

Abstract
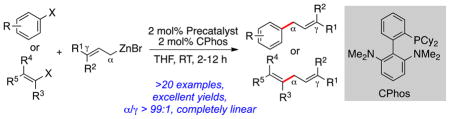
Completely linear: a general and practical palladium-catalyzed linear-selective Negishi coupling of 3,3-disubstituted allylzinc reagents with aryl, heteroaryl and vinyl electrophiles at ambient temperature is described. This method provides an effective means to access a wide range of prenylated arenes or skipped dienes in a completely linear-selective fashion, as demonstrated by a concise synthesis of the anti-HIV natural product siamenol. Finally, DFT calculations shed light on the origin of the excellent regioselectivity observed with the current catalyst system.
Keywords: cross-coupling, palladium, heterocycles, organozinc reagent, prenylation
Prenylated arenes are found in a broad spectrum of naturally occurring bioactive compounds (Scheme 1).[1] Studies have revealed that the inclusion of the prenyl side chain could enhance both the bioactivity and bioavailability of natural products, in part due to the increased protein binding affinity and membrane permeability caused by the lipophilicity of the prenyl group.[2] In Nature, prenylation processes usually involve enzymatic reactions mediated by a range of substrate-specific prenyltransferases (PTases),[3] giving rise to the corresponding prenylated natural products in a highly selective fashion. However, the ability of synthetic chemists to directly introduce prenyl and related 3,3-disubstituted allyl groups onto functionalized aromatic compounds is generally hampered by poor regioselectivity with respect to the unsymmetrical allyl nucleophile.[4,5] Therefore, a general, reliable and practical regioselective prenylation method that emulates the efficiency of Nature’s biosynthetic machinery is highly desirable.[6]
Scheme 1.

Regioselective Negishi Cross-Coupling: Rapid Access to Prenylated Compounds.
Recently, Organ[4] and we[5] have developed conditions for the linear-selective Suzuki-Miyaura coupling of 3,3-disubstituted allylboronates using NHC- or phosphine-based catalysts, respectively. While both protocols were highly selective, relatively high temperatures and extended reaction times were required, and the yields of the desired prenylated products were moderate due to the inevitable formation of homocoupling products.[4,5] In an effort to develop milder and more efficient prenylation methods, we sought to utilize alternative prenyl nucleophiles. Prenyl-type organozinc reagents were our first choice, as we had previously demonstrated the high reactivity as well as functional group compatibility of organozinc reagents in a variety of Negishi coupling processes.[7,8] To date, the cross-coupling of 3,3-disubstituted allylzinc reagents with aryl halides remains rare, presumably due to issues of regioselectivity often experienced with this class of nucleophile.[4,5,9] Herein we report the first general and completely linear-selective conditions for the Negishi coupling of 3,3-disubstituted allylzinc reagents with aryl halides and the application of this methodology in the concise and convergent synthesis of anti-HIV natural product siamenol (1). Computational studies were also carried out to gain a deeper insight into the high level of selectivity observed with the current catalyst system.[10,11]
Using prenylZnBr·LiCl prepared by Knochel’s protocol[12,13] and 1-bromo-4-butylbenzene as model substrates, we commenced our study by examining our recently developed easily activated palladacycle precatalysts derived from biarylphosphine ligands (Table 1).[14] While SPhos (L1) and RuPhos (L2)-based catalysts were effective for the linear-selective prenylation, only moderate conversion was observed (Table 1, entry 1–2). Both the XPhos (L3)- and the CPhos (L5)-based catalyst furnished full conversion of the aryl halide component, affording the α-isomer (4) in good yield with minimal amounts of the γ-isomer or other side products (entry 3 and 5). Ultimately, the catalyst generated from CPhos[15] was identified as the optimal choice for this coupling reaction, affording the α-coupling product in a highly selective manner.[16] Under the optimized reaction conditions, treatment of the aryl bromide with prenylzinc bromide in the presence of 2 mol% CPhos-based catalyst afforded the prenylated product in 94% yield in 30 min at room temperature (entry 5). Complete conversion could also be achieved in ≤ 3 min at 70 °C, further demonstrating the excellent activity of this catalyst system (entry 6). Aryl triflates (entry 7) and iodides (entry 8) were also suitable coupling partners using this protocol. Interestingly, the well-tailored NHC-supported PEPPSI precatalyst[4] (PEPPSI-IPent, 6) proved to be less effective for this transformation (see SI). Other commercially available and frequently used catalysts or ligands such as Pd(PPh3)4, dppm, dppp and dppb were ineffective for this prenylation reaction (see SI for details).
Table 1.
Ligand effect on the palladium catalyzed Negishi cross-coupling of prenylzinc bromide and 1-bromo-4-butylbenzene.
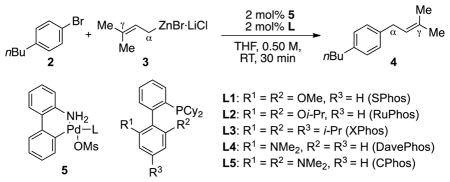
| ||||
|---|---|---|---|---|
| entry | ligand | conversion[a] | yield[a] | α/γ ratio[a] |
| 1 | L1 | 54% | 47% | >99:1 |
| 2 | L2 | 52% | 41% | >99:1 |
| 3 | L3 | 100% | 92% | >99:1 |
| 4 | L4 | 84% | 74% | >99:1 |
| 5 | L5 | 98% | 94% | >99:1 |
| 6[b] | L5 | 100% | 92% | >99:1 |
| 7[c] | L5 | 100% | 94% | >99:1 |
| 8[d] | L5 | 100% | 94% | >99:1 |
Conversions and yields were determined by GC analysis of the crude reaction mixture using dodecane as an internal standard.
70 °C for only 3 min.
1-butyl-4-iodobenzene was used in lieu of 2.
4-butylphenyl triflate was used in lieu of 2.
We next investigated the substrate scope of the method (Scheme 2). It was found that with the CPhos-based catalyst, both electron-donating (7a) and electron-withdrawing (7b–7c) substituents were tolerated with no decrease in the observed regioselectivity. An extremely sterically hindered substrate (7d) could also be successfully transformed. Substrates containing acidic functional groups, such as an acetamide (7e) and a phenol (7f), were also transformed to the desired products. Given the importance of heterocycles in medicinal chemistry,[17] we examined a variety of sterically and electronically diverse heteroaryl halides (Scheme 2 (B)). We found that pyridine (7g), pyrimidine (7h), indoles (7i–7j), azaindole (7k) and pyrazole (7l) all underwent smooth transformation using this protocol.
Scheme 2.

Substrate scope of aryl and heteroaryl halides. Reaction conditions: Ar-X (1.0 mmol), prenylZnBr·LiCl (1.3 mmol), 3 (0.02 mmol), L5 (0.02 mmol), RT, THF, 2 h. Yields are of isolated yield on average of two runs. a. determined by 1H NMR spectroscopy due to the volatility of coupling product. b. 2.3 equiv prenylzinc was used.
One limitation of our previously developed Suzuki-Miyaura coupling was its ability to effectively engage vinyl electrophiles. Lower levels of regioselectivity were often observed with regard to vinyl bromides,[18] and in the case of vinyl triflates, competitive hydrolysis resulted in the formation of low yields of desired coupling products. The current Negishi coupling protocol successfully addressed these problems. Vinyl bromides and triflates were converted to the corresponding “skipped dienes”, which represent key structural motifs in a number of biologically active natural products,[19] in a completely regioselective manner (Scheme 3). Notably, mono- (8a), di- (8b–8d) and trisubstituted (8e–8f) vinyl electrophiles could all be applied in this reaction without noticeable erosion of regioselectivity. Five- (8e) and six-membered (8b–8d) cyclic vinyl triflates represented compatible coupling partners as well.
Scheme 3.

Substrate scope of vinyl halides and pseudohalides. Reaction conditions: vinyl halide (0.5 mmol), prenylZnBr·LiCl (0.65 mmol), 3 (0.01 mmol), L5 (0.01 mmol), RT, THF, 2 h. Yields are of isolated yield on average of two runs.
In an effort to expand the utility of this method, we examined the coupling of various 3,3-unsymmetrically disubstituted allylzinc halides (Scheme 4). In all cases examined, the allylation proceeded smoothly furnishing the linear-coupling product exclusively in excellent yields. While both geranyl- (9a) and farnesylzinc bromides (9b) afforded 75:25 mixtures of olefin stereoisomers, allylzinc halides bearing two substituents of greater steric difference furnished improved stereoselectivity with respect to the trisubstituted olefin moiety (9c–9e). For example, while the use of 3-methyl-3-cyclohexylallylzinc bromide (9c) furnished coupling product as stereoisomeric mixtures (E/Z ratio = 85:15), the coupling of 3-methyl-3-tert-butylallylzinc bromide provided the trisubstituted olefin exclusively with E geometry (9d).
Scheme 4.

Substrate scope of 3,3-disubstituted allylzinc reagents. Reaction conditions: Ar-X (0.50 mmol), allylZnX′·LiCl (0.65 mmol), 3 (0.01 mmol), L5 (0.01 mmol), RT, THF, 2 h. Yields are of isolated yield on average of two runs.
To further showcase the utility of this prenylation methodology in a complex setting, we performed a concise synthesis of siamenol (1), a prenylated natural product isolated from Murraya siamensis and exhibiting anti-HIV activity.[20] Beginning with 4-bromotoluene (10), palladium-catalyzed amination proceeded smoothly to deliver the unsymmetrical diarylamine 12. Subsequently, palladium-catalyzed intramolecular C-H activation and C3-bromination furnished the carbazole 14, which in turn, underwent the completely linear-selective Negishi cross-coupling to afford the prenylated carbazole 15. In the final step, demethylation employing methylmagnesium iodide furnished natural product siamenol (1). Overall, the strategic applications of a series of palladium-catalyzed cross-coupling reactions facilitated by the use of dialkylbiarylphosphine ligands developed in our laboratory have enabled the rapid assembly of prenylated carbazole natural products.
To gain a further understanding into the regiocontrol of this reaction, we computed the reaction coordinate (Figure 1).[21,22] The catalytic cycle begins with the initial complexation of Pd(0) catalyst with the aryl bromide (I). Oxidative addition into the C–Br bond (TS-II) affords the resting state intermediate III. The two possible transmetalation processes involving α-prenylzinc were next investigated, namely a four-membered TS (TS-IV-α-4-mem) and a six-membered TS (TS-IV-γ-6-mem) leading to α- and γ-prenyl palladium intermediate V respectively. Given that the prenylzinc species undergoes rapid 1,3-shift at room temperature,[23] we also evaluated the feasibility of two additional mechanisms utilizing γ-prenylzinc bromide as the transmetalating agent (Figure 1, TS-IV-γ-4-mem and TS-IV-α-6-mem, respectively). We found that there is an energetic preference for the α–4-membered processes over the α–6, but this was reversed in the γcase presumably due to the steric bulk around the forming Pd-C bond in TS-IV-γ-4-mem. The minimum energy pathway involves the TS-IV-α-4-mem, generating the α-prenyl palladium intermediate V, which then undergoes product-determining η1-α reductive elimination TS-VI to afford the product-catalyst complex VII. We also investigated γ reductive elimination leading to the γ-isomer and found that the η3-γ was preferred over the η1-γ. The η3-γ process was found to be disfavored by 12.2 kcal/mol. In a model system in which the prenyl substrate is replaced with an allyl, the η1 reductive elimination is preferred over η3 by 2.2 kcal/mol.[24] In the prenyl case, the steric bulk around the forming C-C bond precludes the γ-reductive elimination from achieving the more stable η1 geometry. The α isomer is favored universally for all steps, including the α-4-mem transmetalation. The high level of linear-selectivity observed with the current catalyst system is controlled by the preference for the α-isomer-forming TS-VI-η1-α reductive elimination.
Figure 1.

Reaction coordinate of Pd(0)-catalyzed cross-coupling of prenylZnBr (3) with p-bromotoluene (above) and transmetalation and reductive elimination transition structures (below).[22] Energies are displayed in kcal/mol and distances in Å. Computed structures are rendered in Pymol.[23]
In summary, we have developed the first general, practical and completely linear-selective Negishi coupling of 3,3-disubstituted allyl organozinc reagents with aryl, heteroaryl and vinyl halides. This reaction features exceptionally mild reaction conditions and is broad in scope with respect to both aryl/vinyl halide and allylzinc coupling partners. This linear-selective allylation methodology provides an effective means for accessing highly functionalized aromatic and heteroaromatic compounds bearing prenyl-type side chains, as illustrated by the concise synthesis of anti-HIV natural product siamenol. Finally, theoretical calculations provided insights into the regiochemical-controlling parameters of these coupling processes, demonstrating the critical choice of the catalyst and the transmetalating reagent in achieving good selectivity for coupling reactions involving 3,3-disubstituted allyl nucleophiles.
Supplementary Material
Scheme 5.
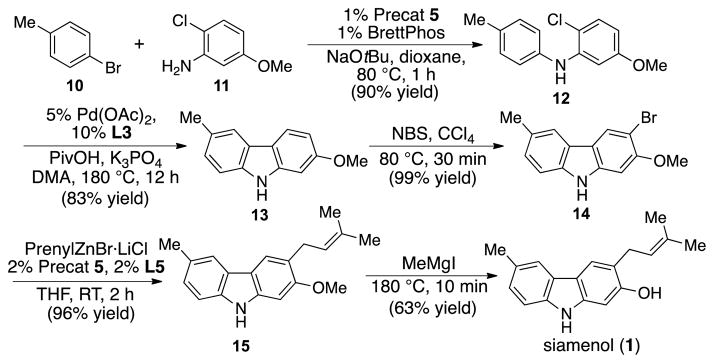
Concise synthesis of siamenol.
Footnotes
We thank the National Institutes of Health (GM46059 for S.L.B.), Oregon State University (P.H.-Y.C.), Vicki and Patrick F. Stone Scholar Funds (P.H.-Y.C.) and David P. Shoemaker Memorial Fellowship (T.J.L.M.) for financial support. MIT has patents on some of the ligands and precatalysts used in this study from which S.L.B. as well as current or former coworkers receive royalty payments.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Yang Yang, Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 (USA).
Thomas J. L. Mustard, Department of Chemistry, Oregon State University, 135 Gilbert Hall, Corvallis, OR 97331 (USA)
Prof. Dr. Paul Ha-Yeon Cheong, Email: paul.cheong@oregonstate.edu, Department of Chemistry, Oregon State University, 135 Gilbert Hall, Corvallis, OR 97331 (USA)
Prof. Dr. Stephen L. Buchwald, Email: sbuchwal@mit.edu, Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 (USA)
References
- 1.For recent reviews, see: Williams RM, Stocking EM, Sanz-Cervera JF. Top Curr Chem. 2000;209:97–173.Li S. Nat Prod Rep. 2010;27:57–78. doi: 10.1039/b909987p.
- 2.(a) Botta B, Delle Monache G, Menendez P, Boffi A. Trends Pharmacol Sci. 2005;26:606–608. doi: 10.1016/j.tips.2005.09.012. [DOI] [PubMed] [Google Scholar]; (b) Botta B, Vitali A, Menendez P, Misiti D, Delle Monache G. Curr Med Chem. 2005;12:717–739. doi: 10.2174/0929867053202241. [DOI] [PubMed] [Google Scholar]
- 3.(a) Tello M, Kuzuyama T, Heide L, Noel JP, Richard SB. Cell Mol Life Sci. 2008;65:1459–1463. doi: 10.1007/s00018-008-7579-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Balibar CJ, Howard-Jones AR, Walsh CT. Nat Chem Biol. 2007;3:584– 592. doi: 10.1038/nchembio.2007.20. [DOI] [PubMed] [Google Scholar]
- 4.Farmer JL, Hunter HN, Organ MG. J Am Chem Soc. 2012;134:17470–17473. doi: 10.1021/ja308613b. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y, Buchwald SL. J Am Chem Soc. 2013;135:10642–10645. doi: 10.1021/ja405950c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For recent reviews on prenylation methods, see: Lindel T, Marsch N, Adla SK. Top Curr Chem. 2012;309:67–130. doi: 10.1007/128_2011_204.. For additional recent examples, see: Kuttruff CA, Zipse H, Trauner D. Angew Chem Int Ed. 2011;50:1402–1405. doi: 10.1002/anie.201006154.
- 7.For palladium-catalyzed sp2-sp2 Negishi cross-coupling reactions, see: Milne JE, Buchwald SL. J Am Chem Soc. 2004;126:13028–13032. doi: 10.1021/ja0474493.Yang Y, Oldenhuis NJ, Buchwald SL. Angew Chem Int Ed. 2013;52:615–619. doi: 10.1002/anie.201207750.
- 8.For palladium-catalyzed sp2-sp3 Negishi cross-coupling reactions, see: Han C, Buchwald SL. J Am Chem Soc. 2009;131:7532–7533. doi: 10.1021/ja902046m.
- 9.For an example where prenylzinc reagents were used in a non-regioselective sense, see: Mitamura Y, Asada Y, Murakami K, Someya H, Yorimitsu H, Oshima K. Chem Asian J. 2010;5:1487–1493. doi: 10.1002/asia.201000068.
- 10.For cross-coupling reactions involving allylzinc halides, see: Nakamura E. Tetrahedron Lett. 1988;29:5155–5156.Baba Y, Toshimitsu A, Matsubara S. Synlett. 2008;13:2061–2063.Xu Z, Negishi E-i. Org Lett. 2008;10:4311–4314. doi: 10.1021/ol8017566.Wang C, Tobrman T, Xu Z, Negishi E-i. Org Lett. 2009;11:4092–4095. doi: 10.1021/ol901566e.
- 11.For palladium-catalyzed coupling reactions involving other allylmetals, see: Si: Hatanaka Y, Ebina Y, Hiyama T. J Am Chem Soc. 1991;113:7076–7077.Hatanaka Y, Goda K, Hiyama T. Tetrahedron Lett. 1994;35:1279–1282.Hatanaka Y, Goda K, Hiyama T. Tetrahedron Lett. 1994;35:6511–6514.Denmark SE, Werner NS. J Am Chem Soc. 2010;132:3612–3620. doi: 10.1021/ja910804u.. Sn: Trost BM, Keinan E. Tetrahedron Lett. 1980;21:2595–2598.Echavarren AM, Stille JK. J Am Chem Soc. 1987;109:5478–5486.Farina V, Krishnan B. J Am Chem Soc. 1991;113:9585–9595.Obora Y, Tsuji Y, Kobayashi M, Kawamura T. J Org Chem. 1995;60:4647–4649.
- 12.(a) Krasovskiy A, Malakhov V, Gavryushin A, Knochel P. Angew Chem Int Ed. 2006;45:6040–6044. doi: 10.1002/anie.200601450. [DOI] [PubMed] [Google Scholar]; (b) Ren H, Dunet G, Mayer P, Knochel P. J Am Chem Soc. 2007;129:5376–5377. doi: 10.1021/ja071380s. [DOI] [PubMed] [Google Scholar]
- 13.We found that the addition of LiCl accelerated the rate of these Negishi cross-coupling reactions. For the role of added LiCl, see: McCann LC, Hunter HN, Clyburne JAC, Organ MG. Angew Chem Int Ed. 2012;51:7024–7027. doi: 10.1002/anie.201203547. and references therein.
- 14.Previous studies showed palladacycle precatalysts (5) allowed the most rapid and quantitative generation of the catalytically active L1Pd(0) species, see: Bruno NC, Tudge MT, Buchwald SL. Chem Sci. 2013;4:916–920. doi: 10.1039/C2SC20903A. and 7b.
- 15.CPhos (CAS# 1160556-64-8) is now commercially available from Aldrich (catalog# 759171).
-
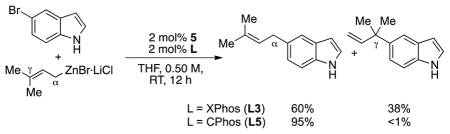
16.For some substrates, the catalyst generated from XPhos (L3) afforded isomeric mixtures of α- and γ-coupling products and thus did not represent the most general system for this reaction. For example, XPhos-based catalyst provided a mixture of linear and branched coupling products for 5-bromoindole, whereas the CPhos (L5)-based catalyst remained highly linear-selective:
- 17.(a) Joule JA, Mills K. Heterocyclic Chemistry. 5. Wiley; Chichester: 2010. [Google Scholar]; (b) Leurs R, Bakker RA, Timmerman H, de Esch IJP. Nat Rev Drug Discovery. 2005;4:107–120. doi: 10.1038/nrd1631. [DOI] [PubMed] [Google Scholar]
- 18.For example, using our previously developed Suzuki-Miyaura coupling protocol, 8a was obtained as a mixture of α - and γ-isomers (α/γ = 93:7) in 67% yield as determined by 1H NMR spectroscopy.
- 19.Macklin TK, Micalizio GC. Nat Chem. 2010;2:638–643. doi: 10.1038/nchem.665. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meragelman KM, McKee TC, Boyd MR. J Nat Prod. 2000;63:427. doi: 10.1021/np990570g. [DOI] [PubMed] [Google Scholar]
- 21.(a) The B3LYP hybrid functional with the 6-31G* (for H, C, N, and P) and LANL2DZ+ECP (for Zn, Br and Pd) basis sets were used Frisch MJ, et al. Gaussian, Inc; Pittsburgh, PA: 2004.
- 22.The PyMOL Molecular Graphics System, version 1.3. Schrödinger, LLC; [Google Scholar]
- 23.(a) Figures and energies for this transformation in the Supporting InformationHoffmann RW. Angew Chem Int Ed. 1982;21:555–566.
- 24.Figures and energies for both the allyl η1 and η3 reductive elimination are presented in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.