

Abstract
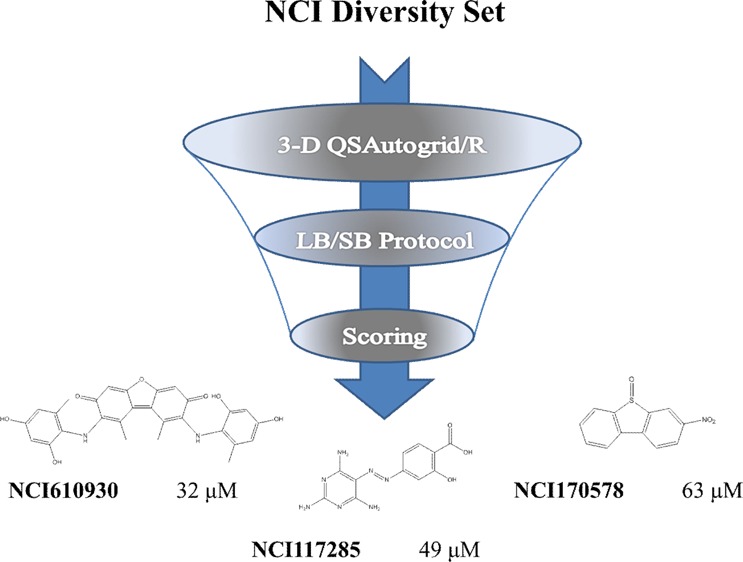
Hsp90 continues to be an important target for pharmaceutical discovery. In this project, virtual screening (VS) for novel Hsp90 inhibitors was performed using a combination of Autodock and Surflex-Sim (LB) scoring functions with the predictive ability of 3-D QSAR models, previously generated with the 3-D QSAutogrid/R procedure. Extensive validation of both structure-based (SB) and ligand-based (LB), through realignments and cross-alignments, allowed the definition of LB and SB alignment rules. The mixed LB/SB protocol was applied to virtually screen potential Hsp90 inhibitors from the NCI Diversity Set composed of 1785 compounds. A selected ensemble of 80 compounds were biologically tested. Among these molecules, preliminary data yielded four derivatives exhibiting IC50 values ranging between 18 and 63 μM as hits for a subsequent medicinal chemistry optimization procedure.
Introduction
Computer-aided virtual screening (VS) represents a powerful in silico technique to discover new bioactive compounds, providing solutions to many high-throughput screening (HTS) problems, such as time and cost, by suggesting what type of compounds should be used for HTS procedures, even when no initial experimental data are available.1 According to the data used, different strategies have been employed in VS: when the structures of experimental three-dimensional (3-D) targets are unknown, quantitative structure–activity relationship (QSAR) and other ligand-based (LB) methods, such 3-D QSAR and pharmacophore-based approaches,2 are used to identify potential hits from chemical libraries; in contrast, in cases where such 3-D information is available, structure-based (SB) protocols that use molecular docking approaches are mainly applied.3 Since the 3-D structures of new target proteins are continuously becoming available, VS is increasingly characterized by molecular docking applications. Acknowledged as one of the fundamental procedures in SB drug discovery, molecular docking, unfortunately, has significant limitation: in fact, no scoring function has been developed yet that can reliably and consistently predict a ligand-protein binding mode and the binding affinity simultaneously. Therefore, a consensus score strategy, based on the synergic use of the two main computer-aided drug design (CADD) methodologies (SB and LB methods), could improve the VS capability in recognizing new bioactive compounds.4
In the present work, such a combination was applied to identify new Hsp90 inhibitors.
Methodology Overview
As shown in Figure 1A, 3-D QSAR models were built and externally validated for Hsp90 inhibitors as reported,5 and they were then employed as a predictive tool in the VS protocol. The procedure was used to rank a set of 1785 compounds (NCI Diversity Set) and prioritize them for biological assay. Since the structures, having unknown 3-D binding conformations, required alignment before testing against the 3-D QSAR models, two different alignment procedures were applied: an LB methodology, using Surflex-sim,6 and an SB methodology, using AutoDock4,7 successfully reported as the molecular docking program for Hsp90.8,9 Both the LB and the SB alignment protocols herein have been tested and validated using a set of 15 compounds (the training set used to build the 3-D QSAR models;5 see Table S1 in the Supporting Information), retrieved from the Protein Data Bank (PDB),10 with known binding modes using either realignment (RA) or cross-alignment (CA) validations (Figure 1B; see the Alignment Rules section). Both alignment methodologies (LB and SB) were applied on the external database to obtain two separate sets of predicted binding conformations used as external prediction sets to feed the 3-D QSAR models5 and yield two sets of predicted pIC50 values. The NCI Diversity Set was virtually screened employing this LB-SB-VS strategy and 80 molecules were selected for enzyme-based biological assays considering both the 3-D QSAR models’ predicted pIC50 values and the predicted free binding energy from the AutoDock4 docking7 (see the Virtual Screening section). Among the tested molecules, four resulted in inhibiting the Hsp90 activity at micromolar levels.
Figure 1.

Overview of (A) the applied procedure and (B) alignment assessment protocol.
Alignment Rules
In those cases where it is possible to perform structure-based (SB) studies on large libraries of compounds, to increase the flexibility of the search method, it may be advantageous to carry out, in parallel, a ligand-based (LB) alignment procedure. In fact, during an LB alignment, the neglecting of proteins’ structural information allows one to extend the alignment’s degrees of freedom (increased search space range), voiding all the possible ligand-protein constraints which can limit, during docking simulations, the ability to find the appropriate poses for certain compounds. Therefore, in the present study, LB and SB alignment methodologies were either assessed (Figure 1B) on the 3-D QSAR’s training set compounds5 and then applied to determine the pose of molecules with unknown binding modes as those comprised in the NCI Diversity Set. The pipeline of the alignment processes was described in detail in a previous work.4 In particular, the LB approach was carried out using the principle of morphological similarity implemented by the Surflex-sim6 program, whereas the SB approach was performed by means of Autodock4.7 The 3-D coordinates of training set compounds,5 used to validate the LB and SB procedure, were taken first from their respective minimized complex (experimental conformation, EC) and second from randomly built conformations (herein random conformation, RC), using the ChemAxon Marvin software (http://www.chemaxon.com), subsequently aligned to the experimental poses (see the LB Alignment Assessment and SB Alignment Assessment sections).
LB Alignment Assessment
The LB-based alignment procedure assessment was carried out as follows: first, each modeled structure (RC) was aligned to its experimental pose (random conformation realignment, RCRA); second, either the experimental (EC) or modeled structure (RC) was aligned to a target list containing all ligands in their binding conformation except itself (experimental conformation cross-alignment (ECCA) and random conformation cross-alignment (RCCA)). The alignment fitness was quantified by evaluating either the root mean square deviaton (RMSD) and the subsequent alignment accuracy (AA, Table 1) values. Similar to the definition of docking accuracy (DA, see below), AA can be used, in an LB alignment, to explain the algorithm’s ability to recognize the ligand’s pose, with respect to those experimentally observed, and can be calculated by the following equation derived from that of the DA:11
| 1 |
where, in the case of docking accuracy, χA = DA, and, in the case of alignment accuracy, χA = AA. χA can range between 0 (no alignment) and 1 (maximum performance of alignment). For comparison purposes, a and b coefficients were chosen as for the docking accuracy,11 following the guidelines indicated by Vieth et al.:12 2 Å and 3 Å were selected as a threshold for correct and partially correct aligned ligands, respectively; structures with RMSD values over 3 Å were not included as they were considered misaligned. Therefore, frmsd ≤ a and frmsd ≤ b represent the fraction of aligned ligands showing an RMSD value less than or equal to 2 and 3 Å, respectively.
Table 1. RMSD and AA Values Calculated between the Random (RC) and Experimental Conformations (EC) in the Realignment and Cross-Alignment Processes (RCRA and RCCA, Respectively).
| RCRA | ECCA | RCCA | ||
|---|---|---|---|---|
| entrya | PDB code | RC vs EC | EC vs all | RC vs all |
| 1 | 1UY8 | 0.46 | 0.37 | 0.52 |
| 2 | 1UYC | 0.47 | 0.45 | 0.49 |
| 3 | 1UYD | 0.7 | 0.36 | 0.43 |
| 4 | 1UYE | 0.69 | 0.55 | 0.6 |
| 5 | 1UYG | 0.41 | 0.74 | 0.65 |
| 6 | 1UYH | 0.46 | 0.38 | 0.49 |
| 7 | 1UYK | 0.62 | 0.26 | 0.56 |
| 13 | 2BT0 | 0.58 | 0.33 | 0.48 |
| 14 | 2CCS | 0.46 | 0.64 | 0.74 |
| 16 | 2CCU | 0.8 | 3.5 | 3.8 |
| 17 | 3B25 | 0.48 | 0.39 | 0.21 |
| 18 | 3B26 | 0.5 | 0.6 | 0.35 |
| 20 | 3B28 | 0.71 | 0.55 | 0.45 |
| 21 | 3OWB | 0.65 | 0.74 | 0.7 |
| 22 | 3OWD | 0.98 | 0.3 | 0.81 |
| AAb | 100 | 93.33 | 93.33 |
Considering RCRA, all of the RMSD values in the training set compounds were below 1 Å, resulting in an AA value equal to 100%, while ECCA and RCCA led to AAs greater than 90% (see Table 1).
The high AA values of the RCCA proved Surflex-Sim’s ability to align randomly built conformations accurately, suggesting that it would exhibit a similar accuracy, even with molecules with unknown binding conformation and, thus, can be considered as a useful tool for LB alignment of Hsp90 inhibitors in a VS protocol.
SB Alignment Assessment
Autodock47 was used for all docking calculations. The AutoDockTools (ADT) package7 was employed to generate the docking input files and analyze the docking results. The grid box was centered on the average mass center of the ligands. A grid box size of 53 × 47 × 51 points, with a spacing of 0.375 Å, was set to accommodate all considered Hsp90 experimental ligands and ATP binding site residues. Autogrid4, as implemented in the Autodock software package,7 was used to generate grid maps. The Lamarckian genetic algorithm (LGA) was employed to generate orientations or conformations of the ligands within the binding site. The global optimization started with a population of 150 randomly positioned individuals, a maximum of 2.5 × 106 energy evaluations and a maximum of 27000 generations. A total of 100 runs were performed, and the cluster analysis was carried out using an RMSD tolerance of 2 Å. The ligands extracted from minimized complexes5 were docked in the known binding site both in the corresponding crystal protein (redocking) and in all crystals (cross-docking) following the procedure previously reported.4
With the purpose to check the reliability of the docking protocol, docking validation was performed using the 15 compounds composing the 3-D QSAR models’ training set,5 as the above-described LB assessment. In particular, experimental and random conformations, EC and RC respectively (see the Alignment Rules section) were docked into the corresponding protein structure (redocking (RD), ECRD, and RCRD). ECRD and RCRD predicted binding energy and RMSD values were calculated, analyzing both best docked (BD, the conformer characterized by the lowest estimated free binding energy) and best cluster (BC, the lowest energy conformer of the most populated cluster) conformations (see Table 2). Interestingly, RMSD values obtained from ECRD and RCRD were generally greater than 2,13 leading to low DA values (Table 2). These results reflect the intrinsic lack of accuracy, of the implemented scoring function, to select the right binding pose in a system (RD) in which the protein is maintained rigid.
Table 2. RMSD and DA Values of Best Docked (BD) and Best Cluster (BC) Conformations of Experimental (EC) and Random Conformations (RC) Calculated after Superimposition on Experimental Conformation (EC) in Redocking (ECRD, RCRD) and Cross-Docking (ECCD, RCCD) Procedures.
| ECRD |
RCRD |
ECCD |
RCCD |
||||||
|---|---|---|---|---|---|---|---|---|---|
| entrya | PDB code | BD | BC | BD | BC | BD | BC | BD | BC |
| 1 | 1UY8 | 5.63 | 4.55 | 4.79 | 4.13 | 2.11 | 2.14 | 3.09 | 2.01 |
| 2 | 1UYC | 5.18 | 4.21 | 4.66 | 4.80 | 1.67 | 1.67 | 2.01 | 2.01 |
| 3 | 1UYD | 2.67 | 4.42 | 2.59 | 2.59 | 1.77 | 1.77 | 1.28 | 1.28 |
| 4 | 1UYE | 3.34 | 4.41 | 4.90 | 4.90 | 1.31 | 1.31 | 1.76 | 1.76 |
| 5 | 1UYG | 7 | 5.39 | 4.86 | 4.86 | 1.5 | 1.5 | 1.8 | 1.8 |
| 6 | 1UYH | 5.45 | 4.44 | 5.69 | 3.69 | 1.56 | 1.56 | 2.75 | 2.07 |
| 7 | 1UYK | 6.17 | 5.4 | 5.83 | 5.83 | 1.7 | 1.7 | 2.05 | 2.05 |
| 13 | 2BT0 | 1.12 | 1.12 | 0.88 | 0.88 | 1.4 | 1.4 | 1.75 | 1.75 |
| 14 | 2CCS | 0.82 | 0.82 | 2.7 | 0.49 | 1.40 | 1.40 | 1.37 | 1.37 |
| 16 | 2CCU | 2.21 | 2.21 | 2.14 | 2.14 | 3.91 | 3.91 | 5.69 | 3.94 |
| 17 | 3B25 | 2.7 | 2.7 | 3.36 | 3.8 | 1.7 | 1.7 | 1.3 | 1.3 |
| 18 | 3B26 | 5.17 | 5.17 | 4.18 | 4.90 | 2.53 | 2.53 | 2.71 | 2.71 |
| 20 | 3B28 | 4 | 4 | 3.6 | 3.24 | 2.1 | 2.1 | 1.8 | 1.8 |
| 21 | 3OWB | 4.73 | 4.44 | 5.57 | 4.46 | 2.76 | 2.14 | 2.87 | 2.27 |
| 22 | 3OWD | 3.32 | 3.32 | 4.90 | 4.92 | 1.75 | 1.75 | 2.69 | 2.77 |
| docking accuracy, DAb | 23 | 20 | 17 | 20 | 80 | 80 | 67 | 70 | |
To implicitly account for conformational protein flexibility, cross docking (CD) was applied, with the purpose to improve DA, on each EC (experimental conformation cross-docking, ECCD) and RC (random conformation cross-docking, RCCD) considering all the available receptors excluding the native one (see Table 2). All the obtained poses were merged in a single file and clustered; finally, the BD and BC conformations were considered.
The CD procedure was first applied on the compounds of the training set5 to verify the accuracy and the reproducibility of the method. DA values were greater than those obtained from redocking procedures, reaching DA values ranging from 67% to 80% (see Table 2). Because of the greater DA values in both RCRD and RCCD, with the latter being much more accurate, the RCCD using the BC conformation was adopted as a SB tool in the VS screening herein. Note that a DA reduction is obtained when RC is used in place of EC. This could be the reflection that the reproducibility of any docking methods depends on the starting conformation; however, in a VS application, the EC is unknown and this is, of course, a source of error that is likely related to the lack of the complementary protein conformation.
Ordered water molecules play an important role in protein–ligand recognition, either being displaced on ligand binding or bridging groups to stabilize the complex. In the common practice and also in the herein application, water molecules are ignored during the docking simulations to reduce the time-consuming calculations. The inclusion of experimental water molecules in the Hsp90 inhibitors binding site could have a significant impact only in the case of a redocking procedure. In fact, simulating a real application, the redocking of 1UY6 (not included in the training set) with experimental complexed water molecules, within 5 Ǻ around the ligand, improved the DA by decreasing the RMSD value from 2.7 (without waters) to 0.75. On the other hand, cross-docking the 1UY6 ligand into the 15 training set Hsp90 binding sites led to a RMSD value of 1.7 thus demonstrating the improvement of accuracy through the application of cross-docking protocol. A cross-docking protocol is not applicable if the experimental water molecules are retained, preventing the correct docking of different scaffold compounds.
Virtual Screening (VS)
Based on AA and DA values, RCCA and RCCD (considering BC conformations) procedures were applied on the 1785 molecules containing the NCI Diversity Set to obtain two conformations for each compound. Therefore, two external prediction sets (herein called Autodock/3-D QSAR and Surflex/3-D QSAR, respectively; see Table S2 in the Supporting Information) were obtained and predicted by the selected 3-D QSAR models: A, N, OA, and MP.5 For both Autodock/and Surflex/3-D QSAR prediction sets, a score was derived by listing the average predicted pIC50 values obtained from the four selected 3-D QSAR models. These scores were then used, together with the corresponding predicted free binding energy from the AutoDock4 docking,7 to compose a hit parade. Considering these three factors, according to rank-by-rank strategy,14 the top 80 compounds (see Table S2 in the Supporting Information) were selected and tested. Biological activities of selected compounds were determined by applying a previously described procedure.15
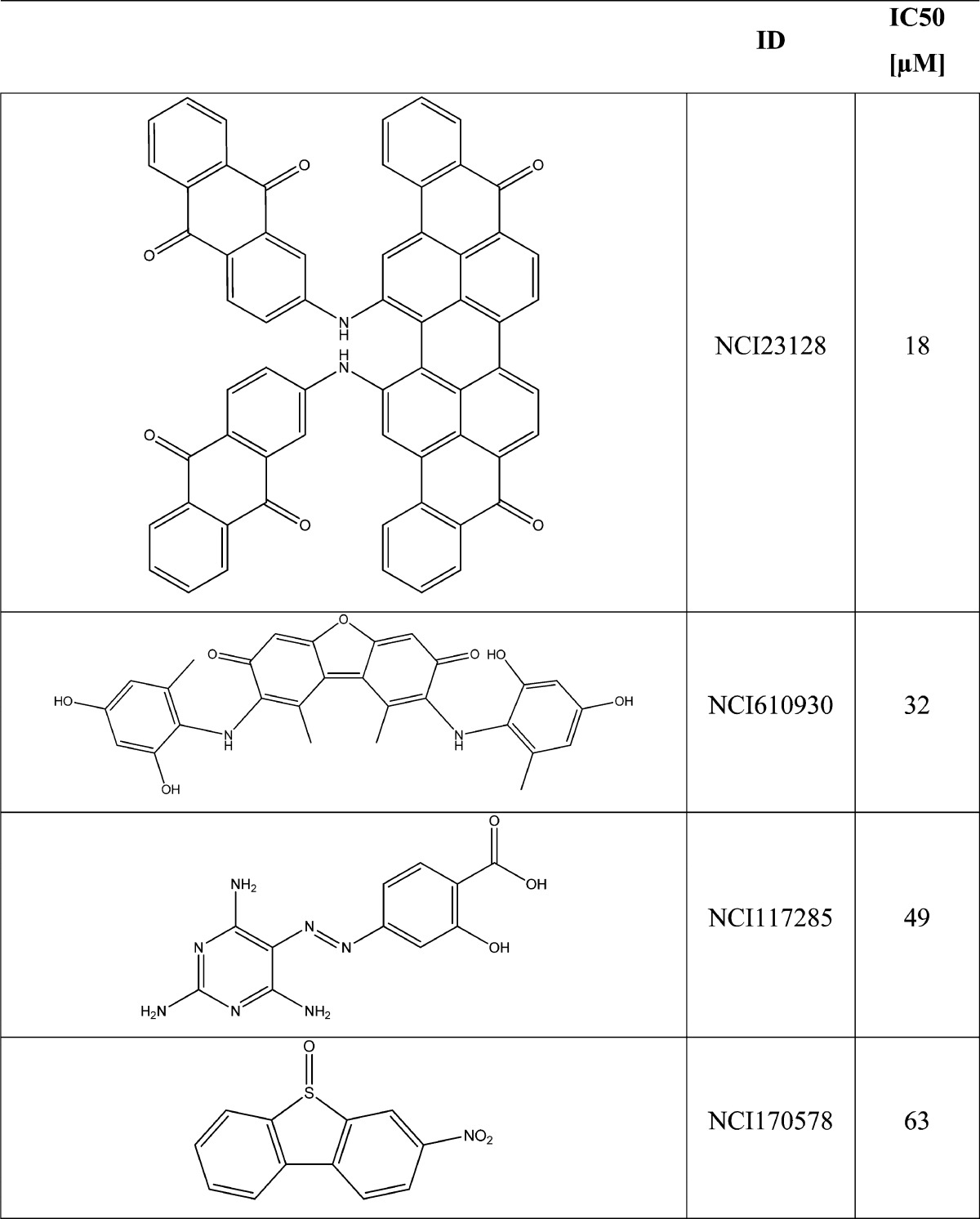
The preliminary data yielded nine compounds with detectable inhibitory activity (see Table S3 in the Supporting Information): four of these compounds (NCI23128, NCI23128, NCI117285, and NCI170578) showed IC50 values between 18 μM and 63 μM (see Table 3). The activity values are not comparable to those of the training set; they could reflect the effects of a limited covered chemical space that characterize the NCI Diversity Set. As a matter of fact, all of the results of the applied strategy were quite consistent to disclose new Hsp90 inhibitors.
Table 3. Molecular Structure and Biological Activity of the Most Active Compounds Selected by the VS Protocol.
Binding Mode Analysis of New Hsp90 Inhibitors
The binding mode analysis of the four most active compounds (see Table 3 and Figure 2) was conducted to define the crucial interactions within the Hsp90 binding site and point out potential molecular transformations to increase inhibitory activity. Among the most active screened compounds, attention was focused on NCI610930, NCI170578, and NCI117285, which exhibit possible interesting new scaffolds for Hsp90 inhibition. By using the newly introduced activity contribution prediction feature5 on the prediction set molecules, it is possible to represent how the quantitative models predict the effects of prediction set molecules three dimensionally. It is interesting to note that, starting from the most active compound to the least active, a positive predicted activity contribution area (green surface, Figure 2), in the proximity of LYS58, ILE96, and GLY97, decreases in magnitude jointly with the biological response, while the results considering NCI170578 and NCI117285 (the least active compounds) near LEU48, VAL186, THR 184, ASP93, SER 52, and LEU48 appear to be unchanged, confirming the importance of these two residues’ series, as previously reported.5 Moreover, NCI610930 and NCI170578 are, respectively, dibenzofurandione and dibenzothiophene derivatives that could be ascribed to the tricyclic series of Hsp90 inhibitors recently identified and also characterized by X-ray.16 From these, a ternary complex (PDB code 2YK2) that exhibits two binding subpockets into the Hsp90 N-domain was reported.16 As shown in Figure 3A, the docked pose of NCI610930 occupies both the binding areas of Hit 116 and Hit 2,16 suggesting that this characteristic, together with the interactions with LYS58, ILE96, and GLY97 (Figure 2), could determine its greater inhibitory activity than NCI170578 and NCI117285 (see Figures 3B and 3C). This argument is also supported by considering the higher inhibitory potency of Hit 8 (PDB code 2YKI),16 which is a compound specifically designed, as reported,16 to occupy both areas. By overlapping the binding poses of NCI610930 and Hit 816 (see Figure 4), it is possible to recognize that the latter extends its two 3H-imidazo[4,5-c]pyridinil groups in two areas, the first characterized by PHE170, TRP162, LEU107, and GLN23, and the second characterized by ASP93, SER52, ASN51, THR184, LEU48, and VAL186, respectively uncovered and partially overlapped by NCI610930. On the other hand, NCI610930 can establish, with the 2,4-dihydroxy-6-methylanilyl moiety, stronger interactions with LYS58, ASP54, ILE96, GLY97, and ALA55. Although they have these differences, each compound can be seen as the “volumetric extension” of the other, suggesting that the integration of the characteristics of the two molecules could improve the overall inhibitory activity.
Figure 2.

Predicted activity contribution plots (solid: 75%, positive: green, negative: yellow), overlapped with PLS-coefficients plots (mesh: 65%, positive: red, negative: blue) obtained from the used 3-D QSAR models at the selected PC,5 for the most active screened compounds in their BC system (protein and pose): NCI23128 in 1UY6, NCI610930 in 1UYC, NCI117285 and NCI170578 in 1UY8.
Figure 3.

The most active screened compounds in their BC system (protein and pose) overlapped with 2YK2(16) (Hit 1 in orange, Hit 2 in cyan, protein in cyan): (A) NCI610930 in 1UYC (ligand and protein in green), (B) NCI117285 in 1UY8 (ligand in yellow, protein in magenta), and (C) NCI170578 in 1UY8 (ligand and protein in magenta).
Figure 4.
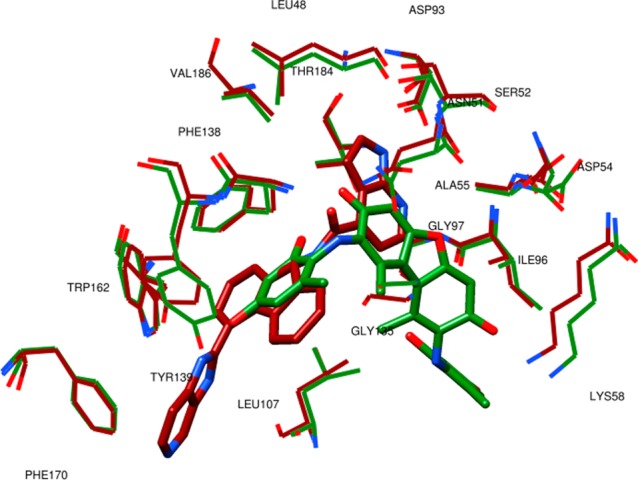
Depiction of the most active screened compound NCI610930 in its BC system (ligand and protein in green) overlapped with 2YKI(16) (brown colored).
Conclusion
The structure-based 3-D QSAR model for Hsp90 inhibitors was successfully coupled with both ligand-based (LB) and structure-based (SB) validated alignment methods. The protocol was extensively assessed for both alignment and predictive ability. A virtual screening (VS) application using the NCI Diversity Set led to the discovery of new Hsp90 inhibitors compounds with activity in the micromolar range. Two of the active compounds, NCI610930 and NCI170578, are characterized by a tricyclic ring, similar to an already-recognized interesting scaffold for Hsp90 inhibitors. Further molecular modeling studies, such as a focused VS approach on NCI610930, are on due course for lead optimization to discover new and more-potent Hsp90 inhibitors.
Acknowledgments
F.B. acknowledges Sapienza Università di Roma (“Progetti per Avvio alla Ricerca” Grant No. C26N12JZCT). R.B.W. acknowledges Washington University in St. Louis (uSTAR Fellowship). R.R. acknowledges Italian Minister of University and Research (MIUR) (PRIN Grant No. 20105YY2HL_003 and FIRB Grant No. RBFR10ZJQT). The authors thank the Developmental Therapeutics Program (DTP, http://dtp.cancer.gov) at the NCI/NIH for providing the 80-compound set biologically tested in this study.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ A.C. and F.B. contributed equally to this work.
The authors declare no competing financial interest.
Dedication
Dedicated to Prof. Marino Artico for his 80th birthday.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ragno R.; Gioia U.; Laneve P.; Bozzoni I.; Mai A.; Caffarelli E. Identification of small-molecule inhibitors of the XendoU endoribonucleases family. ChemMedChem 2011, 6, 1797–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggner C.; Wolber G.; Kirchmair J.; Schuster D.; Langer T.. Pharmacophore-based Virtual Screening in Drug Discovery. In Chemoinformatics Approaches to Virtual Screening; The Royal Society of Chemistry: London, 2008; Chapter 3, pp 76–119. [Google Scholar]
- Kitchen D. B.; Decornez H.; Furr J. R.; Bajorath J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discovery 2004, 3, 935–949. [DOI] [PubMed] [Google Scholar]
- Musmuca I.; Caroli A.; Mai A.; Kaushik-Basu N.; Arora P.; Ragno R. Combining 3-D quantitative structure-activity relationship with ligand based and structure based alignment procedures for in silico screening of new hepatitis C virus NS5B polymerase inhibitors. J. Chem. Inf. Model. 2010, 50, 662–676. [DOI] [PubMed] [Google Scholar]
- Ballante F.; Caroli A.; Wickersham R. B. III; Ragno R.. Hsp90 Inhibitors, Part 1: Definition of 3-D QSAutogrid/R Models as a Tool for Virtual Screening. J. Chem. Inf. Model. 2014, DOI: 10.1021/ci400759t. [DOI] [PubMed] [Google Scholar]
- Jain A. N. Ligand-based structural hypotheses for virtual screening. J. Med. Chem. 2004, 47, 947–961. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta S.; Pilla S.; Paul S. Design of novel Geldanamycin analogue Hsp90 alpha-inhibitor in silico for breast cancer therapy. Med. Hypotheses 2013, 81, 463–469. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee B.; Vijayasarathy S.; Karunakar P.; Chatterjee J. Comparative reverse screening approach to identify potential anti-neoplastic targets of saffron functional components and binding mode. Asian Pac J Cancer Prev. 2012, 13, 5605–5611. [DOI] [PubMed] [Google Scholar]
- Bernstein F. C.; Koetzle T. F.; Williams G. J.; Meyer E. F. Jr.; Brice M. D.; Rodgers J. R.; Kennard O.; Shimanouchi T.; Tasumi M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [DOI] [PubMed] [Google Scholar]
- Ragno R.; Frasca S.; Manetti F.; Brizzi A.; Massa S. HIV-reverse transcriptase inhibition: inclusion of ligand-induced fit by cross-docking studies. J. Med. Chem. 2005, 48, 200–212. [DOI] [PubMed] [Google Scholar]
- Vieth M.; Hirst J. D.; Kolinski A.; Brooks C. L. Assessing energy functions for flexible docking. J. Comput. Chem. 1998, 19, 1612–1622. [Google Scholar]
- Morris G. M.; Lim-Wilby M.. Molecular Docking. In Molecular Modeling of Proteins, Vol. 443; Kukol A., Ed.; Humana Press: Totowa, NJ, 2008; pp 365–382. [DOI] [PubMed] [Google Scholar]
- Markovic D.; Darvas O. Inborn errors of pentose and hexose metabolism and current methods of their discovery. Med. Arh. 1975, 29, 31–33. [PubMed] [Google Scholar]
- Barker J. J.; Barker O.; Courtney S. M.; Gardiner M.; Hesterkamp T.; Ichihara O.; Mather O.; Montalbetti C. A.; Muller A.; Varasi M.; Whittaker M.; Yarnold C. J. Discovery of a novel Hsp90 inhibitor by fragment linking. ChemMedChem 2010, 5, 1697–1700. [DOI] [PubMed] [Google Scholar]
- Vallee F.; Carrez C.; Pilorge F.; Dupuy A.; Parent A.; Bertin L.; Thompson F.; Ferrari P.; Fassy F.; Lamberton A.; Thomas A.; Arrebola R.; Guerif S.; Rohaut A.; Certal V.; Ruxer J. M.; Gouyon T.; Delorme C.; Jouanen A.; Dumas J.; Grepin C.; Combeau C.; Goulaouic H.; Dereu N.; Mikol V.; Mailliet P.; Minoux H. Tricyclic series of heat shock protein 90 (Hsp90) inhibitors, Part I: Discovery of tricyclic imidazo[4,5-c]pyridines as potent inhibitors of the Hsp90 molecular chaperone. J. Med. Chem. 2011, 54, 7206–7219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.