

Summary
The protein C pathway provides important biological activities to maintain the fluidity of the circulation, prevent thrombosis, and protect the integrity of the vasculature in response to injury. Activated protein C (APC), in concert with its cofactors and cell receptors, assembles in specific macromolecular complexes to provide efficient proteolysis of multiple substrates that result in anticoagulant and cytoprotective activities. Numerous studies on APC’s structure-function relation with its cofactors, cell receptors, and substrates provide valuable insights into the molecular mechanisms and presumed assembly of the macromolecular complexes that are responsible for APC’s activities. These insights allow for molecular engineering approaches specifically targeting the interaction of APC with one of its substrates or cofactors. Thus far, these approaches resulted in several anticoagulant-selective and cytoprotective-selective APC mutants, which provide unique insights into the relative contributions of APC’s anticoagulant or cytoprotective activities to the beneficial effects of APC in various murine injury and disease models. Because of its multiple physiological and pharmacological activities, the anticoagulant and cytoprotective protein C pathway have important implications for the (patho)physiology of vascular disease and for translational research exploring novel therapeutic strategies to combat complex medical disorders such as thrombosis, inflammation, ischemic stroke and neurodegenerative disease.
Keywords: activated protein C, anticoagulant, factor V, factor VIII, protein S, endothelial protein C receptor, protease activated receptor 1
Introduction
The hemostatic system safeguards the patency of the vasculature. Platelet aggregation, coagulation, and fibrinolysis operate in concert with the endothelium and other vascular cells to arrest bleeding and prevent thrombosis. The coagulation pathway contributes to regulation of the hemostatic balance via multiple mechanisms and pathways that ensure a balanced and confined hemostatic response at the site of injury. Despite these feedback mechanisms to fine-tune the hemostatic response, genetic and/or acquired defects often tilt the balance sufficiently to increase the risk for thrombophilia.
In the current model of coagulation,1,2 clot formation is initiated by the extrinsic pathway with little or no role for the contact system in the initiation of physiological coagulation, although platelet-derived polyphosphates may provide an endogenous activation mechanism for FXII.3,4 Thrombin formation occurs during two mechanistically different phases. In the first phase, referred to as primary thrombin formation, the extrinsic pathway generates the clot initiated by the tissue factor/FVIIa complex. However, primary thrombin formation is short-lived due to rapid inactivation of the tissue factor/FVIIa initiator complex by tissue factor pathway inhibitor (TFPI). In the second phase, when sufficient thrombin is generated to initiate FXI activation, secondary thrombin formation will continue inside the clot via thrombin-mediated FXI activation and amplification by the intrinsic pathway.5 This secondary thrombin formation contributes to clot strength and conveys resistance to fibrinolysis via the activation of FXIII and thrombin Activatable finbrinolysis inhibitor (TAFI).6,7
Control of coagulation is generally provided by three different mechanisms. First, the γ-carboxyglutamic acid (Gla)-domain, common to most coagulation factors, requires the presence of negatively charged phosphatidylserine for calcium-dependent binding to lipid surfaces.8 Thus, assembly of the tenase (FIXa, FX, and FVIIIa) and prothrombinase (FXa, FII, and FVa) complex is limited by the presence of negatively charged lipid surfaces (such as on activated platelets).8,9 Second, serine protease inhibitors (SERPINs) rapidly inhibit activated coagulation factors, thereby blunting coagulation and preventing the escape of active coagulation factors in the circulation.10 Finally, the protein C pathway actively inhibits coagulation by proteolysis of the tenase and prothrombinase complex cofactors, FVa and FVIIIa, thereby providing a dynamic regulation of coagulation.11–15
The focus of this review we will be on the protein C pathway. Because activated protein C (APC) inactivates both FVa and FVIIIa, it has important effects on the down-regulation of both primary and secondary thrombin formation that manifest as potent anti-thrombotic effects in vivo. Furthermore, APC’s relatively new activities on cells provide physiological and pharmacological relevant protective effects on the endothelium and the vasculature. Thus APC conveys multiple activities that require assembly of macromolecular complexes with different cofactors, cell receptors and substrates. Structure-function analysis of these APC complexes, discussed in the next sections, provides a unique understanding of how a single enzyme can mediate multiple biologically and therapeutically relevant activities.
The protein C pathway
The protein C pathway provides important contributions to maintain the fluidity of the circulation, prevent thrombosis, and protect the integrity of the vasculature in response to injury. The reactions of the protein C pathway encompass protein C activation on endothelial cells, the anticoagulant protein C pathway on activated platelets, the cytoprotective protein C pathway on cell membranes, and inactivation of APC by plasma serine protease inhibitors (SERPINs) in the fluid phase (Figure 1). Each of these aspects of the protein C pathway will be discussed in the sections below.
Figure 1. Reaction of the protein C pathway.
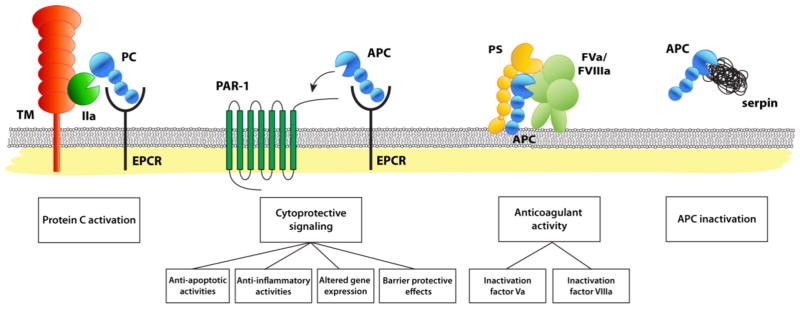
Schematic representation of the protein C pathway reactions, from left to right: protein C activation, the cytoprotective protein C pathway, the anticoagulant protein C pathway and the inactivation of APC by serine protease inhibitors (SERPINs). Protein C activation: physiologic activation of protein C (PC) by the thrombin (IIa)-thrombomodulin (TM) complex occurs on the surface of endothelial cell membranes when protein C is bound to the endothelial receptor (EPCR). Since protein C and APC have a similar affinity for EPCR, after activation APC can remain bound to EPCR to initiate cytoprotective signaling. The cytoprotective protein C pathway: APC’s direct effects of on endothelial cells require the cellular receptors EPCR and PAR1. These cellular activities of APC include anti-apoptotic and anti-inflammatory activities, alteration of gene expression profiles, and protection of endothelial barrier functions and are collectively referred to as APC’s cytoprotective activities. The anticoagulant protein C pathway: APC anticoagulant activities involve proteolytic cleavages of FVa and FVIIIa. Different protein cofactors, such as protein S (PS), FV, and various lipid cofactors (e.g. phosphatidylserine, phosphatidylethanolamine cardiolipin, glucosylceramide, etc.), enhance the inactivation of FVa and FVIIIa by APC. APC inactivation: Inactivation of APC in plasma by serine protease inhibitors (SERPINs) is slow, which contributes to a remarkably long circulation half-life of APC (~ 20 min). Most important inhibitors of APC in plasma are protein C inhibitor (PCI), plasminogen activator inhibitor-1 (PAI-1), and α1-antitrypsin and, to a lesser extent, α2-macroglobulin and α2-antiplasmin.
The physiologic importance of the protein C system is best illustrated by the manifestation of massive thrombotic complications in infants with protein C deficiency.16,17 Neonatal purpura fulminans, a rapidly progressing hemorrhagic necrosis of the skin due to microvascular thrombosis, inflammation, and disseminated intravascular coagulation (DIC), is typically observed in severe protein C deficiency, whereas heterozygous protein C deficiency in adults carries a significantly increased risk for venous thrombosis.18–20 A rare complication referred to as warfarin-induced skin necrosis with clinical symptoms similar to that of purpura fulminans, may present within days after initiation of oral anticoagulant therapy with coumarin derivatives. This is due to a temporary functional protein C deficiency caused by the shorter circulation half-life of protein C (8 hr) compared to the other procoagulant coagulation factors (24–72 hr).17,18,21 Acquired protein C deficiency is also found in patients with severe infection and sepsis, most likely due to consumption and poor synthesis in the liver, and low protein C levels correlate with poor clinical outcome and death.22
Protein C Activation
The protein C zymogen is synthesized in the liver and circulates in plasma at 4 μg/ml, which is equivalent to ~70 nM based on a molecular weight of 62,000 Da. The domain topology of protein C is typical of vitamin K-dependent coagulation factors.23 The N-terminal protein C light chain contains nine γ-carboxylated Glu residues (Gla-domain) and two epidermal growth factor (EGF)-like domains. The C-terminal heavy chain contains an N-terminal acidic protein C activation peptide that is removed upon activation and the protease domain with a typical His211 (mature protein C numbering), Asp257 and Ser360 active site triad (residues His57, Asp102 and Ser195 in chymotrypsin nomenclature, for a conversion table see24).
Protein C is activated by thrombin through limited proteolysis at Arg169. Physiological activation of protein C on the endothelial cell surface requires binding of thrombin to thrombomodulin (TM) and binding of protein C to the endothelial protein C receptor (EPCR) (Figure 1).14,25–27 The binding surface for TM on protein C shows a partial overlap with the exosite for interactions with FVa, and includes residues in loop 37 (Lys191 and Lys192), loop 60 (Lys217 and Lys218), loop 70–80 (Arg229 and Arg230), and possibly loop 20 (Lys174, Arg177, and Arg178) although the direct interaction of these latter residues with TM remains controversial (loops are referred to by their chymotrypsin numbering24).28–30
Protein C activation by thrombin in the absence of TM is very inefficient and is inhibited by calcium. Presumably, this limitation ensures that APC generation is initiated only when the clot covers the intact endothelium and thrombin comes in contact with TM.14 Several residues surrounding the Arg169 activation site in protein C (i.e. P3–P9′ residues relative to Arg169 denoted as P1)31 are responsible for the inhibitory effect of calcium on the activation of protein C by free thrombin. Mutation of these residues allows for efficient protein C activation by thrombin in the presence of calcium that is no longer dependent on the presence of TM.32–34 In vivo proof-of-principal that TM-independent protein C activation by thrombin results in enhanced APC generation was provided by a transgenic mouse (named the “APChigh” mouse) expressing human protein C with mutations of the P3 and P3′ residues (Asp167Phe/Asp172Lys).33,35 Interestingly, increased blood loss after tail amputation in these mice suggest that uncoupling of protein C activation from TM disrupts the regulation of normal thrombus formation.
Inactivation of APC
Inactivation of APC in plasma is driven by serine protease inhibitors (SERPINs) of which protein C inhibitor (PCI), plasminogen activator inhibitor-1 (PAI-1), α1-antitrypsin and, to a lesser extent, α2-macroglobulin and α2-antiplasmin are most relevant for APC (Figure 1).36 Even though heparin and vitronectin accelerate the reaction of APC with PCI and PAI-1 several orders of magnitude, the reaction of APC with SERPINs is relatively slow, resulting in a ~20 min half-life of APC in the circulation.36 APC exosites required for interactions of APC with the various inhibitors and heparin largely overlap with those required for interaction with FVa and include residues in loop 37 and the autolysis loop.36,37 Interestingly, some residues that affect interactions with SERPINs are not shared with FVa and these include Leu194 in loop 37, Lys217 and Lys218 in loop 60, Thr254, and Ser336.36,38,39
The anticoagulant protein C pathway
The protein C pathway is best known for its anticoagulant activity that involves proteolytic inactivation of FVa and FVIIIa on negatively charged phospholipid membranes and that is enhanced by cofactors protein S and FV (Figure 1).40–42 Because APC inactivates both FVa and FVIIIa, it has important effects on the down-regulation of both primary and secondary thrombin formation. Inhibition of primary thrombin formation results in delayed clot formation, whereas inhibition of secondary thrombin formation results in diminished activation of TAFI and subsequently in an enhanced susceptibility of the clot to fibrinolysis. The latter effects of APC on secondary thrombin formation are also referred to as APC’s profibrinolytic effects.43
APC’s anticoagulant activity requires binding of the Gla-domain to negatively charged phospholipid membranes. Although membranes containing phosphatidylethanolamine in addition to phosphatidylserine generally improve APC’s lipid-dependent functions, binding of APC to negatively charged phospholipids is relatively poor compared to other vitamin K-dependent coagulation factors.44 Therefore, anticoagulant activity of APC can be enhanced by strategies aimed at improving APC’s affinity for membranes, such as Gla-domain swaps or targeted mutagenesis of the Gla-domain.45,46
Inactivation of FVa and FVIIIa
FVa is a non-covalent heterodimeric complex composed of a heavy chain (domains A1-A2) and a light chain (domains A3-C1-C2).47 Since FVa enhances prothrombinase ~10,000-fold, inactivation of FVa by APC effectively shuts down thrombin formation.40,48 Inactivation of FVa involves APC-mediated cleavages at Arg306 and Arg506. The rapid cleavage at Arg506 is kinetically favored over cleavage at Arg306, but results only in partial inactivation of FVa, whereas the slower cleavage at Arg306 results in a complete loss of FVa function.40,41 The importance of APC-mediated FVa inactivation is clear from the increased risk for thrombosis associated with mutations of the APC cleavage sites in FV (Arg506Gln a.k.a. FVLeiden, Arg306Thr a.k.a. FVCambridge and Arg306Gly a.k.a. FVHong Kong). In fact, FVLeiden is the most common identifiable hereditary risk factor for venous thrombosis among Caucasians.47,49
Mutagenesis studies have identified several positively charged exosites on the APC protease domain surface that are required for rapid inactivation of FVa (Figure 2B).36,37,50,51 This extended FVa exosite on APC is comprised of residues in loop 37 (Lys191, Lys192, and Lys193), loop 60 (Asp214, Glu215 and Arg222), loop 70 (Arg229 and Arg230) and the autolysis loop (Lys306, Lys311, Arg312 and Arg314).36,37,50,51 Remarkably, these residues primarily contribute to APC cleavage of FVa at Arg506 but have little effect on APC-mediated cleavage at Arg306, suggesting that the FVa Arg306 cleavage site does not rely on APC exosite interactions or that the exosite for Arg306 has not been found yet.50,51 Instead, protein S enhances APC-mediated cleavage at Arg306 (see below).
Figure 2. Schematic structural model of the anticoagulant protein C pathway.
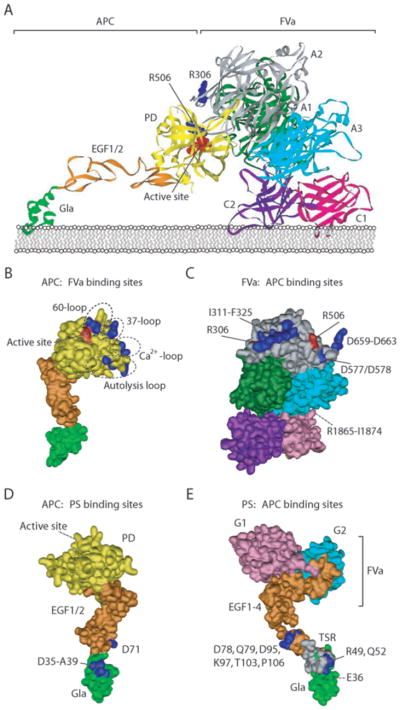
(A) Proposed model for the APC-mediated inactivation of FVa at Arg506 based on the interaction of an extended basic exosite on the protease domain (PD) of APC with negatively charged residues on the surface of FVa (adapted from Pellequer et al.70). In this model, the protease domain of APC (yellow) interacts with the A1 (green) and A2 (gray) domains of FVa. The interactions between APC and FVa position Arg506 (blue) of FVa in the active site pocket (red) of APC. The Gla-domain of APC (green) and the C1 (pink) and C2 (purple) domains of FVa interact with the phospholipid layer. Although biochemical data strongly support additional interactions between the APC Gla-domain and the FVa light chain (A3-C1-C2), especially residues within the A3 (light blue) sequence Arg1865-Ile1874, the extended projection of these domains in this model illustrate the need for a more flexible orientation of the APC protease domain to accommodate this (see text for additional details). (B). Schematic overview of the extended exosite (blue) on the surface of the APC protease domain, comprised of residues in loop 37, loop 60, the calcium-binding loop (loop70–80) and the autolysis loop that are required for interactions with FVa to accommodate cleavage at Arg506. (C) Schematic overview of the residues on the FVa surface (blue) that are involved in interactions of FVa with APC. Important residues are located on the A2 domain surrounding the Arg306 and Arg506 cleavage sites (red) and between the A1 and A3 domain at the back of the protein (not visible). (D) A schematic overview of APC residues implicated in the interaction of APC with protein S. Important residues (blue) in the Gla-domain (Asp35-Ala39) and the EGF1-domain (Asp71) are highlighted. (E) Schematic overview of the protein S residues (blue) implicated in the interaction of protein S with APC. Important residues (blue) in the Gla-domain (Glu36), TSR (Arg49 and Gln52), and EGF1-domain (Asp78, Gln79, Asp95, Lys97, Thr103 and Pro106) are highlighted.
In the circulation, tight non-covalent binding (KD ~0.5 nM) of FVIII to von Willebrand factor (vWF) prevents the incorporation of factor VIII into the tenase complex.52 FVIIIa dissociates from vWF after activation and enhances FXa formation by the tenase complex approximately 200,000-fold.53 Despite a domain topology similar to that of FVa, FVIIIa circulates as a heterotrimer and not a heterodimer due to different cleavage patterns that cause their respective activations. As a consequence of being a heterotrimer, FVIIIa is quite unstable with a half-life of only 2 min due to spontaneous dissociation of the A2-domain.52 Nevertheless, several observations, including stabilization of FVIIIa by FIXa in the tenase complex, support a role for APC in the inactivation of FVIIIa.54 Homologous to inactivation of FVa, inactivation of FVIIIa by APC occurs upon cleavage at Arg336 and Arg562 but in contrast to FVa, cleavage of FVIIIa at either site results in a complete loss of cofactor activity.55,56 Both protein S and FV but not FVa enhance inactivation of FVIIIa by APC.56,57
Protein S
Protein S is best known for its function as a non-enzymatic cofactor to APC in the inactivation of FVa and FVIIIa. In addition, protein S has APC-independent anticoagulant effects and also has direct (cytoprotective) effects on cells that are independent of its anticoagulant functions but instead require interactions with receptors on cells (the reader is referred to the references provided as a starting point for a more detailed discussion of these protein S activities).58,59
The important anticoagulant contributions of protein S are clear from the thrombotic complications and increased risk of venous thromboembolism associated with homozygous and heterozygous protein S deficiency.60 Functionally, five distinct domains can be identified that include an N-terminal Gla domain, a thrombin sensitive region (TSR), a repeat of four EGF-like domains, and a sex hormone-binding globulin (SHBG) domain composed of two laminin G-type domains.23 Protein S predominantly stimulates FVa cleavage at Arg306 by APC but also neutralizes the FXa-mediated protection of FVa against APC cleavage at Arg506.61,62 Molecular mechanisms for enhanced APC-mediated cleavage of FVa at Arg306 by protein S have been partially elucidated and involve a protein S-induced change in the geometry of APC relative to the membrane. Presumably, protein S binding lowers APC’s active site closer to the membrane, placing it in a better position to cleave Arg306.63 This provides a molecular explanation for how APC can inactivate FVa at Arg306 without extensive exosite interactions between APC and FVa. APC residues that are implicated in interactions with protein S (Figure 2D) include Gla-domain residues 35 to 39 (in particular Leu38), Asp71 that contains a post-translational β-hydroxyaspartic acid modification in EGF1, and potentially the C-terminus of the light-chain.64–67
Molecular interaction between APC, protein S, and FVa
Since limited structural information is available, the perceived assembly of APC with protein S and the interactions with FVa remain highly speculative.24,68–70 An APC-FVa model for cleavage at Arg506 (Figure 2A), based on the interaction of the extended positively charged exosite of APC (Figure 2B) with a negatively charged region on FVa that includes Asp513, Asp577, and Asp578 in the A2-domain and Asp659, Asp660, Glu661, Glu662, and Asp663 that follows the A2-domain (Figure 2C), projects the APC Gla-domain rather far away from FVa.51,70 In complex with protein S and FVa, the APC Gla-EGF1 domains are anticipated to be orientated in closer proximity to FVa, with a flexible conformation of the APC protease domain that bends down to Arg506 (or Arg306). This hypothesis is consistent with biochemical data that indicates binding of APC to the FVa light chain.71,72 Thus, protein S is likely to have important implications for the spatial orientation APC in the ternary APC-protein S-FVa complex.
The APC-cofactor function of protein S involves a complex set of interactions of protein S with APC and factor Va.73 The minimal structure of protein S to support APC cofactor activity requires the Gla-domain, the TSR and EGF1 (~30% compared with native protein S) although EGF2 and part of the SHBG domain provide additional interactions with APC and FVa and are required for full protein S cofactor activity.74–76 Direct APC-binding to protein S seems contained to protein S EGF1 with important contributions of Asp78, Gln79, Asp95, and Thr103 (Figure 2E).75,77 The protein S Gla-domain, TSR and EGF-2 are unlikely to contribute to direct interactions with APC but rather play a supporting structural role. Important residues in these supporting domains identified thus far include Glu36 in the Gla-domain and Arg49 and Gln52 in the TSR.69,74–78
The SHBG domain of protein S is projected to extend above the protease domain of APC since protein S contains two additional EGF-like domains compared to APC. The contributions of the SHBG-domain to protein S cofactor activity seem contained to the C-terminus of the SHBG laminin G2-domain and are likely derived from mediating interactions with FVa rather than APC.79 Especially residues Lys630, Lys631, and Lys633 in the G2-domain seem to provide important contributions for binding of protein S to FVa, which possibly facilitates directing the APC protease domain to the FVa cleavage sites consistent with protein S decreasing the distance of the APC active site to the membrane.63,80
The cytoprotective protein C pathway
In addition to its anticoagulant activity, APC can convey direct effects on cells, collectively referred to as APC cytoprotective activities that require EPCR and PAR1.11,81–84 Dependent on the cell type and injury, these cellular activities of APC include anti-apoptotic and anti-inflammatory activities, alteration of gene expression profiles and protection of endothelial barrier function. Other receptors may also contribute to APC-initiated signaling such as sphingosine-1-phosphate receptor 1 (S1P1), apolipoprotein E receptor 2 (ApoER2), CD11b/CD18 (αMβ2; Mac-1; CR3), PAR-3, and Tie2, whereas APC’s ability to inactivate extracellular histones is presumably independent from APC’s cell signaling effects.85,86
The currently prevailing paradigm for APC’s direct cytoprotective actions on endothelial cells is that when PAR1 and EPCR are colocalized in caveolin-1 enriched lipid rafts or caveolae, APC binding to EPCR permits non-canonical PAR1 activation at Arg46 to initiate cytoprotective signaling.11,87–89 It is important to realize that there are several fundamental distinctions between PAR1 activation by APC and thrombin on endothelial cells. Foremost, the functional outcome is different. Thrombin activation of PAR1 results in proinflammatory and endothelial barrier disruptive effects, whereas PAR1 activation by APC results in cytoprotective actions that include endothelial barrier stabilization. The reasons for this functional contrast become evident when taking the fundamental differences in PAR1 activation and signaling between these two proteases in consideration. PAR1 activation by thrombin occurs after cleavage of the canonical Arg41 site after which the tethered-ligand starting at Ser42 promotes G-protein dependent signaling that includes activation of barrier disruptive Ras homolog gene family member A (RhoA). In contrast, activation of PAR1 by APC occurs through cleavage of the non-canonical Arg46 site after which the tethered-ligand starting at Asn47 mediates β-arrestin 2-dependent barrier protective Ras-related C3 botulinum toxin substrate 1 (Rac1) activation (Figure 3).89,90 Synthetic peptides representing the sequences of the different tethered ligands exposed after cleavage of PAR1 at Arg41 (thrombin receptor activating peptide (TRAP)) or Arg46 (TR47) also recapitulate the remarkable differences in PAR1 signaling. TRAP induces typical phosphorylation of ERK1/2 but TR47 does not. Instead, TR47 but not TRAP induces phosphorylation of Akt in endothelial cells that is linked to cytoprotective functions.89 The fact that different ligands induce different signaling pathways via the same receptor of which one employs G protein-dependent signaling (TRAP) and the other initiates β-arrestin 2-dependent signaling (TR47) is highly indicative that PAR1 can induce biased signaling (Figure 3).91
Figure 3. Induction of biased signaling by canonical and non-canonical activation of PAR1.

Schematic representation of the fundamental and functional differences between APC and thrombin-mediated PAR1 activation. APC cleavage of PAR1 at Arg46 and the tethered-ligand sequence generated by this cleavage starting at Asn47 (TR47) induces a subset of PAR1 conformations that prefer signaling mediated by β-arrestin-2 involving activation of the PI3K-Akt pathway and that result in activation of barrier protective Rac1. In contrast, thrombin cleavage of PAR1 at Arg46 and the tethered-ligand sequence generated by this cleavage starting at Ser42 (TRAP) induces a subset of PAR1 conformations that prefer G protein-mediated signaling involving activation of the ERK1/2 and that result in activation of barrier disruptive RhoA. Thus, depending on the activating ligand, PAR1 can recruit different signaling pathways that result in different functional outcomes, which has been labeled “biased agonism.” The agonist bias is thus directly related to the cleavage sites of the tethered-ligand and the new N-terminal sequence as represented by the TRAP peptide that exists after cleavage at Arg41 or the TR47 peptide that exists after cleavage at Arg46. This figure was originally published in Blood.89 L.O. Mosnier, R.K. Sinha, L. Burnier, E.A. Bouwens, J.H. Griffin. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 2012;120:5237-46. © the American Society of Hematology.
Presumably the thrombin generated TRAP-like tethered-ligand induces a subset of PAR1 conformations that preferentially employs G protein-dependent signaling, whereas the APC generated TR47-like tethered-ligand induces a different spectrum of PAR1 conformations that recruit β-arrestin 2-dependent signaling. Thus, non-canonical activation of PAR1 by APC at Arg46 and canonical activation of PAR1 by thrombin at Arg41 can understandably mediate the often opposite effects of thrombin and APC because each protease generates different tethered-ligand agonists that utilize different signaling pathways with different functional consequences.
Relative contributions of APC’s anticoagulant and cytoprotective activities
Because of its multiple biologic activities, APC and the protein C pathway components have important roles in complex and challenging medical disorders and provide potential opportunities for pharmacologic treatment strategies in thrombosis, inflammation, and ischemic stroke.15,92,93 Although APC conveys beneficial effects in numerous different in vivo disease models, not all APC activities are necessarily beneficial. Based on the notion that the substrates and cofactors for APC’s anticoagulant activity (phospholipids, protein S and FVa) differ from APC’s cytoprotective effects (EPCR and PAR1), engineered APC mutants with cytoprotective-selective activities or anticoagulant-selective activities allowed the interrogation of the differential requirements for APC beneficial activities in vivo.66,94,95 Targeted disruption of the interaction of APC with protein S (Leu38Asp-APC) or with FVa results in cytoprotective-selective APC mutants such as, Arg222Cys/Asp237Cys (stabilizing the 70–80 loop, Arg229Ala/Arg230Ala-APC, Lys191Ala/Lys192Ala/Lys193Ala (a.k.a. 3K3A-APC) or a combination of the latter two (a.k.a. 5A-APC) (Figure 4).11,64,94–97 Targeted disruption of APC binding to EPCR while leaving phospholipid binding relatively intact (Leu8Gln-APC) or disruption of a region on APC that is required for cleavage of PAR1 (Glu330Ala and Glu333Ala) yields anticoagulant-selective APC mutants.98,99 Glu149Ala-APC, another anticoagulant-selective mutant provides a challenging test of our current understanding of the cytoprotective protein C pathway as its lack of cytoprotective activities remains enigmatic.66 Cytoprotective-selective but not anticoagulant-selective APC mutants provide beneficial effects in models of inflammation, sepsis, and ischemic stroke, whereas anticoagulant-selective but not cytoprotective-selective APC mutants prevent thrombosis, generally consistent with the concept that APC’s cytoprotective activities protect cells and APC’s anticoagulant activities prevent occlusive thrombosis.66,93,94 For instance, cytoprotective-selective 3K3A-APC or 5A-APC reduce mortality in bacteremia and LPS-induced endotoxemia but anticoagulant-selective Glu149Ala-APC fails to reduce mortality in these settings, indicating that the anticoagulant activity of APC that contributes to bleeding is dispensable for mortality reduction in sepsis.66,94 Comparable results are obtained for APC protection in ischemic stroke and neurodegenerative disease.93 In contrast, cytoprotective-selective 5A-APC fails to delay time to first occlusion in an acute carotid artery thrombosis model, whereas anticoagulant-selective Glu149Ala-APC effectively delays time to first occlusion as anticipated.66 Interestingly, and to contrast expectations, anticoagulant-selective Glu149Ala-APC but not cytoprotective-selective 5A-APC mitigates toxicity induced by lethal total body irradiation.100 Thus, depending on insult or disease model some activities of APC mediate beneficial effects, whereas other APC activities are dispensable or even harmful.
Figure 4. Schematic representation of the different structural requirements for APC’s anticoagulant and cytoprotective activities.
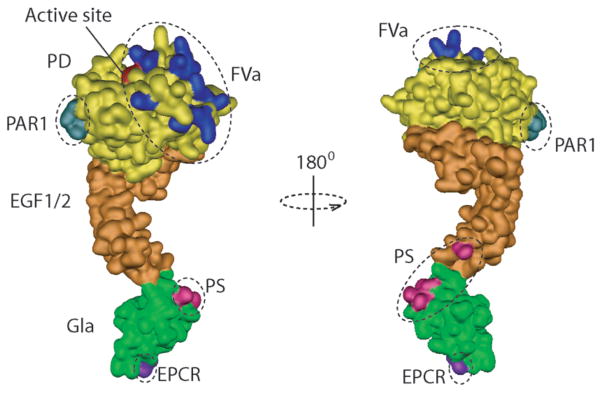
Anticoagulant activity of APC requires binding of the Gla-domain (green) to phospholipid surfaces, interaction with protein S (PS) mediated by residues on the Gla-domain and EGF1-domain (pink), and interactions of exosite residues (blue) on the APC protease domain (PD) with FVa. In contrast, cytoprotective activity of APC requires binding of the Gla-domain to EPCR (indicated by Leu8 (purple)), and interactions of a region on the opposite side of the FVa exosite on protease domain of APC that involves residues Glu330 and Glu333.
Concluding remarks
A concerted effort by many has resulted in several important discoveries for the protein C pathway in the last decade. These important advances include novel insights into the structure-function relation of APC with its multiple cofactors, receptors and substrates. The discovery of a novel cytoprotective protein C pathway that is independent of APC anticoagulant activities and conveys activities directly on cells through interactions with cellular receptors such as EPCR and PAR1 exemplifies another major advance in the last decade. The subsequent search for molecular mechanisms to explain these remarkable effects of APC on cells has provided some initial clues on the fundamental differences and contrasting functional effects between APC and thrombin-mediated initiation of PAR1-dependent cell signaling. Nevertheless, many unanswered questions still remain. The notion that PAR1 can initiate biased signaling with different and often opposite outcomes provides an intriguing challenge for ongoing and future basic and translational research on the protein C pathway and PAR1 structure-function. Biased signaling by PAR1 is especially relevant and helpful for interpretation of recent outcomes of large phase III clinical trials that evaluated the anti-thrombotic effects of PAR1 antagonists as these PAR1 antagonists were associated with increased bleeding, especially intracranial bleeding.101 In this regard second generation PAR1 compounds that antagonize PAR1-dependent G protein-mediated signaling but not β-arrestin 2-dependent signaling may provide therapeutic relevant entities, especially since APC, although an anticoagulant, prevented bleeding in the brain associated with profibrinolytic therapy.102
Perhaps the most striking advancement in the last decade that impacts our current view of APC’s multiple activities and the regulation thereof relates to the structure-function analysis of the protein C pathway. The notion that APC’s anticoagulant activity requires the APC Gla-domain to bind phospholipids and the APC protease domain to interact with FVa and FVIIIa aided by protein S versus APC’s cytoprotective activities that require the APC Gla-domain to bind to EPCR and the APC protease domain to interact with PAR1, led to new interrogations of the extended exosite on the protease domain of APC that is required for interactions with FVa and FVIIIa. Observations that the FVa exosite on APC is not required for APC interactions with PAR1 but instead that APC interaction with PAR1 requires a negatively charged region on the other side of the protease domain provided a way to separate APC anticoagulant activities from its cytoprotective activities. Pharmacological applications of these activity-selective APC mutants have allowed for unique insights into the relative contributions and requirements of anticoagulant versus cytoprotective activities of APC for its beneficial effects in numerous in vivo injury and disease models. In addition, activity-selective APC mutants allow for the exploration of new avenues in translation, preclinical and clinical research. For instance, the cytoprotective-selective APC 3K3A mutant has recently entered phase I clinical testing for applications in ischemic stroke.103
In summary, APC has multiple activities that require assembly of APC in macromolecular complexes supported by interactions of APC with cofactors and by exosite interactions on the protease domain of APC with its different substrates. These exosites interactions are overlapping or partially overlapping for some substrates, whereas for other substrates they are unique and non-overlapping. Although the novel advances of the last decade provide unique insights into how a single enzyme can mediate multiple biologically and therapeutically relevant activities, information on spatial orientation of the various ternary APC-cofactor-substrate complexes is limited and much remains unknown. Overall, the protein C pathway provides plentiful opportunities for basic research on the structure-function and molecular mechanisms of its multiple activities, as well as exciting avenues for translational research with potential therapeutic applications in complex diseases, such as the treatment of thrombosis, ischemic stroke, inflammatory disease, atherosclerosis, and vascular disease.
Learning Goals.
The protein C pathway provides multiple important functions to maintain a regulated balance between hemostasis and host defense systems.
APC’s anticoagulant activities prevent thrombosis whereas APC’s cytoprotective activities protect cells.
APC’s different activities require assembly of different macromolecular complexes with different cofactors that can be targeted by mutagenesis to obtain activity-selective APC mutants.
Anticoagulant-selective and cytoprotective-selective APC mutants allow insights into the relative contributions of these APC activities to beneficial effects in various murine injury and disease models.
Acknowledgments
We apologize to our colleagues whose work could not be cited due to space limitations. This work was supported by an American Heart Association Western States Affiliate postdoctoral fellowship (E.A.B.) and National Institutes of Health (NHLBI) grant HL104165 (L.O.M.).
Abbreviations
- APC
activated protein C
- ApoER2
apolipoprotein E receptor 2
- EPCR
endothelial protein C receptor
- FVa
activated factor V
- FVIIa
activated factor VIIa
- FVIIIa
activated factor VIII
- PAR
protease activated receptor
- PS
protein S
- Rac1
Ras-related C3 botulinum toxin substrate 1
- RhoA
Ras homolog gene family member A
- S1P1
S1P receptor 1
- SHBG
sex hormone-binding globulin domain
- TAFI
thrombin Activatable fibrinolysis inhibitor
- TRAP
thrombin receptor activating peptide
- TSR
thrombin sensitive region
Footnotes
Conflict of Interest disclosure
L.O. Mosnier is a co-inventor for intellectual property owned by The Scripps Research Institute that is related to concepts discussed here. The other authors have nothing to disclose.
Contributor Information
Fabian Stavenuiter, Email: fabianst@scripps.edu.
Eveline A. M. Bouwens, Email: evelineb@scripps.edu.
References
- 1.Gailani D, Broze GJ., Jr Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–12. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 2.Bouma BN, von dem Borne PA, Meijers JC. Factor XI and protection of the fibrin clot against lysis--a role for the intrinsic pathway of coagulation in fibrinolysis. Thromb Haemost. 1998;80:24–7. [PubMed] [Google Scholar]
- 3.Renne T, Schmaier AH, Nickel KF, Blomback M, Maas C. In vivo roles of factor XII. Blood. 2012;120:4296–303. doi: 10.1182/blood-2012-07-292094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morrissey JH, Choi SH, Smith SA. Polyphosphate: an ancient molecule that links platelets, coagulation, and inflammation. Blood. 2012;119:5972–9. doi: 10.1182/blood-2012-03-306605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosnier LO, Bouma BN. Regulation of fibrinolysis by thrombin activatable fibrinolysis inhibitor, an unstable carboxypeptidase B that unites the pathways of coagulation and fibrinolysis. Arterioscler Thromb Vasc Biol. 2006;26:2445–53. doi: 10.1161/01.ATV.0000244680.14653.9a. [DOI] [PubMed] [Google Scholar]
- 6.Bajzar L, Manuel R, Nesheim ME. Purification and characterization of TAFI, a thrombin-activable fibrinolysis inhibitor. J Biol Chem. 1995;270:14477–84. doi: 10.1074/jbc.270.24.14477. [DOI] [PubMed] [Google Scholar]
- 7.von dem Borne PA, Bajzar L, Meijers JC, Nesheim ME, Bouma BN. Thrombin-mediated activation of factor XI results in a thrombin-activatable fibrinolysis inhibitor-dependent inhibition of fibrinolysis. J Clin Invest. 1997;99:2323–7. doi: 10.1172/JCI119412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zwaal RF, Comfurius P, Bevers EM. Lipid-protein interactions in blood coagulation. Biochim Biophys Acta. 1998;1376:433–53. doi: 10.1016/s0304-4157(98)00018-5. [DOI] [PubMed] [Google Scholar]
- 9.Rosing J, van Rijn JL, Bevers EM, van Dieijen G, Comfurius P, Zwaal RF. The role of activated human platelets in prothrombin and factor X activation. Blood. 1985;65:319–32. [PubMed] [Google Scholar]
- 10.Huntington JA. Serpin structure, function and dysfunction. J Thromb Haemost. 2011;9 (Suppl 1):26–34. doi: 10.1111/j.1538-7836.2011.04360.x. [DOI] [PubMed] [Google Scholar]
- 11.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–72. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 12.Dahlbäck B, Villoutreix BO. Regulation of blood coagulation by the protein C anticoagulant pathway. Novel insights into structure-function relationships and molecular recognition. Arterioscler Thromb Vasc Biol. 2005;25:1311–20. doi: 10.1161/01.ATV.0000168421.13467.82. [DOI] [PubMed] [Google Scholar]
- 13.Rezaie AR. Regulation of the protein C anticoagulant and antiinflammatory pathways. Curr Med Chem. 2010;17:2059–69. doi: 10.2174/092986710791233706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esmon CT. Molecular events that control the protein C anticoagulant pathway. Thromb Haemost. 1993;70:29–35. [PubMed] [Google Scholar]
- 15.Esmon CT. Protein C anticoagulant system--anti-inflammatory effects. Semin Immunopathol. 2012;34:127–32. doi: 10.1007/s00281-011-0284-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffin JH, Evatt B, Zimmerman TS, Kleiss AJ, Wideman C. Deficiency of protein C in congenital thrombotic disease. J Clin Invest. 1981;68:1370–3. doi: 10.1172/JCI110385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Branson HE, Katz J, Marble R, Griffin JH. Inherited protein C deficiency and coumarin-responsive chronic relapsing purpura fulminans in a newborn infant. Lancet. 1983;2:1165–8. doi: 10.1016/s0140-6736(83)91216-3. [DOI] [PubMed] [Google Scholar]
- 18.Chalmers E, Cooper P, Forman K, Grimley C, Khair K, Minford A, et al. Purpura fulminans: recognition, diagnosis and management. Arch Dis Child. 2011;96:1066–71. doi: 10.1136/adc.2010.199919. [DOI] [PubMed] [Google Scholar]
- 19.Reitsma PH, Poort SR, Allaart CF, Briet E, Bertina RM. The spectrum of genetic defects in a panel of 40 Dutch families with symptomatic protein C deficiency type I: heterogeneity and founder effects. Blood. 1991;78:890–4. [PubMed] [Google Scholar]
- 20.Dahlbäck B. The protein C anticoagulant system: inherited defects as basis for venous thrombosis. Thromb Res. 1995;77:1–43. doi: 10.1016/0049-3848(94)00138-4. [DOI] [PubMed] [Google Scholar]
- 21.Nazarian RM, van Cott EM, Zembowicz A, Duncan LM. Warfarin-induced skin necrosis. J Am Acad Dermatol. 2009;61:325–32. doi: 10.1016/j.jaad.2008.12.039. [DOI] [PubMed] [Google Scholar]
- 22.Yan SB, Helterbrand JD, Hartman DL, Wright TJ, Bernard GR. Low levels of protein C are associated with poor outcome in severe sepsis. Chest. 2001;120:915–22. doi: 10.1378/chest.120.3.915. [DOI] [PubMed] [Google Scholar]
- 23.Mosnier LO, Griffin JH. Protein C, protein S, thrombomodulin and the endothelial protein C receptor pathways. In: Marder VJ, Aird WC, Bennett JS, Schulman S, White GC, Colman RW, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Philadelphia: Lippincott Williams & Wilkins; 2013. pp. 300–313. [Google Scholar]
- 24.Mather T, Oganessyan V, Hof P, Huber R, Foundling S, Esmon CT, et al. The 2.8 Å crystal structure of Gla-domainless activated protein C. EMBO J. 1996;15:6822–31. [PMC free article] [PubMed] [Google Scholar]
- 25.Esmon CT, Owen WG. Identification of an endothelial cell cofactor for thrombin catalyzed activation of protein C. Proc Natl Acad Sci U S A. 1981;78:2249–52. doi: 10.1073/pnas.78.4.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukudome K, Esmon CT. Identification, cloning, and regulation of a novel endothelial cell protein C/activated protein C receptor. J Biol Chem. 1994;269:26486–91. [PubMed] [Google Scholar]
- 27.Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci U S A. 1996;93:10212–6. doi: 10.1073/pnas.93.19.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grinnell BW, Gerlitz B, Berg DT. Identification of a region in protein C involved in thrombomodulin-stimulated activation by thrombin: potential repulsion at anion-binding site I in thrombin. Biochem J. 1994;303 ( Pt 3):929–33. doi: 10.1042/bj3030929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gale AJ, Griffin JH. Characterization of a thrombomodulin binding site on protein C and its comparison to an activated protein C binding site for factor Va. Proteins. 2004;54:433–41. doi: 10.1002/prot.10627. [DOI] [PubMed] [Google Scholar]
- 30.Knobe KE, Berntsdotter A, Shen L, Morser J, Dahlbäck B, Villoutreix BO. Probing the activation of protein C by the thrombin-thrombomodulin complex using structural analysis, site-directed mutagenesis and computer modeling. Proteins. 1999;35:218–34. doi: 10.1002/(sici)1097-0134(19990501)35:2<218::aid-prot8>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 31.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27:157–62. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 32.Ehrlich HJ, Grinnell BW, Jaskunas SR, Esmon CT, Yan SB, Bang NU. Recombinant human protein C derivatives: altered response to calcium resulting in enhanced activation by thrombin. EMBO J. 1990;9:2367–73. doi: 10.1002/j.1460-2075.1990.tb07411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richardson MA, Gerlitz B, Grinnell BW. Enhancing protein C interaction with thrombin results in a clot-activated anticoagulant. Nature. 1992;360:261–4. doi: 10.1038/360261a0. [DOI] [PubMed] [Google Scholar]
- 34.Pozzi N, Barranco-Medina S, Chen Z, Di CE. Exposure of R169 controls protein C activation and autoactivation. Blood. 2012;120:664–70. doi: 10.1182/blood-2012-03-415323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isermann B, Vinnikov IA, Madhusudhan T, Herzog S, Kashif M, Blautzik J, et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–58. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- 36.Rezaie AR. Exosite-dependent regulation of the protein C anticoagulant pathway. Trends Cardiovasc Med. 2003;13:8–15. doi: 10.1016/s1050-1738(02)00191-3. [DOI] [PubMed] [Google Scholar]
- 37.Wildhagen KC, Lutgens E, Loubele ST, Ten CH, Nicolaes GA. The structure-function relationship of activated protein C. Lessons from natural and engineered mutations. Thromb Haemost. 2011;106:1034–45. doi: 10.1160/TH11-08-0522. [DOI] [PubMed] [Google Scholar]
- 38.Berg DT, Gerlitz B, Shang J, Smith T, Santa P, Richardson MA, et al. Engineering the proteolytic specificity of activated protein C improves its pharmacological properties. Proc Natl Acad Sci U S A. 2003;100:4423–8. doi: 10.1073/pnas.0736918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen L, Villoutreix BO, Dahlbäck B. Involvement of lys 62(217) and lys 63(218) of human anticoagulant protein C in heparin stimulation of inhibition by the protein C inhibitor. Thromb Haemost. 1999;82:72–9. [PubMed] [Google Scholar]
- 40.Kalafatis M, Rand MD, Mann KG. The mechanism of inactivation of human factor V and human factor Va by activated protein C. J Biol Chem. 1994;269:31869–80. [PubMed] [Google Scholar]
- 41.Nicolaes GAF, Tans G, Thomassen MC, Hemker HC, Pabinger I, Varadi K, et al. Peptide bond cleavages and loss of functional activity during inactivation of factor Va and factor VaR506Q by activated protein C. J Biol Chem. 1995;270:21158–66. doi: 10.1074/jbc.270.36.21158. [DOI] [PubMed] [Google Scholar]
- 42.Smirnov MD, Esmon CT. Phosphatidylethanolamine incorporation into vesicles selectively enhances factor Va inactivation by activated protein C. J Biol Chem. 1994;269:816–9. [PubMed] [Google Scholar]
- 43.Bajzar L, Nesheim ME, Tracy PB. The profibrinolytic effect of activated protein C in clots formed from plasma is TAFI-dependent. Blood. 1996;88:2093–100. [PubMed] [Google Scholar]
- 44.McDonald JF, Shah AM, Schwalbe RA, Kisiel W, Dahlbäck B, Nelsestuen GL. Comparison of naturally occurring vitamin K-dependent proteins: correlation of amino acid sequences and membrane binding properties suggests a membrane contact site. Biochemistry. 1997;36:5120–7. doi: 10.1021/bi9626160. [DOI] [PubMed] [Google Scholar]
- 45.Smirnov MD, Safa O, Regan L, Mather T, Stearns-Kurosawa DJ, Kurosawa S, et al. A chimeric protein C containing the prothrombin Gla domain exhibits increased anticoagulant activity and altered phospholipid specificity. J Biol Chem. 1998;273:9031–40. doi: 10.1074/jbc.273.15.9031. [DOI] [PubMed] [Google Scholar]
- 46.Shen L, Shah AM, Dahlbäck B, Nelsestuen GL. Enhancement of human protein C function by site-directed mutagenesis of the gamma-carboxyglutamic acid domain. J Biol Chem. 1998;273:31086–91. doi: 10.1074/jbc.273.47.31086. [DOI] [PubMed] [Google Scholar]
- 47.Camire RM. A new look at blood coagulation factor V. Curr Opin Hematol. 2011;18:338–42. doi: 10.1097/MOH.0b013e3283497ebc. [DOI] [PubMed] [Google Scholar]
- 48.Nesheim ME, Taswell JB, Mann KG. The contribution of bovine Factor V and Factor Va to the activity of prothrombinase. J Biol Chem. 1979;254:10952–62. [PubMed] [Google Scholar]
- 49.Bertina RM, Koeleman BPC, Koster T, Rosendaal FR, Dirven RJ, de Ronde H, et al. Mutations in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–7. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 50.Friedrich U, Nicolaes GAF, Villoutreix BO, Dahlbäck B. Secondary substrate-binding exosite in the serine protease domain of activated protein C important for cleavage at Arg-506 but not at Arg-306 in factor Va. J Biol Chem. 2001;276:23105–8. doi: 10.1074/jbc.M103138200. [DOI] [PubMed] [Google Scholar]
- 51.Gale AJ, Tsavaler A, Griffin JH. Molecular characterization of an extended binding site for coagulation factor Va in the positive exosite of activated protein C. J Biol Chem. 2002;277:28836–40. doi: 10.1074/jbc.M204363200. [DOI] [PubMed] [Google Scholar]
- 52.Lenting PJ, van Mourik JA, Mertens K. The life cycle of coagulation Factor VIII in view of its structure and function. Blood. 1998;92:3983–96. [PubMed] [Google Scholar]
- 53.van Dieijen G, Tans G, Rosing J, Hemker HC. The role of phospholipid and factor VIIIa in the activation of bovine factor X. J Biol Chem. 1981;256:3433–42. [PubMed] [Google Scholar]
- 54.Regan LM, Lamphear BJ, Huggins CF, Walker FJ, Fay PJ. Factor IXa protects factor VIIIa from activated protein C. Factor IXa inhibits activated protein C-catalyzed cleavage of factor VIIIa at Arg562. J Biol Chem. 1994;269:9445–52. [PubMed] [Google Scholar]
- 55.Manithody C, Fay PJ, Rezaie AR. Exosite-dependent regulation of factor VIIIa by activated protein C. Blood. 2003;101:4802–7. doi: 10.1182/blood-2003-01-0126. [DOI] [PubMed] [Google Scholar]
- 56.O’Brien LM, Mastri M, Fay PJ. Regulation of factor VIIIa by human activated protein C and protein S: inactivation of cofactor in the intrinsic factor Xase. Blood. 2000;95:1714–20. [PubMed] [Google Scholar]
- 57.Cramer TJ, Gale AJ. The anticoagulant function of coagulation factor V. Thromb Haemost. 2012;107:15–21. doi: 10.1160/TH11-06-0431. [DOI] [PubMed] [Google Scholar]
- 58.Hafizi S, Dahlbäck B. Gas6 and protein S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. FEBS J. 2006;273:5231–44. doi: 10.1111/j.1742-4658.2006.05529.x. [DOI] [PubMed] [Google Scholar]
- 59.Hackeng TM, Maurissen LF, Castoldi E, Rosing J. Regulation of TFPI function by protein S. J Thromb Haemost. 2009;7:165–8. doi: 10.1111/j.1538-7836.2009.03363.x. [DOI] [PubMed] [Google Scholar]
- 60.ten Kate MK, van der Meer J. Protein S deficiency: a clinical perspective. Haemophilia. 2008;14:1222–8. doi: 10.1111/j.1365-2516.2008.01775.x. [DOI] [PubMed] [Google Scholar]
- 61.Rosing J, Hoekema L, Nicolaes GAF, Thomassen MC, Hemker HC, Varadi K, et al. Effects of protein S and factor Xa on peptide bond cleavages during inactivation of factor Va and factor Va R506Q by activated protein C. J Biol Chem. 1995;270:27852–8. doi: 10.1074/jbc.270.46.27852. [DOI] [PubMed] [Google Scholar]
- 62.Norstrom EA, Tran S, Steen M, Dahlback B. Effects of factor Xa and protein S on the individual activated protein C-mediated cleavages of coagulation factor Va. J Biol Chem. 2006;281:31486–94. doi: 10.1074/jbc.M606441200. [DOI] [PubMed] [Google Scholar]
- 63.Yegneswaran S, Wood GM, Esmon CT, Johnson AE. Protein S alters the active site location of activated protein C above the membrane surface. A fluorescence resonance energy transfer study of topography. J Biol Chem. 1997;272:25013–21. doi: 10.1074/jbc.272.40.25013. [DOI] [PubMed] [Google Scholar]
- 64.Harmon S, Preston RJ, Ainle FN, Johnson JA, Cunningham MS, Smith OP, et al. Dissociation of activated protein C functions by elimination of protein S cofactor enhancement. J Biol Chem. 2008;283:30531–9. doi: 10.1074/jbc.M802338200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Preston RJ, Ajzner E, Razzari C, Karageorgi S, Dua S, Dahlbäck B, et al. Multifunctional specificity of the protein C/activated protein C GLA domain. J Biol Chem. 2006;281:28850–7. doi: 10.1074/jbc.M604966200. [DOI] [PubMed] [Google Scholar]
- 66.Mosnier LO, Zampolli A, Kerschen EJ, Schuepbach RA, Banerjee Y, Fernandez JA, et al. Hyper-antithrombotic, non-cytoprotective Glu149Ala-activated protein C mutant. Blood. 2009;113:5970–8. doi: 10.1182/blood-2008-10-183327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ohlin AK, Landes G, Bourdon P, Oppenheimer C, Wydro R, Stenflo J. Beta-hydroxyaspartic acid in the first epidermal growth factor-like domain of protein C. Its role in Ca2+ binding and biological activity. J Biol Chem. 1988;263:19240–8. [PubMed] [Google Scholar]
- 68.Adams TE, Hockin MF, Mann KG, Everse SJ. The crystal structure of activated protein C-inactivated bovine factor Va: Implications for cofactor function. Proc Natl Acad Sci U S A. 2004;101:8918–23. doi: 10.1073/pnas.0403072101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Villoutreix BO, Teleman O, Dahlback B. A theoretical model for the Gla-TSR-EGF-1 region of the anticoagulant cofactor protein S: from biostructural pathology to species-specific cofactor activity. J Comput Aided Mol Des. 1997;11:293–304. doi: 10.1023/a:1007912929828. [DOI] [PubMed] [Google Scholar]
- 70.Pellequer JL, Gale AJ, Getzoff ED, Griffin JH. Three-dimensional model of coagulation factor Va bound to activated protein C. Thromb Haemost. 2000;84:849–57. [PubMed] [Google Scholar]
- 71.Walker FJ, Scandella D, Fay PJ. Identification of the binding site for activated protein C on the light chain of factors V and VIII. J Biol Chem. 1990;265:1484–9. [PubMed] [Google Scholar]
- 72.Krishnaswamy S, Williams EB, Mann KG. The binding of activated protein C to factors V and Va. J Biol Chem. 1986;261:9684–93. [PubMed] [Google Scholar]
- 73.Dahlbäck B, Villoutreix BO. The anticoagulant protein C pathway. FEBS Lett. 2005;579:3310–6. doi: 10.1016/j.febslet.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 74.Saller F, Villoutreix BO, Amelot A, Kaabache T, Le Bonniec BF, Aiach M, et al. The gamma-carboxyglutamic acid domain of anticoagulant protein S is involved in activated protein C cofactor activity, independently of phospholipid binding. Blood. 2005;105:122–30. doi: 10.1182/blood-2004-06-2176. [DOI] [PubMed] [Google Scholar]
- 75.He XH, Shen L, Villoutreix BO, Dahlbäck B. Amino acid residues in thrombin-sensitive region and first epidermal growth factor domain of vitamin K-dependent protein S determining specificity of the activated protein C cofactor function. J Biol Chem. 1998;273:27449–58. doi: 10.1074/jbc.273.42.27449. [DOI] [PubMed] [Google Scholar]
- 76.Hackeng TM, Yegneswaran S, Johnson AE, Griffin JH. Conformational changes in activated protein C caused by binding of the first epidermal growth factor-like module of protein S. Biochem J. 2000;349:757–64. doi: 10.1042/bj3490757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Andersson HM, Arantes MJ, Crawley JT, Luken BM, Tran S, Dahlbäck B, et al. Activated protein C cofactor function of protein S: a critical role for Asp95 in the EGF1-like domain. Blood. 2010;115:4878–85. doi: 10.1182/blood-2009-11-256610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ahnstrom J, Andersson HM, Canis K, Norstrom E, Yu Y, Dahlbäck B, et al. Activated protein C cofactor function of protein S: a novel function for a {gamma}-carboxyglutamic acid residue. Blood. 2011;117:6685–93. doi: 10.1182/blood-2010-11-317099. [DOI] [PubMed] [Google Scholar]
- 79.Evenas P, Garcia de FP, Nicolaes GA, Dahlback B. The second laminin G-type domain of protein S is indispensable for expression of full cofactor activity in activated protein C-catalysed inactivation of factor Va and factor VIIIa. Thromb Haemost. 2000;84:271–7. [PubMed] [Google Scholar]
- 80.Heeb MJ, Kojima Y, Rosing J, Tans G, Griffin JH. C-terminal residues 621-635 of protein S are essential for binding to factor Va. J Biol Chem. 1999;274:36187–92. doi: 10.1074/jbc.274.51.36187. [DOI] [PubMed] [Google Scholar]
- 81.Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein C defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- 82.Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest. 2003;111:25–7. doi: 10.1172/JCI17564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–2. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 84.Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, et al. High-resolution crystal structure of human protease-activated receptor 1. Nature. 2012;492:387–92. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weiler H. Multiple receptor-mediated functions of activated protein C. Hamostaseologie. 2011;31:185–95. doi: 10.5482/ha-1166. [DOI] [PubMed] [Google Scholar]
- 87.Rezaie AR. The occupancy of endothelial protein C receptor by its ligand modulates the par-1 dependent signaling specificity of coagulation proteases. IUBMB Life. 2011;63:390–6. doi: 10.1002/iub.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Russo A, Soh UJ, Paing MM, Arora P, Trejo J. Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc Natl Acad Sci U S A. 2009;106:6393–7. doi: 10.1073/pnas.0810687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by non-canonical cleavage at Arg46. Blood. 2012;120:5237–46. doi: 10.1182/blood-2012-08-452169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Soh UJ, Trejo J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through beta-arrestin and dishevelled-2 scaffolds. Proc Natl Acad Sci U S A. 2011;108:E1372–E1380. doi: 10.1073/pnas.1112482108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular Mechanism of β-Arrestin-Biased Agonism at Seven-Transmembrane Receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–97. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Danese S, Vetrano S, Zhang L, Poplis VA, Castellino FJ. The protein C pathway in tissue inflammation and injury: pathogenic role and therapeutic implications. Blood. 2010;115:1121–30. doi: 10.1182/blood-2009-09-201616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zlokovic BV, Griffin JH. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci. 2011;34:198–209. doi: 10.1016/j.tins.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204:2439–48. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bae JS, Yang L, Manithody C, Rezaie AR. Engineering a disulfide bond to stabilize the calcium binding loop of activated protein C eliminates its anticoagulant but not protective signaling properties. J Biol Chem. 2007;282:9251–9. doi: 10.1074/jbc.M610547200. [DOI] [PubMed] [Google Scholar]
- 96.Mosnier LO, Gale AJ, Yegneswaran S, Griffin JH. Activated protein C variants with normal cytoprotective but reduced anticoagulant activity. Blood. 2004;104:1740–5. doi: 10.1182/blood-2004-01-0110. [DOI] [PubMed] [Google Scholar]
- 97.Mosnier LO, Yang XV, Griffin JH. Activated protein C mutant with minimal anticoagulant activity, normal cytoprotective activity, and preservation of thrombin activable fibrinolysis inhibitor-dependent cytoprotective functions. J Biol Chem. 2007;282:33022–33. doi: 10.1074/jbc.M705824200. [DOI] [PubMed] [Google Scholar]
- 98.Yang L, Bae JS, Manithody C, Rezaie AR. Identification of a specific exosite on activated protein C for interaction with protease activated receptor 1. J Biol Chem. 2007;282:25493–500. doi: 10.1074/jbc.M702131200. [DOI] [PubMed] [Google Scholar]
- 99.Preston RJ, Villegas-Mendez A, Sun YH, Hermida J, Simioni P, Philippou H, et al. Selective modulation of protein C affinity for EPCR and phospholipids by Gla domain mutation. FEBS J. 2005;272:97–108. doi: 10.1111/j.1432-1033.2004.04401.x. [DOI] [PubMed] [Google Scholar]
- 100.Geiger H, Pawar SA, Kerschen EJ, Nattamai KJ, Hernandez I, Liang HP, et al. Pharmacological targeting of the thrombomodulin-activated protein C pathway mitigates radiation toxicity. Nat Med. 2012;18:1123–9. doi: 10.1038/nm.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366:1404–13. doi: 10.1056/NEJMoa1200933. [DOI] [PubMed] [Google Scholar]
- 102.Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, et al. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med. 2006;12:1278–85. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- 103.Williams PD, Zlokovic BV, Griffin JH, Pryor KE, Davis TP. Preclinical safety and pharmacokinetic profile of 3K3A-APC, a novel, modified activated protein C for ischemic stroke. Curr Pharm Des. 2012;18:4215–22. doi: 10.2174/138161212802430413. [DOI] [PMC free article] [PubMed] [Google Scholar]