NOX to SwH-NOX/SwHaCE), calculated as {[(ΔN/5)2 + (ΔH)2]/2}1/2, are plotted against the primary sequence residue number. The cutoff shift was kept at 0.02, and shifts above 0.02 (dashed line) were considered significant. The secondary structure of SwH-NOX is shown as a cartoon in which the rectangles represent α-helices and the solid arrows represent β-strands. The color code corresponds to the ribbon diagram in panel A.
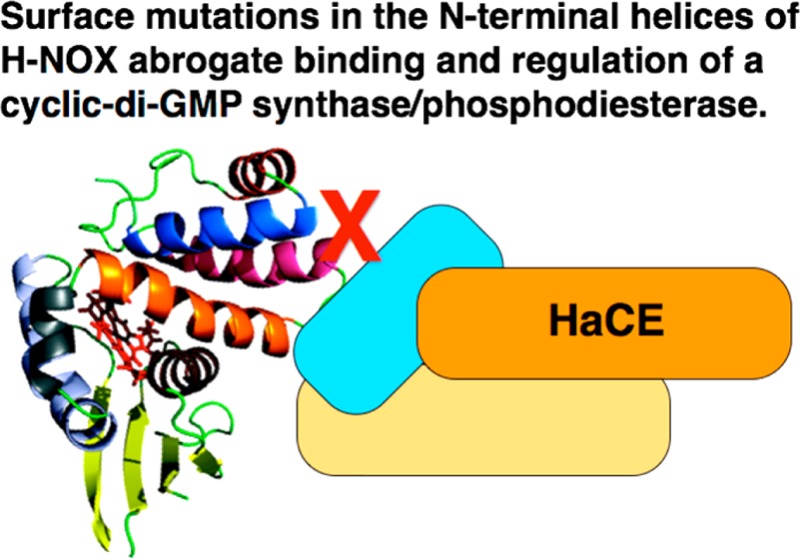
We were particularly interested in a set of five residues (amino acids 16–20) in the N-terminal helices of SwH-NOX that displayed the maximal shift. These residues form a cluster in N-terminal helices αB and αC on the surface of SwH-NOX, suggesting this contributes to the binding site for SwHaCE. To test this hypothesis, we constructed three SwH-NOX point mutants (highlighted in Figure 4), E16K, E20K, and F17A. We reasoned that charge reversal mutants (E16K and E20K) would be expected to significantly perturb binding if these residues play a key role and that reducing the bulk of F17 might weaken hydrophobic interactions during binding.
Figure 4.
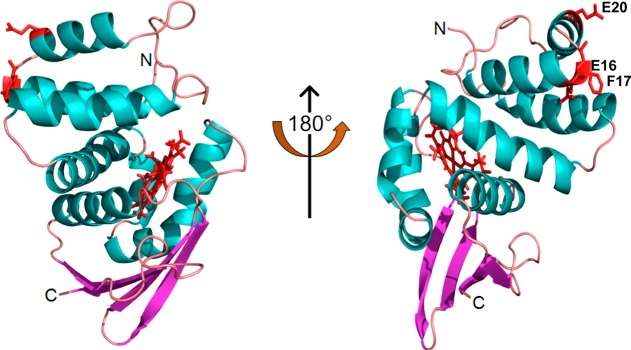
SwH-NOX surface residues identified from chemical shift mapping. The SwH-NOX residues with the maximal perturbation (E16, F17, and E20) upon incubation with SwHaCE are colored red. The heme cofactor is also colored red. These residues likely represent the binding patch for SwHaCE.
Characterization of ligand binding (binding of CO and NO to ferrous SwH-NOX and binding of CN– to ferric SwH-NOX) and NO dissociation kinetics were conducted for the WT and the surface mutants of SwH-NOX. The Soret values and koff(NO) calculated for H-NOX surface mutants were similar to those of WT H-NOX (Table S1 of the Supporting Information), confirming that mutating these surface residues does not affect the overall structure or heme properties of the SwH-NOX protein.
Fluorescence Quenching Assays Indicate That Binding of SwHaCE Is Significantly Weaker for Surface Mutants E16K, F17A, and E20K Than for WT SwH-NOX
SwHaCE has seven Trp residues, while SwH-NOX has none; thus, we reasoned that binding of SwH-NOX to SwHaCE might perturb the Trp fluorescence intensity of SwHaCE. Therefore, the steady-state fluorescence of 1 μM SwHaCE was followed at 370 nm in the presence of increasing concentrations of NO-bound H-NOX to assess SwH-NOX/SwHaCE binding. The SwH-NOX Fe(II)–NO complex (WT or mutant, separately; ∼1–10 μM) was titrated into the SwHaCE solution while it was being stirred constantly until the maximal change in fluorescence had been achieved. We observed a quenching of the fluorescence signal as a function of an increasing concentration of the SwH-NOX Fe(II)–NO complex in solution (Figure 5). Here, to have positive numbers, the difference (F0 – F) is plotted as a function of the concentration of NO-bound H-NOX in solution for each SwH-NOX construct. The data were corrected for dilution and inner filter effects and plotted against SwH-NOX Fe(II)–NO complex concentration. The apparent dissociation constant (KD,app) for the complex was determined to be 2.5–3 μM from three independent runs. This likely overestimates the true KD, as the concentrations used were high and not under pseudo-first-order conditions, to account for the sensitivity of the detector. Nonetheless, the effect of SwH-NOX surface mutations on binding to SwHaCE could be easily determined in this assay.
Figure 5.
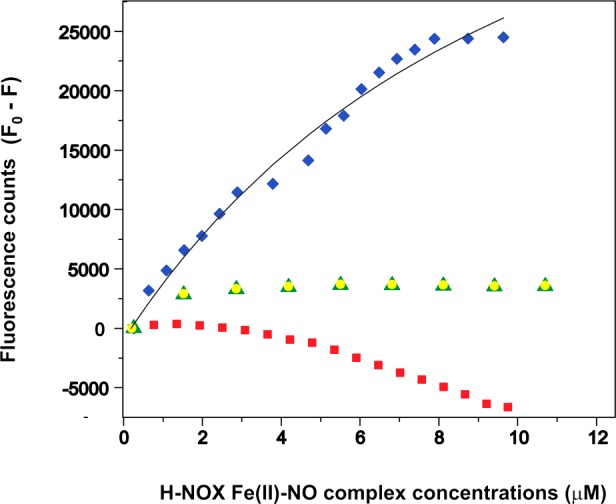
Tryptophan fluorescence quenching experiments for assessing SwH-NOX/SwHaCE binding. The Fe(II)–NO complex of SwH-NOX [WT (blue diamonds), E16K (red squares), F17A (green triangles), and E20K (yellow circles)] was titrated into a SwHaCE solution (1 μM) to determine the amount of SwHaCE Trp quenching. The difference in fluorescence (F0 – F) with and without SwH-NOX is plotted as a function of SwH-NOX Fe(II)–NO complex concentration. WT SwH-NOX caused significant quenching of SwHaCE fluorescence. In general, the SwH-NOX mutants do not quench SwHaCE fluorescence, indicating a loss of binding. From these data, the apparent equilibrium dissociation constant for the SwH-NOX/SwHaCE complex was determined to be ∼2.5 μM (the gray line shows the fit used to determine KD,app). Each titration was conducted in triplicate.
For the titrations with the Fe(II)–NO complex of the E16K mutant of SwH-NOX, the fluorescence signal for SwHaCE did not decrease over time and actually increased slightly (Figure 5). This indicates that the E16K mutation abolished binding of SwH-NOX to SwHaCE. The slight increase in the magnitude of the fluorescence signal toward the end of the titration could be due to conformational changes resulting from interactions of the SwH-NOX E16K mutant with other regions of SwHaCE at high H-NOX concentrations. For the SwH-NOX F17A and E20K mutants, we observed significantly less quenching of the SwHaCE signal than for the WT, suggesting a reduced binding affinity for each of these mutants for SwHaCE (Figure 5). These results confirm that surface residues E16, F17, and E20 in N-terminal helices αB and αC of SwH-NOX each contribute to SwHaCE binding and likely are part of a binding surface for interaction with SwHaCE.
Sedimentation Equilibrium Indicates That SwHaCE Forms a Dimer in Solution and the SwH-NOX/SwHaCE Complex Forms a Heterotetramer
On the basis of the NMR and fluorescence titration experiments described above, we concluded that SwH-NOX and SwHaCE form a protein/protein complex. We used sedimentation equilibrium measurements to determine the stoichiometry of this complex. Sedimentation equilibrium experiments are extensively used to determine the molecular masses of proteins and protein complexes in solution.31 On the basis of previously characterized diguanylate cyclases, it is expected that active diguanylate cyclases exist as dimers in solution.32−34 Because SwHaCE is an active cyclase enzyme,24 we wanted to determine if SwHaCE also forms oligomers (dimers) in solution, and we wanted to determine if the oligomerization state changes upon formation of a complex with SwH-NOX.
Sedimentation equilibrium experiments were conducted with SwHaCE alone, SwH-NOX [as the Fe(II)–NO or Fe(II)-unligated complex, separately] alone, and a mixture of SwHaCE and SwH-NOX [as the Fe(II)–NO or Fe(II)-unligated complex, separately] at a ratio of 1:1.5. These data are summarized in Table 1. For SwHaCE, HeteroAnalysis indicates a molecular mass of 144.3 kDa, which is close to the theoretical molecular mass expected for a dimer, 149 kDa (Figure 6A and Table 1). For WT SwH-NOX, as either the Fe(II)-unligated or Fe(II)–NO complex, our analysis indicated a molecular mass of 23 kDa, which is close to the theoretical molecular mass of 22.5 kDa (Figure S1 of the Supporting Information and Table 1). Thus, WT SwH-NOX alone exists as a monomer and SwHaCE as a dimer.
Table 1. Summary of Sedimentation Equilibrium Results and Controls.
| protein/protein complex | calculated molecular mass (theoretical molecular mass) (kDa)f |
|---|---|
| HaCE by itselfa,b | 144.3 (149.0, dimer) |
| HaCE with WT H-NOX Fe(II)–NOa,c | 197.2 (193.0, 2+2 heterotetramer) |
| HaCE with WT H-NOX Fe(II)a,d | 198.0 (193.0, 2+2 heterotetramer) |
| HaCE with E16K H-NOX Fe(II)–NOa,c | 76.0 (−) |
| WT H-NOX Fe(II)–NOa,c | 75.0 (−) |
| WT H-NOX Fe(II)–NOe,c | 23.3 (22.5, monomer) |
| WT H-NOX Fe(II)e,d | 23.0 (22.5, monomer) |
| E16K H-NOX Fe(II)–NOe,c | 23.8 (22.5, monomer) |
The sedimentation equilibrium run was conducted at three rotor speeds calculated for the molecular mass of SwHaCE (74.5 kDa): 9000 rpm (6520g), 14000 rpm (15800g), and 18000 rpm (26100g).
The scans for the sedimentation equilibrium runs were collected at 280 nm.
The scans for the sedimentation equilibrium runs were collected at 400 nm.
The scans for the sedimentation equilibrium runs were collected at 430 nm.
The sedimentation equilibrium run was conducted at three rotor speeds calculated for the molecular mass of SwH-NOX (22.5 kDa): 17000 rpm (23300g), 27000 rpm (58700g), and 34000 rpm (93100g).
HeteroAnalysis and Sednterp were used to calculate the molecular mass.
Figure 6.

(A) Sedimentation equilibrium experiments with SwHaCE in solution. Using rotor speeds of 9000, 14000, and 18000 rpm (which are equal to 6500g, 15800g, and 26100g, respectively), and following protein at 280 nm, the calculated molecular mass of SwHaCE is 144.3 kDa, indicating a dimer in solution at equilibrium. (B) Sedimentation equilibrium experiment for SwHaCE in solution with the SwH-NOX Fe(II)–NO complex. Using rotor speeds of 9000, 14000, and 18000 rpm (which are equal to 6500g, 15800g, and 26100g, respectively), and following protein complexes at 400 nm, the calculated molecular mass for the complex is 197.2 kDa at equilibrium. This matches the molecular mass expected for a heterotetramer of two SwH-NOX proteins and two SwHaCE proteins. Each experiment was conducted in triplicate.
The molecular mass for the SwH-NOX/SwHaCE complex, which was monitored at 400 nm (to follow heme absorbance), was calculated to be 197.2 kDa when SwH-NOX was run as the Fe(II)–NO complex (Figure 6B and Table 1) and 198.0 kDa when SwH-NOX was run as the Fe(II)-unligated complex (monitored at 430 nm to follow heme absorbance) (Figure S2 of the Supporting Information and Table 1), both of which are close to the theoretical molecular mass of 193 kDa for a heterotetrameric complex of four molecules comprised of two SwHaCE monomers and two SwH-NOX monomers. All of the molecular masses computed from the AUC experiments are within 3% of the theoretical value, which is within the experimental error of the instrument. Therefore, we conclude that SwHaCE and SwH-NOX form a 2:2 heterotetrameric protein complex in solution, regardless of whether NO is bound to SwH-NOX. Because SwH-NOX and SwHaCE are cocistronic, these data support our model in which H-NOX and HaCE likely form a complex upon production in bacterial cells.24 This is the first H-NOX/HaCE protein complex stoichiometry to have been determined.
The E16K SwH-NOX mutant [as the Fe(II)–NO complex] was analyzed both by itself and as a complex with SwHaCE. The mutant was determined to be a monomer by itself [calculated molecular mass of 23 kDa (Figure S1 of the Supporting Information and Table 1)], similar to WT SwH-NOX. The E16K SwH-NOX/SwHaCE protein mixture, followed at the heme absorbance, did not form a complex, as suggested by the calculated molecular mass of 76.5 kDa (Figure S2 of the Supporting Information and Table 1). In support of this, when SwH-NOX was run without SwHaCE at the optimal rotor speeds to analyze the 193 kDa SwH-NOX/SwHaCE complex, rather than the 22.5 kDa SwH-NOX species alone, we obtained a calculated molecular mass of 75 kDa (Figure S2 of the Supporting Information and Table 1). The molecular mass of SwH-NOX is not accurately calculated at the rotor speeds used to analyze the SwH-NOX/SwHaCE complex because higher angular velocities are required for lower-molecular mass solutes to balance the sedimentation and diffusion forces within the radius of the centrifuge cell. This result again suggests a loss of SwH-NOX/SwHaCE binding due to this SwH-NOX surface mutation.
Enzyme Assays Confirm the SwH-NOX/SwHaCE Binding Interface
Next we tested the effect of SwH-NOX and SwH-NOX surface mutants on the regulation of SwHaCE activity. We have previously reported that the diguanylate cyclase activity for SwHaCE increases in the presence of the WT SwH-NOX Fe(II)-unligated complex, whereas the phosphodiesterase activity is enhanced in the presence of the WT SwH-NOX Fe(II)–NO complex.24 In these studies, we used a new reaction end point assay for cyclase activity, based on the detection of phosphate (Pi) in solution with the dye Malachite green.
In a diguanylate cyclase-catalyzed reaction, two molecules of GTP are condensed to generate PPi (inorganic pyrophosphate) as a byproduct along with cyclic-di-GMP (2GTP → cyclic-di-GMP + 2PPi).35 In the presence of the enzyme inorganic pyrophosphatase (IPP), PPi is hydrolyzed to Pi (inorganic phosphate) in solution (2PPi → 2Pi).36 Malachite green is a commercially available reagent, which is used to determine the concentration of free Pi in solution.37 This end point assay can be used to quantify diguanylate cyclase activity by measuring the amount of Pi generated from PPi produced during the reaction. Because this is an end point assay, the SwHaCE enzyme was boiled before IPP was added to the solution to ensure that IPP does not interfere with the activity of the enzyme. To confirm that IPP is not the rate-limiting enzyme in the reaction, the concentration of IPP was doubled to confirm that the same amount of Pi was generated (Table S2 of the Supporting Information). Because we are detecting the amount of PPi produced in the reaction, the turnover of cyclic-di-GMP into pGpG by the phosphodiesterase domain of SwHaCE does not affect the measurement. As a control, the amount of Pi generated was determined at various concentrations of SwHaCE (Figure S3 of the Supporting Information). A linear correlation was observed between the enzyme concentration and the amount of Pi produced, confirming that only SwHaCE is the rate-limiting enzyme in the reaction.
This assay was used to determine the concentration of Pi generated during the reaction of SwHaCE with GTP in the presence of the WT, E16K, F17A, and E20K SwH-NOX Fe(II)-unligated complexes. The specific activity values are reported in Figure 7 as concentrations of Pi (micromolar per minute per milligram). It was observed that the WT SwH-NOX Fe(II)-unligated complex (20 μM) increased the initial velocity of the cyclase activity of SwHaCE (1 μM) by 2-fold in the presence of 200 μM GTP, as expected from our previous work24 (Figure 7). In general, addition of the SwH-NOX mutants led to a loss of this enhanced effect, indicating a loss of SwH-NOX binding and regulation of SwHaCE cyclase activity. For the E16K and E20K SwH-NOX mutants, the observed initial velocity was lower than that of SwHaCE with WT SwH-NOX but slightly higher than that of the enzyme by itself, indicating some residual binding and regulation by SwH-NOX, but significantly less than that observed with WT SwH-NOX. For F17A SwH-NOX, we observed the same initial velocity as SwHaCE without SwH-NOX. These data, like all of the experiments described above, also indicate that SwH-NOX/SwHaCE binding has been disrupted by the surface mutations and support our conclusion that SwH-NOX binds to SwHaCE, at least in part, using the N-terminal helical region of H-NOX.
Figure 7.
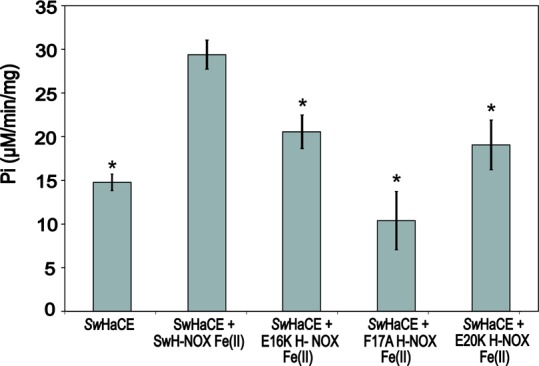
Regulation of SwHaCE diguanylate cyclase activity by SwH-NOX. Addition of 20 μM Fe(II)-unligated complex of SwH-NOX to SwHaCE causes a significant increase in the specific activity of SwHaCE. Addition of any of the Fe(II)-unligated SwH-NOX surface mutants (E16K, F17A, and E20K, all at 20 μM), however, had a weaker effect or no effect on SwHaCE activity, indicating weakened binding to SwHaCE. All assays contained 1 μM SwHaCE and 200 μM GTP as substrate. Error bars represent three independent trials on different days. A Student’s p test was conducted to determine significance. Asterisks indicate p ≤ 0.006. Each data set was compared to the activity of SwHaCE with WT SwH-NOX Fe(II).
Discussion
The spatiotemporal regulation of intracellular cyclic-di-GMP is central to many important cellular processes like virulence, biofilm formation, quorum sensing, motility, and cell division.38,39 Diguanylate cyclase and phosphodiesterase enzymes, which maintain the intracellular pool of cyclic-di-GMP, are usually found to be associated with regulatory domains and/or proteins that modulate their activity. However, very few of these enzymes have been characterized with their regulatory partner protein.
NO has been shown to play an important role in bacterial signaling, and recent studies have indicated that low concentrations of NO disperse biofilms in bacteria through the regulation of cyclic-di-GMP concentrations.40 We have previously reported that NO disperses biofilms in the marine, bioluminescent, biofilm-dwelling bacterium S. woodyi through H-NOX by regulating the production of cyclic-di-GMP by HaCE.24 It was shown that SwH-NOX and NO differentially modulate the enzymatic activities of both the diguanylate cyclase and phosphodiesterase domains of SwHaCE. These results necessitate a model in which H-NOX and HaCE are associated in a manner that is independent of the ligation state of H-NOX. Data presented in this study confirm that SwH-NOX and SwHaCE form a complex independent of the ligation state of H-NOX.
This study investigates the structural and biophysical details of the association between SwH-NOX and SwHaCE. We have used NMR chemical shift perturbation analysis along with peak assignments for SwH-NOX to identify the critical H-NOX surface residues that are involved in binding to SwHaCE. The tremendous change in the chemical environment of a patch of N-terminal residues, including residues E16, F17, and E20, indicates a binding region for SwHaCE on SwH-NOX (Figure 3A,B). We also identified other residues (approximately six or seven) with significant chemical shift perturbations upon binding to SwHaCE that are spread out on other parts of SwH-NOX. These chemical shift perturbations may indicate additional binding surface area or, alternatively, a conformational rearrangement of SwH-NOX that is triggered upon binding to SwHaCE.
To determine the role of identified SwH-NOX N-terminal amino acids (E16, F17, and E20) in binding SwHaCE, we made mutants of each and investigated the effect of mutation in several binding and enzymatic assays. Fluorescence binding assays indicate a significant loss of binding (Figure 5) and alteration of enzyme regulation (Figure 7) for individual mutants of these surface residues (E16, F17, and E20). Hence, we can conclude that these residues mediate interaction of SwH-NOX with SwHaCE. Sedimentation equilibrium experiments reveal the WT SwH-NOX/SwHaCE protein complex to be a heterotetramer (Figure 6B), in both the Fe(II)-unligated and Fe(II)–NO-bound states, whereas SwH-NOX alone and all the surface mutants in the presence of SwHaCE were determined to be monomeric in solution.
On the basis of our results, we developed a model for the SwH-NOX/SwHaCE protein complex. Because SwH-NOX and SwHaCE are cocistronic,24 we hypothesize that they are expressed together in the cell and always associate to form this heterotetrameric complex. This association likely occurs via dimerization of SwHaCE, followed by binding of two molecules of SwH-NOX to the SwHaCE dimer, as illustrated in Figure 8. Upon expression, presumably, SwH-NOX is in the Fe(II)-unligated state, and hence, the cyclase activity of SwHaCE is enhanced to generate more cyclic-di-GMP. When the signal (NO) binds, there must be a change in the conformation of the heterotetrameric protein complex, leading to a change in the activity of SwHaCE that results in less cyclic-di-GMP produced, ultimately causing biofilm dispersal. If monomeric SwHaCE also binds SwH-NOX, there may be a complex equilibrium involving dimerization of SwHaCE, monomeric association of the SwH-NOX/SwHaCE complex, and two monomers of SwH-NOX binding to the SwHaCE dimer to form the final heterotetrameric species. However, sedimentation equilibrium cannot be used to determine the ratio of these species in solution.
Figure 8.

Possible mechanism of formation of the complex between SwHaCE and SwH-NOX. Monomeric SwHaCE is likely in equilibrium with dimeric SwHaCE, with the equilibrium lying toward the dimer. Monomeric or dimeric SwHaCE may associate with one or two molecules of SwH-NOX, respectively. The most abundant species at equilibrium is the heterotetrameric complex (2:2), containing two molecules each of SwHaCE and SwH-NOX.
Furthermore, the data presented here indicate that N-terminal helices αA−αC mediate binding to SwHaCE. This result is supported by several previous studies that also point to the role of the N-terminal helices in transmitting the signal of NO binding to H-NOX partner proteins. In crystal structures of the H-NOX domain from Nostoc punctiforme,41 the N-terminal helices have been shown to be maximally displaced upon binding of the signaling ligands NO and CO, suggesting that movement in helices αA−αC constitutes part of the signal transfer mechanism. Furthermore, a recent study from the Marletta lab describes the important higher-order interactions in sGC, elucidating the mechanism of NO-regulated stimulation of activity.42 They found that in the N-terminal region of sGC, H-NOX participates in the signal transfer mechanism by binding to the PAS domain. Mutational studies confirm that NO binding triggers these local interactions, leading to global conformational changes, ultimately increasing activity.
Further evidence that the N-terminal helices of H-NOX are important for interaction with partner proteins and signal propagation comes from studies of the structure of the heme cofactor of H-NOX. The heme unit in H-NOX is highly distorted from planarity,43 but structural studies have indicated that mutation of a conserved proline residue in the proximal heme pocket causes substantial heme flattening.44 The conformational change caused by heme flattening propagates to the surface of H-NOX, causing a shift in the position of the N-terminal helices. We have shown in SwH-NOX that the heme-flattened mutant affects SwHaCE activity exactly the same as binding of NO to WT SwH-NOX,45 indicating heme flattening causes a structural change similar to that of binding of the SwH-NOX/SwHaCE complex to NO. These data suggest that transmission of the NO signal involves the N-terminal helices, which is highly supportive of our results reported here, indicating the N-terminal helices are involved in binding HaCE, which is required for translation of NO binding to the enzymatic active sites.
Taken together, there is strong evidence that ligand binding events in bacterial H-NOX domains cause conformational changes that are relayed to the N-terminal helices, which serve as a binding site for interaction with partner enzymes such as HaCEs or HahKs (H-NOX-associated histidine kinases), ultimately resulting in functional changes within the cell.
In conclusion, NO and cyclic-di-GMP are becoming increasingly important as signaling molecules that direct bacteria on how to make the choice between motile and sessile lifestyles. This study is the first of its kind that provides a structural understanding of how a bacterial NO-sensing protein, H-NOX, can regulate its associated enzyme activity (HaCE and HahK), to control the downstream functions such as biofilm formation. Such studies improve our understanding of how small molecule signaling regulates downstream functions and how enzymes that regulate the cyclic-di-GMP concentration in the cell are functionally regulated by a signal-sensing protein.
Acknowledgments
We thank the staff of the New York Structural Biology Center (NYSBC) for help with the NMR experiments. We also thank Prof. Roger Johnson and members of the Boon group for useful comments and advice.
Glossary
Abbreviations
- NO
nitric oxide
- H-NOX
heme-nitric oxide/oxygen-binding
- HaCE
H-NOX-associated cyclic-di-GMP processing enzyme
- DGC
diguanylate cyclase
- PDE
phosphodiesterase
- GTP
guanosine triphosphate
- c-di-GMP or cyclic-di-GMP
bis-(3′–5′)-cyclic dimeric guanosine monophosphate
- pGpG
5′-phosphoguanylyl-(3′–5′)-guanosine
- Sw
S. woodyi
- GGDEF
conserved amino acids in the catalytic sites of diguanylate cyclases (the domain containing these conserved residues is often termed a GGDEF domain)
- EAL
conserved amino acids in the catalytic site of phosphodiesterases (the domain containing these conserved residues is often termed an EAL domain)
- sGC
soluble guanylate cyclase (a eukaryotic H-NOX domain-containing protein)
- SwHaCE
wild-type S. woodyi diguanylate cyclase/phosphodiesterase protein used in this study
- SwH-NOX
wild-type H-NOX protein used in this study
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- OD
optical density
- Tris
tris(hydroxymethyl)aminomethane
- PMSF
phenylmethanesulfonyl fluoride
- BME
β-mercaptoethanol
- NMR
nuclear paramagnetic resonance spectroscopy
- BSA
bovine serum albumin
- TROSY
transverse relaxation-optimized spectroscopy
- HSQC
heteronuclear single-quantum coherence
- HNCO, HNCACB, CBCACONH, and HNCA
triple-resonance nuclear magnetic resonance spectroscopy to assign protein backbone atoms
- AUC
analytical ultracentrifugation
- MG
Malachite green
- IPP
inorganic pyrophosphatase
- PPi
inorganic pyrophosphate.
Supporting Information Available
Soret band maxima and NO dissociation rate constants for Fe(II), Fe(II)–NO, and Fe(II)–CO species for WT, E16K, F17A, and E20K SwH-NOX; a standard curve for PPi concentration; data showing that generation of Pi from PPi by IPP is not the rate-limiting step in our kinetic assays; a plot of the amount of Pi generated during the diguanylate cyclase reaction of SwHaCE using GTP as a substrate at different SwHaCE concentrations; and sedimentation equilibrium experiments with WT SwH-NOX, E16K SwH-NOX, and the E16K SwH-NOX/SwHaCE mixture at different speeds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by Office of Naval Research Grant N00014-10-1-0099 to E.M.B., National Science Foundation Grants CHE-0910771 to E.M.B. and MCB-1330259 to D.P.R., and National Institutes of Health Grant NIH-T32 GM092714 to T.L.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Galperin M. Y. (2004) Bacterial signal transduction network in a genomic perspective. Environ. Microbiol. 6(6), 552–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatan E.; Watnick P. (2009) Signals, regulatory networks, and materials that build and break bacterial biofilms. Microbiol. Mol. Biol. Rev. 73(2), 310–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Argenio D. A.; Miller S. I. (2004) Cyclic-di-GMP as a bacterial second messenger. Microbiology 150(Part 8), 2497–2502. [DOI] [PubMed] [Google Scholar]
- Jenal U. (2004) Cyclic di-guanosine-monophosphate comes of age: A novel secondary messenger involved in modulating cell surface structures in bacteria. Curr. Opin. Microbiol. 7(2), 185–191. [DOI] [PubMed] [Google Scholar]
- Jenal U.; Malone J. (2006) Mechanisms of cyclic-di-GMP signaling in bacteria. Annu. Rev. Genet. 40, 385–407. [DOI] [PubMed] [Google Scholar]
- Cotter P. A.; Stibitz S. (2007) C-di-GMP-mediated regulation of virulence and biofilm formation. Curr. Opin. Microbiol. 10(1), 17–23. [DOI] [PubMed] [Google Scholar]
- Tamayo R.; Pratt J. T.; Camilli A. (2007) Roles of cyclic diguanylate in the regulation of bacterial pathogenesis. Annu. Rev. Microbiol. 61, 131–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge R. (2009) Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 7(4), 263–273. [DOI] [PubMed] [Google Scholar]
- Aldridge P.; Paul R.; Patrick G.; Rainey P.; Jenal U. (2003) Role of the GGDEF regulator PleD in polar development of Caulobacter crescentus. Mol. Microbiol. 47(6), 1695–1708. [DOI] [PubMed] [Google Scholar]
- Iyer L.; Anantharaman V.; Aravind L. (2003) Ancient conserved domains shared by animal soluble guanylyl cyclases and bacterial signaling proteins. BMC Genomics 4(1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryjenkov D. A.; Tarutina M.; Moskvin O. V.; Gomelsky M. (2005) Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: Insights into biochemistry of the GGDEF protein domain. J. Bacteriol. 187(5), 1792–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen M.; Christen B.; Folcher M.; Schauerte A.; Jenal U. (2005) Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J. Biol. Chem. 280(35), 30829–30837. [DOI] [PubMed] [Google Scholar]
- Schmidt A. J.; Ryjenkov D. A.; Gomelsky M. (2005) The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: Enzymatically active and inactive EAL domains. J. Bacteriol. 187(14), 4774–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan R. P.; Fouhy Y.; Lucey J. F.; Crossman L. C.; Spiro S.; He Y. W.; Zhang L. H.; Heeb S.; Camara M.; Williams P.; Dow J. M. (2006) Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. U.S.A. 103(17), 6712–6717. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Denninger J. W.; Marletta M. A. (1999) Guanylate cyclase and the NO/cGMP signaling pathway. Biochim. Biophys. Acta 1411, 334–350. [DOI] [PubMed] [Google Scholar]
- Boon E. M.; Huang S. H.; Marletta M. A. (2005) A molecular basis for NO selectivity in soluble guanylate cyclase. Nat. Chem. Biol. 1(1), 53–59. [DOI] [PubMed] [Google Scholar]
- Barraud N.; Schleheck D.; Klebensberger J.; Webb J. S.; Hassett D. J.; Rice S. A.; Kjelleberg S. (2009) Nitric oxide signaling in Pseudomonas aeruginosa biofilms mediates phosphodiesterase activity, decreased cyclic di-GMP levels, and enhanced dispersal. J. Bacteriol. 191(23), 7333–7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S.; Wu J.; Zhang L. H. (2010) Modulation of Pseudomonas aeruginosa biofilm dispersal by a cyclic-di-GMP phosphodiesterase with a putative hypoxia-sensing domain. Appl. Environ. Microbiol. 76(24), 8160–8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukl E. T.; Ioanoviciu A.; Sivaramakrishnan S.; Nakano M. M.; Ortiz de Montellano P. R.; Moenne-Loccoz P. (2011) Nitric oxide dioxygenation reaction in DevS and the initial response to nitric oxide in Mycobacterium tuberculosis. Biochemistry 50(6), 1023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon E. M.; Marletta M. A. (2005) Ligand discrimination in soluble guanylate cyclase and the H-NOX family of heme sensor proteins. Curr. Opin. Chem. Biol. 9(5), 441–446. [DOI] [PubMed] [Google Scholar]
- Arora D. P.; Boon E. M. (2012) Nitric oxide regulated two-component signaling in Pseudoalteromonas atlantica. Biochem. Biophys. Res. Commun. 421(3), 521–526. [DOI] [PubMed] [Google Scholar]
- Liu N.; Pak T.; Boon E. M. (2010) Characterization of a diguanylate cyclase from Shewanella woodyi with cyclase and phosphodiesterase activities. Mol. BioSyst. 6(9), 1561–1564. [DOI] [PubMed] [Google Scholar]
- Carlson H. K.; Vance R. E.; Marletta M. A. (2010) H-NOX regulation of c-di-GMP metabolism and biofilm formation in Legionella pneumophila. Mol. Microbiol. 77(4), 930–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N.; Xu Y.; Hossain S.; Huang N.; Coursolle D.; Gralnick J. A.; Boon E. M. (2010) Nitric oxide regulation of cyclic di-GMP synthesis and hydrolysis in Shewanella woodyi. Biochemistry 51(10), 2087–2099. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Erbil W. K.; Price M. S.; Wemmer D. E.; Marletta M. A. (2009) A structural basis for H-NOX signaling in Shewanella oneidensis by trapping a histidine kinase inhibitory conformation. Proc. Natl. Acad. Sci. U.S.A. 106(47), 19753–19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Goddard T. D., and Kneller D. G. (2008) SPARKY 3, University of Calfornia, San Francisco. [Google Scholar]
- Schuck P. (2000) Size-Distribution Analysis of Macromolecules by Sedimentation Velocity Ultracentrifugation and Lamm Equation Modeling. Biophys. J. 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon E. M.; Davis J. H.; Tran R.; Karow D. S.; Huang S. H.; Pan D.; Miazgowicz M. M.; Marletta M. A. (2006) Nitric oxide binding to prokaryotic homologs of the soluble guanylate cyclase β1 H-NOX domain. J. Biol. Chem. 281(31), 21892–21902. [DOI] [PubMed] [Google Scholar]
- Cole J. L.; Lary J. W.; Moody T.; Laue T. M. (2008) Analytical Ultracentrifugation: Sedimentation Velocity and Sedimentation Equilibrium. Methods Cell Biol. 84, 143–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone J. G.; Williams R.; Christen M.; Jenla U.; Spiers A. J.; Rainey P. B. (2007) The structure-function relationship of WspR, a Pseudomonas fluorescens response regulator with a GGDEF output domain. Microbiology 153(Part 4), 980–994. [DOI] [PubMed] [Google Scholar]
- Paul R.; Abel S.; Wassmann P.; Beck A.; Heerklotz H.; Jenal U. (2007) Activation of the Diguanylate Cyclase PleD by Phosphorylation-mediated Dimerization. J. Biol. Chem. 282(40), 29170–29177. [DOI] [PubMed] [Google Scholar]
- De N.; Pirruccello M.; Krasteva P. V.; Bae N.; Raghavan R. V.; Sondermann H. (2008) Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biol. 6(3), e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C.; Paul R.; Samoray D.; Amiot N. C.; Giese B.; Jenal U.; Schirmer T. (2004) Structural basis of activity and allosteric control of diguanylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 101(49), 17084–17089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upson R. H.; Haugland R. P.; Malekzadeh M. N.; Haugland R. P. (1996) A Spectrophotometric Method to Measure Enzymatic Activity in Reactions That Generate Inorganic Pyrophosphate. Anal. Biochem. 243(1), 41–45. [DOI] [PubMed] [Google Scholar]
- Carter S. G.; Karl D. W. (1982) Inorganic phosphate assay with malachite green: An improvement and evaluation. J. Biochem. Biophys. Methods 7(1), 7–13. [DOI] [PubMed] [Google Scholar]
- Mills E.; Pultz I. S.; Kulasekara H. D.; Miller S. I. (2011) The bacterial second messenger c-di-GMP: Mechanisms of signalling. Cell. Microbiol. 13(8), 1122–1129. [DOI] [PubMed] [Google Scholar]
- Krasteva P. V.; Giglio K. M.; Sondermann H. (2012) Sensing the messenger: The diverse ways that bacteria signal through c-di-GMP. Protein Sci. 21(7), 929–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougald D.; Rice S. A.; Barraud N.; Steinberg P. D.; Kjelleberg S. (2012) Should we stay or should we go: Mechanisms and ecological consequences for biofilm dispersal. Nat. Rev. Microbiol. 10(1), 39–50. [DOI] [PubMed] [Google Scholar]
- Ma X.; Sayed N.; Beuve A.; van den Akker F. (2007) NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 26, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underbakke E. S.; Iavarone A. T.; Marletta M. A. (2013) Higher-order interactions bridge the nitric oxide receptor and catalytic domains of soluble guanylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 110(17), 6777–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicena P.; Karow D. S.; Boon E. M.; Marletta M. A.; Kuriyan J. (2004) Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc. Natl. Acad. Sci. U.S.A. 101(35), 12854–12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olea C.; Boon E. M.; Pellicna P.; Kuriyan J.; Marletta M. A. (2008) Probing the function of heme distortion in the H-NOX family. ACS Chem. Biol. 3(11), 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan S.; Boon E. M. (2012) Heme flattening is sufficient for signal transduction in the H-NOX family. J. Am. Chem. Soc. 134(4), 2044–2046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.