

Abstract
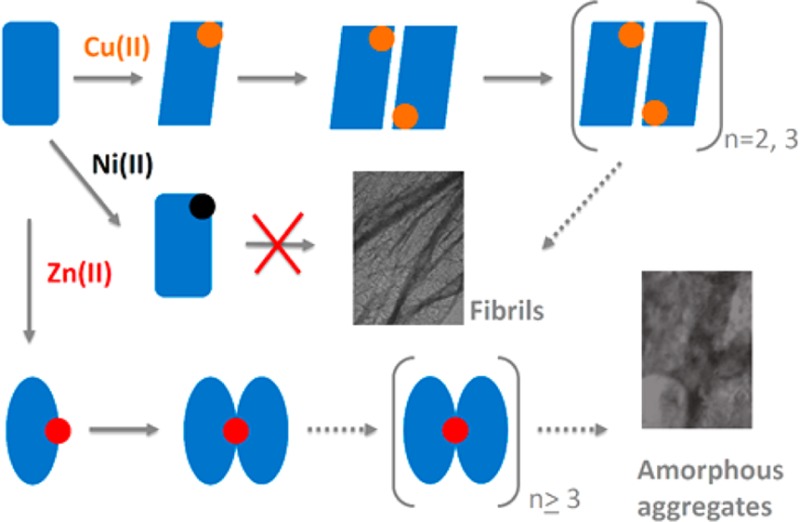
β-2-Microglobulin (β2m) forms amyloid fibrils in the joints of patients undergoing hemodialysis treatment as a result of kidney failure. In the presence of stoichiometric amounts of Cu(II), β2m self-associates into discrete oligomeric species, including dimers, tetramers, and hexamers, before ultimately forming amyloid fibrils that contain no copper. To improve our understanding of whether Cu(II) is unique in its ability to induce β2m amyloid formation and to delineate the coordinative interactions that allow Cu(II) to exert its effect, we have examined the binding of Ni(II) and Zn(II) to β2m and the resulting influence that these metals have on β2m aggregation. We find that, in contrast to Cu(II), Ni(II) does not induce the oligomerization or aggregation of β2m, while Zn(II) promotes oligomerization but not amyloid fibril formation. Using X-ray absorption spectroscopy and new mass spectrometry-related techniques, we find that different binding modes are responsible for the different effects of Ni(II) and Zn(II). By comparing the binding modes of Cu(II) with Ni(II), we find that Cu(II) binding to Asp59 and the backbone amide between the first two residues of β2m are important for allowing the formation of amyloid-competent oligomers, as Ni(II) appears not to bind these sites on the protein. The oligomers formed in the presence of Zn(II) are permitted by this metal’s ability to bridge two β2m units via His51. These oligomers, however, are not able to progress to form amyloid fibrils because Zn(II) does not induce the required structural changes near the N-terminus and His31.
β-2-Microglobulin (β2m) is a monomeric protein with 99 residues and is essential for the correct folding, assembly, and cell surface expression of the class I major histocompatibility complex (MHC-1).1 During normal turnover, β2m is released from the MHC-1 complex and transported to the kidney where it is degraded. For patients who are suffering from kidney disease and undergo long-term dialysis treatment, β2m forms amyloid fibrils and deposits in the joints and connective tissues, leading to a condition known as dialysis-related amyloidosis (DRA).2 The circulating concentration of β2m in DRA patients increases up to ∼60 times above the normal level of 0.1 μM, but elevated β2m concentrations alone are not sufficient to trigger fibrillogenesis.3,4 β2m amyloid formation must therefore result from other factors related to hemodialysis, but the exact mechanism in vivo is not known. In vitro, β2m oligomerization and fibril formation can be generated several ways, including by incubation of the protein under acidic conditions,5 by truncation of the first six N-terminal amino acids,6 by mixing the protein with collagen at pH 6.4,7 by sonication of the protein with sodium dodecyl sulfate at pH 7.0,8 and by incubation of the protein under physiological conditions in the presence of stoichiometric amounts of Cu(II).9,10
Initiation of β2m amyloid formation by Cu(II) is particularly interesting because clinical studies have shown that dialysis patients treated with Cu(II)-free membranes have a >50% reduced incidence of DRA compared to patients who are exposed to traditional Cu(II)-containing dialysis membranes.11,12 Additionally, it seems that Cu(II), more than any other, can influence the amyloid formation of a variety of other protein systems.13 For example, Cu(II) binds α-synuclein and enhances the formation of oligomers and amyloid fibrils by this protein.14,15 In the presence of substoichiometric levels of Cu(II), Aβ1–42, which is associated with Alzheimer’s disease, can also assemble into amyloid fibrils;16 however, excess Cu(II) concentrations cause the formation of spherical oligomers and amorphous aggregates that are unable to seed fibril formation of this peptide.17 Cu(II) has even been implicated in the misfolding and fibril formation of the prion protein as Cu(II) binding causes structural changes and induces endocytosis of the protein.18 Cu(II) has also been shown to induce amyloid formation of the immunoglobulin light chain19 as well as the Huntington protein.20 In contrast, other transition metal ions can induce amyloid formation in some cases, but more often they tend to initiate non-amyloid aggregation or perhaps even inhibit amyloid formation. For instance, Zn(II) can promote α-synuclein fibril formation in vitro but promotes amorphous aggregation of Aβ1–40 only on modified surfaces21 while actually inhibiting fibril formation at equivalent concentrations in solution.16
In analogy with many other amyloid-forming proteins, β2m can be induced to form amyloids in the presence of Cu(II), whereas other metals do not; only a few isolated experiments have been conducted to study the effect of different metals on β2m.22,23 In this work, we describe a more detailed set of experiments to delineate how other transition metals influence β2m oligomerization and aggregation. Specifically, we chose Ni(II) and Zn(II) for comparison to Cu(II) primarily because they have binding preferences (e.g., coordination numbers and geometries) different from those of Cu(II), while maintaining a similar ionic radius. In our experiments, we found that Ni(II) does not induce β2m oligomerization or aggregation, while Zn(II) facilitates oligomerization and aggregation but not amyloid fibril formation. Using a variety of tools, we elucidate the different ways that these metals bind β2m, thereby identifying key features of the β2m–Cu(II) interaction that are essential for allowing this protein to form amyloid fibrils.
Materials and Methods
Materials
Human β2m purified from urine was purchased from Lee Biosolutions (St. Louis, MO). Using mass spectrometry-based sequencing experiments, we find that ∼100% of the purchased protein has its disulfide intact. In addition, there are no modifications to the protein, except for a small fraction (∼15–20%) of the protein that shows oxidation at Met99. l-Ascorbate acid, D2O, dithiothreitol (DTT), glacial acetic acid, 3-morpholinopropanesulfonic acid (MOPS), potassium acetate, potassium bromide, urea, and Zn(II) sulfate were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile, ammonium acetate, sodium persulfate, Cu(II) sulfate, and Ni(II) sulfate were purchased from Thermo Fisher Scientific (Waltham, MA). Immobilized trypsin and chymotrypsin (digestion buffer with triethylamine included) were purchased from Princeton Separations (Adelphia, NJ). Amicon molecular weight cutoff (MWCO) filters were purchased from Millipore (Burlington, MA). Deionized water was prepared with a Millipore Simplicity 185 water purification system.
Formation of β2m Oligomers and Amyloid Fibrils
A sample solution containing 100 μM β2m, the metal of interest at the desired concentration, 150 mM potassium acetate, 500 mM urea, and 25 mM MOPS (pH 7.4) was incubated at 37 °C. Control experiments without the metal were also performed in which 1 mM EDTA was added to prevent any association between trace metals and β2m. In these control experiments, no oligomers or fibrils are formed under any of the conditions used; the urea concentration of 500 mM is higher than the concentration found in uremic patients (50 mM)24 undergoing dialysis, but this relatively high concentration was used so that β2m oligomerization and fibril formation could be seen in a reasonable time period.
Thioflavin T Fluorescence
Fluorescence experiments for monitoring β2m amyloid formation were performed using a QuantaMaster 4 SE spectrofluorometer (Photon Technology International, Lawrenceville, NJ). A solution containing 100 μM β2m, 500 mM urea, 150 mM potassium acetate, and 25 mM MOPS (pH 7.4) was initially equilibrated at 37 °C, and after the addition of the metal of interest, the fluorescence of ThT was monitored at an emission wavelength of 483 nm, using an excitation wavelength of 437 nm.
Transmission Electron Microscopy (TEM)
TEM images were obtained on a JEOL JEM-100CX electron microscope operated at 100 keV. Before analysis, a solution containing β2m aggregates was centrifuged (12500 rpm) for 5 min and decanted. The solid material was then suspended in 1 mL of deionized water, applied to carbon-coated grids (Electron Microscopy Sciences, Hatfield, PA), stained with 1% phosphotungstic acid (pH 7.4), air-dried overnight, and then analyzed.
Size Exclusion Chromatography (SEC)
Incubated solutions of β2m were separated using a TSK-gel SuperSW2000 column (Tosoh bioscience, King of Prussia, PA) installed on an HP1100 series high-performance liquid chromatography system (Agilent, Santa Clara, CA). Before the analysis of any given sample, the SEC column was first equilibrated with a 150 mM ammonium acetate mobile phase (pH 6.8) at a flow rate of 0.35 mL/min for 1 h. Twenty microliters of the incubated sample or calibration standard was injected for analysis, and the variable-wavelength detector was set at 214 nm. A solution containing 5 μM bovine serum albumin (MW = 66000), 5 μM ovalbumin (MW = 45000), 5 μM carbonic anhydrase (MW = 29040), and 5 μM β2m (MW = 11731) was used for molecular weight calibration.
Metal-Catalyzed Oxidation (MCO) Reactions
The MCO reactions for the β2m–Cu(II) complex were performed in the presence of 5 μM β2m, 5 μM CuSO4, 0.1 mM ascorbate, 0.1 mM persulfate, 150 mM potassium acetate, and 25 mM MOPS (pH 7.4) at room temperature for 10 min. The MCO reactions for the β2m–Ni(II) complex were performed in the presence of 200 μM β2m, 250 μM NiSO4, 5 mM ascorbate, 0.5 mM persulfate, 150 mM potassium acetate, and 25 mM MOPS (pH 7.4) at room temperature for 20 min. The MCO substitution reactions for the β2m–Cu(II)–Zn(II) complex were performed in the presence of 5 μM β2m, 5 μM CuSO4, 0–200 μM ZnSO4, 0.5 mM ascorbate, 25 μM persulfate, 150 mM potassium acetate, and 25 mM MOPS (pH 7.4) at room temperature for 1 min. The reactions were initiated by the addition of ascorbate and were quenched by the addition of 2% acetic acid. The samples were then immediately desalted using a 10000 MWCO filter and reconstituted in 50 mM triethylamine (pH 8.0) for proteolytic digestion.
Hydrogen–Deuterium Exchange of the C2 Hydrogens of Histidine25
A solution containing 100 μM β2m, 150 mM potassium acetate, and 25 mM MOPS with and without 100 μM ZnSO4 was incubated in D2O (95%) at pH 7.4 and 37 °C. After a given reaction time, an aliquot of the sample was diluted 20-fold into H2O, desalted, reconcentrated, digested by immobilized chymotrypsin, and then analyzed by liquid chromatography and mass spectrometry (LC–MS). The total time in H2O before analysis was kept constant at 3.5 h to ensure back-exchange to hydrogen for the fast exchanging amide groups on the backbone and side chains of the protein. This step was important to ensure that deuteriums remained only at the C2 position of the His residues. The deuterium content of each His-containing peptide, after proteolytic digestion, was determined by calculating the weighted average mass of its isotopic peaks. For dissociation constant (Kd) measurements of the β2m–Zn(II) complex, 75 μM β2m was incubated with 0, 25, 50, 75, 110, and 135 μM ZnSO4 in 150 mM potassium acetate and 25 mM MOPS in D2O at pH 7.4 and 37 °C for 3 days. Aliquots of the samples were analyzed by LC–MS in a manner similar to that described above.
Hydrogen–Deuterium Exchange of the Amide Hydrogens
The β2m global exchange experiments were conducted by taking a 750 μM solution of β2m in buffered H2O and diluting it 20-fold into D2O that was buffered with 25 mM MOPS, 150 mM potassium acetate, and the metal of interest. The final protein solution in D2O contained 6 mM NiSO4, 150 μM ZnSO4, 75 μM CuSO4, or no metal. The exchange time was varied between 10 s and 3 h. After the allotted exchange time, the reaction was quenched by bringing the sample to pH 2.6 and 0 °C. The sample was then immediately injected into an LC–MS instrument, where the deuterium uptake was measured.
Proteolytic Digestion
Immobilized trypsin and immobilized chymotrypsin were used to proteolytically digest β2m after the MCO and histidine-based hydrogen–deuterium exchange reactions. An 80 μL solution of the protein was first incubated with 15 μL of acetonitrile at 45 °C for 30 min, and then 7.5 mM DTT was added and allowed to react with the protein at 37 °C for an additional 30 min. The immobilized enzymes were added to yield a final enzyme:substrate ratio of 1:10. The protein samples were digested in a shaking water bath (VWR, Radnor, PA) at 37 °C for 2 h. After the enzymes had been inactivated via the addition of 2 μL of acetic acid, the samples oxidized by the MCO reactions were frozen at −10 °C and analyzed within 24 h, whereas the hydrogen–deuterium exchange samples were analyzed immediately.
Liquid Chromatography and Mass Spectrometry
A Bruker (Billerica, MA) AmaZon quadrupole ion trap mass spectrometer coupled with an HP1100 series high-performance liquid chromatography system (Agilent) was used for all MS analyses. Typically, the electrospray needle voltage was kept at 4–4.5 kV, and the capillary temperature was set to 200 °C. Tandem mass spectra were recorded using an isolation width of 2.0–4.0 Da and excitation voltages of 0.5–0.8 V. Peptide sequences were determined from tandem MS data via de novo sequencing.
X-ray Absorption Spectroscopy
Samples of β2m were prepared at varying concentrations (1.0–3.2 mM) in a buffer containing 25 mM MOPS buffer and 150 mM KBr (pH 7.4). These relatively high concentrations of protein are common in X-ray absorption experiments to obtain sufficient signal. KBr was used in the buffer to distinguish binding of buffer anions, because Cl– binding cannot be unambiguously distinguished from an S donor ligand from extended X-ray absorption fine structure (EXAFS) analysis. Such an approach has been used extensively in the past.26,27 In separate experiments, the addition of KBr had no significant effect on the amyloid formation of β2m. Indeed, as compared to 150 mM potassium acetate, which is typically used in our amyloid formation reactions, KBr changes the rate of amyloid formation by only <15%, as indicated by ThT fluorescence experiments (data not shown). Separate solutions of metal sulfates were also prepared in the same buffer system to act as a control in the X-ray absorption experiments. The metal solution and β2m were combined with glycerol (final concentration of 10% by volume) and mixed thoroughly. Solutions were run down a desalting column to remove nonspecifically bound metal ions. The solutions were then injected via syringe into polycarbonate holders and frozen in liquid nitrogen. On the basis of the binding affinity of the metal for β2m, the final concentrations of the metal and the protein were combined such that the resulting solutions contained (1) 3.2 mM β2m and 1.0 mM NiSO4 [β2m–Ni(II) complex], (2) 1.0 mM β2m and 1.0 mM CuSO4 [β2m–Cu(II) complex], and (3) 2 mM β2m and 1.2 mM ZnSO4 [β2m–Zn(II) complex]. The concentrations of the metal were determined by inductively coupled plasma atomic emission spectroscopy (ICP-OES) and were found to be stoichiometric or slightly substoichiometric (>85% loadings in each case) with respect to the protein in the sample.
Nickel and zinc K-edge XAS data were collected as previously described28 under dedicated ring conditions at the National Synchrotron Light Source (NSLS, 2.8 GeV ring, 120–300 mA) of the Brookhaven National Laboratory on beamline X3B using a sagitally focusing Si(111) double-crystal monochromator. X-ray fluorescence was collected using a 30-element Ge fluorescence detector (Canberra) on samples held at ∼14 K in a He displex cryostat. Scattering was minimized by placing a Z-1 element filter [Co(II) or Cu(II)] between the sample chamber and the detector. Internal energy calibration was performed by collecting spectra simultaneously in transition mode on the corresponding metal foil to determine the first inflection point on the edge, which was set to 8331.6 eV [Ni(II)] or 9660.7 eV [Zn(II)]. X-ray absorption near-edge spectroscopy (XANES) data were collected from −200 to 200 eV relative to the metal K-edge. EXAFS was collected to 13.5–15k above the edge energy (E0), depending on the signal:noise at high k values.
Copper K-edge X-ray fluorescence data were similarly collected at the Stanford Synchrotron Radiation Laboratory (SSRL) beamline 7-3 using a 30-element fluorescence detector (Canberra) on samples held at 10 K using a liquid helium cryostat (Oxford Instruments). Beamline optics include a Si(220) double-crystal monochromator and a single rhodium-coated mirror for harmonic rejection. Scattering was minimized by placing a set of Soller slits with a Z-1 element filter (Ni) between the sample chamber and the detector. Internal energy calibration was performed by collecting spectra simultaneously in transition mode on a copper metal foil to determine the first inflection point on the edge (8980.3 eV). X-ray absorption near-edge spectroscopy (XANES) data were collected from −200 to 200 eV relative to the metal’s K-edge. EXAFS was collected to 15k above E0. Because the β2m–Cu(II) sample is photoreduced in the beam, the sample was moved after each scan such that the incident X-ray beam irradiated a fresh section on the sample to obtain the spectrum of the Cu(II) complex. This was done using two cuvettes, yielding six scans, and these scans were averaged. Samples held in place for 10 scans showed edge energy shifts of as much as 3 eV. The scans used for the average spectrum obtained by moving the sample after each scan were essentially superimposable.
Data analysis was performed as previously described29 using SIXpack30 and the Horae (Artemis) software package.31 The data were converted to k space using the relationship k = {[2me(E – E0)]/(ℏ2)}1/2, where me is the mass of the electron, ℏ is Plank’s constant divided by 2π, and E0 is the threshold energy of the absorption edge. Data were loaded, averaged after removal of bad detector elements, background corrected, normalized, and calibrated using SIXpack. The Artemis fitting software package, which builds on the IFEFFIT engine, was used to fit EXAFS data during model refinement.32 The k3-weighted data were fit in r space over a k range of 2–12.5 Å–1 (uncorrected for phase shifts) using an S0 value of 0.9, and a Kaiser–Bessel window where dk = 1. Separate sets of Δreff and σ2 for the sulfur, nitrogen, and bromide ligands were used, with a universal E0 [initially set to be 8340 eV for Ni(II), 8990 eV for Cu(II), and 9670 eV for Zn(II)]. Initial input metal–ligand distances were 2.0 Å for the M–N(O) bond, 2.3 Å for the M–S bond, and 2.4 Å for the M–Br bond. Single-scatter fits were generated using an R′ space range of 1–2.5 Å, and multiple-scattering contributions from histidines were explored for the best single-scattering fits using a data range of 1–4.5 Å (uncorrected for phase shifts) in r space. Single-scatter and multiple-scatter fits were performed using the general EXAFS equation
For multiple scattering arising from histidine imidazole ligands, average values and bond lengths obtained from crystallographic data were used to construct rigid imidazole rings.33 The distance of the five non-hydrogen atoms in the imidazole rings from the metal center was fit in terms of a single metal–ligand bond distance (reff) for various angles α (0–10°), around an axis perpendicular to the plane of the ring and going through the coordinating nitrogen.29a,34
To assess the goodness of fit from different fitting models, the fit parameters reduced χ2 and Rfactor were minimized. Increasing the number of adjustable parameters is generally expected to improve the Rfactor; however, reduced χ2 may go through a minimum and then increase, indicating the model is overfitting the data. These parameters are defined as follows:35
where Nidp is the number of independent data points defined as
where Δr is the fitting range in r space, Δk is the fitting range in k space, Npts is the number of points in the fitting range, Nvar is the number of variables floating during the fit, ε is the measurement of uncertainty, the Re term is the real part of the EXAFS Fourier-transformed data and theory functions, the Im term is the imaginary part of the EXAFS Fourier-transformed data and theory functions, X(Ri) is the Fourier-transformed data or theory function, and
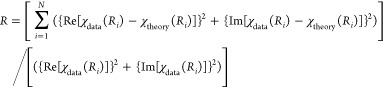 |
Single-scattering fits were calculated for models containing an integer number of ligands from two to seven, using all possible combinations of N and S donors, and including Br– ligands as needed. The best single-scattering fits were further refined using multiple-scattering parameters to account for EXAFS arising from the imidazole side chain of histidine ligands, by replacing integer numbers of N/O donors in the single-scattering fits with imidazoles (see Tables S1–S9 of the Supporting Information). The best fits listed in Table 1 were determined by minimizing the values of Rfactor and reduced χ2 for fits with reasonable values of σ2.
Table 1. Best Fits with Multiple Scattering for EXAFS Fits from Cu(II)–, Ni(II)–, and Zn(II)−β2m Complexes.
| radius R′ (Å) | σ2 (Å2) | E0 shift (eV) | %Rfactor | ||
|---|---|---|---|---|---|
| Cu(II) | 3 N/O | 1.95(1) | 18(5) | –2(1) | 7.8 |
| 1 Im | 1.95(1) | 2(1) | |||
| 1 Br | 2.40(2) | 12(2) | |||
| Ni(II) | 4 N/O | 2.06(8) | 2(1) | 0(2) | 6.4 |
| 1 Im | 2.06(8) | 1(4) | |||
| 1 Br | 2.44(3) | 9(2) | |||
| Zn(II) | 2 N/O | 1.99(1) | 4(2) | –8(2) | 6.2 |
| 2 Im | 1.99(1) | 5(2) | |||
| 1 Br | 2.38(1) | 6(1) |
Results
Distinct Effects of Cu(II), Ni(II), and Zn(II) on β2m Oligomerization and Fibril Formation
To test the specificity of Cu(II) for inducing β2m amyloid formation, we explored the aggregation properties of β2m in the presence of Zn(II) and Ni(II). Oligomerization and amyloid formation of β2m were monitored by four methods: (i) thioflavin T (ThT) fluorescence, (ii) size exclusion chromatography (SEC), (iii) sodium dodecyl sulfate (SDS) dissolution, and (iv) transmission electron microscopy (TEM). An increase in ThT fluorescence at 483 nm is a common means of monitoring the formation of amyloid-like species in solution.36 When each metal is added to a solution with β2m and ThT, we find that Cu(II) and Zn(II) cause a change in the dye’s fluorescence, suggesting amyloid formation, but Ni(II) does not (Figure 1). Similarly, when SEC is used to follow the formation of β2m oligomeric species, we find that no β2m oligomers are formed in the presence of Ni(II) (Figure 2a). In contrast, discrete oligomeric species are formed in the presence of Zn(II) and Cu(II) (panels b and c, respectively, of Figure 2). Curiously, only dimers and hexamers are formed in the presence of Zn(II), whereas dimers, tetramers, and hexamers are formed when Cu(II) is present. The progression of oligomers in the presence of Cu(II) is consistent with previous reports by our group and others.23,37 The absence of tetramers when Zn(II) is added clearly indicates that Zn(II) has a different effect on β2m aggregation.
Figure 1.
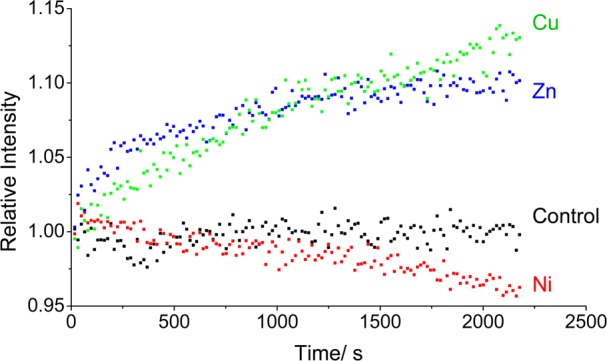
Changes in ThT fluorescence at 483 nm over time in the absence (black) and presence of Cu(II) (green), Zn(II) (blue), and Ni(II) (red). Each solution contained 100 μM β2m, 150 μM CuSO4 or ZnSO4 or 3 mM NiSO4, 500 mM urea, 150 mM potassium acetate, 25 mM MOPS (pH 7.4), and 80 μM ThT.
Figure 2.
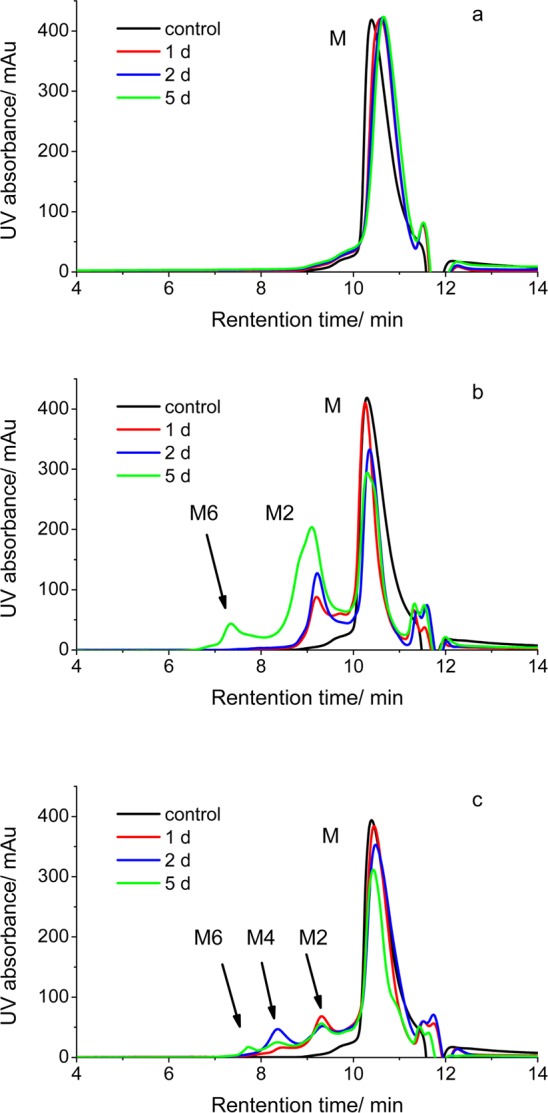
SEC analyses of 100 μM β2m incubated with (a) 4 mM Ni(II), (b) 150 μM Zn(II), and (c) 150 μM Cu(II). In each case, the protein and indicated metal were mixed with 500 mM urea, 150 mM potassium acetate, and 25 mM MOPS (pH 7.4) at 37 °C. The control sample is 100 μM β2m incubated with 5 mM EDTA in 150 mM potassium acetate and 25 mM MOPS (pH 7.4) at 37 °C. M, M2, M4, and M6 indicate the β2m monomer, dimer, tetramer, and hexamer, respectively.
The ability of the metals to induce the formation of amyloid fibrils was assessed using SDS dissolution and TEM measurements after incubation for 1.5 months with all three metals. Consistent with the data in Figures 1 and 2, no insoluble aggregates of β2m are found in the presence of Ni(II) for this time period. The Zn(II)- and Cu(II)-containing samples, however, do form precipitates. We had previously found that β2m amyloid fibrils do not dissolve after exposure to a 2% SDS solution at 37 °C for 24 h. As we observed previously, the aggregates in the sample incubated with Cu(II) did not dissolve. In contrast, the insoluble aggregates formed with Zn(II) dissolve in 2% SDS within 1 h, suggesting that amyloids are formed with Cu(II) but not with Zn(II). TEM of the aggregates is consistent with this conclusion as amorphous aggregates are formed with Zn(II) present, while long thin fibrils are formed with Cu(II) present (Figure 3). Interestingly, the fact that the ThT fluorescence increases with Zn(II) present, even though no β2m amyloid species are formed, demonstrates that ThT fluorescence may be a poor indicator of the presence of β2m amyloid-like species, an observation that has been made previously.23,37 Taken as a whole, the ThT fluorescence, SEC, SDS, and TEM data reveal that Cu(II) is unique in inducing the amyloid formation of β2m. Moreover, Ni(II) and Zn(II) influence the protein in very distinct ways. To further understand the unique role of Cu(II), we set out to characterize the nature of binding of Cu(II), Zn(II), and Ni(II) to β2m.
Figure 3.

TEM images obtained after incubation of β2m with Cu(II) (left) and Zn(II) (right) for 1.5 months.
Binding of Cu(II) to β2m
We previously reported that Cu(II) binds β2m via the N-terminal amine, the backbone amide between Ile1 and Gln2, His31, and Asp59.38 Further insight into the structural basis of Cu(II)’s unique effect on β2m amyloidosis comes from X-ray absorption spectroscopy (XAS). XANES analysis provides information about the coordination number and oxidation state of the metal ion.
The XANES spectra of the β2m–Cu(II) samples show the development of a peak centered around 8984 eV, intensifying with each subsequent scan. This peak is associated with a 1s → 4p transition of Cu(I) centers. The occurrence of Cu(I) is due to the photoreduction caused by the incident X-ray beam. For this reason, the samples were shifted after each scan such that a fresh spot on the sample was being examined. These scans were averaged and showed no indications of photoreduction (edge energy shift).
The EXAFS region of an XAS spectrum provides information about the metal center’s coordination environment, such as the types of donor atoms and their bonding distance, and provides a second measure of the coordination number. The best fit to the spectrum (Figure 4 and Table 1) obtained for the β2m–Cu(II) complex shows that the ligands are primarily N/O donors with one bromide anion. The bromide is a ligand derived from the buffer, which contained 150 mM NaBr. The spectra exhibit an intense feature at ∼1.7 Å (without phase correction) in the Fourier-transformed spectrum and smaller peaks in the 2.0–4.0 Å range. The latter peaks are consistent with the presence of ligands that give rise to multiple scattering, which in biological samples generally indicate the presence of histidine imidazole ligands. The best fit for the β2m–Cu(II) complex (Figure 4 and Table 1) is a five-coordinate model in which the coordinating species can be best described as three N/O donors, one imidazole donor (from histidine), and one bromide ligand. This model is consistent with our previous measurements of the Cu(II)–protein binding site.38 Prior results indicated that the metal has four protein-based ligands, including one imidazole, which is presumably His31, as identified previously. The other three protein-based ligands were identified as the N-terminal amine, the backbone amide of Gln2, and Asp59.38 The presence of the Br– ligand in the EXAFS analysis indicates the presence of a vacant or labile coordination site on the Cu(II), such as an aqua ligand.
Figure 4.
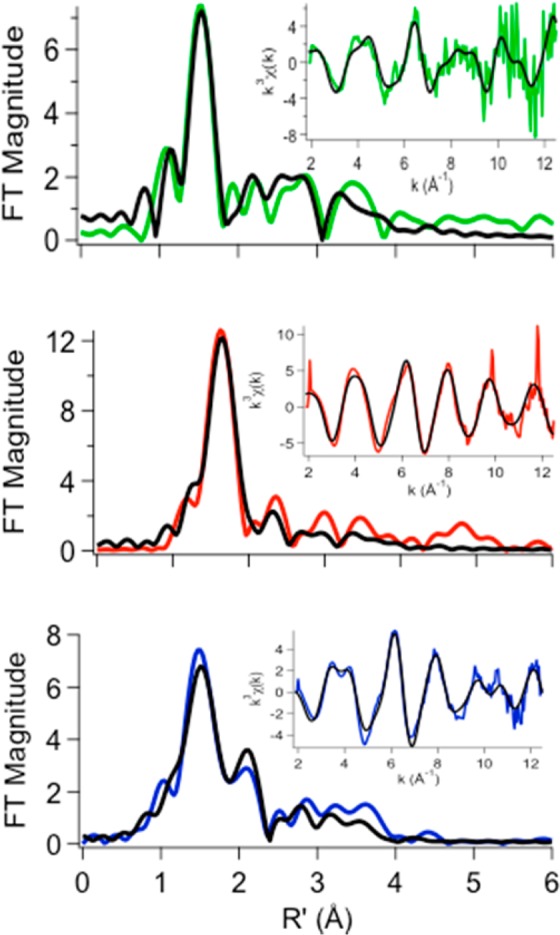
Fourier-transformed XAS data (colored lines) and best fits (black lines) from Table 1 for metal complexes of β2m in 150 mM KBr, 25 mM MOPS (pH 7.4), and 10% glycerol in the presence of Cu(II) (green), Ni(II) (red), or Zn (blue). Insets are unfiltered k3-weighted EXAFS spectra and fits. The radial distance (R′) has not been phase-corrected.
Binding of Ni(II) to β2m
The analysis of EXAFS spectra obtained for the β2m–Ni(II) complex shows that the best fit is for a six-coordinate site comprised of primarily nitrogen or oxygen donor ligands with one bromide. A comparison of the XAS data for the β2m–Cu(II) and β2m–Ni(II) complexes indicates that one significant difference is that Ni(II) is bound to β2m in a hexacoordinate environment. As a result, the ligands are further from the metal than in the case of Cu(II). The β2m–Ni(II) XANES spectrum shows a small peak associated with the 1s → 3d transition located at ∼8333 eV (Figure S1 and Table S10 of the Supporting Information). The peak area of this transition [5.2(7) × 10–2 eV] and the absence of a 1s → 4pz transition reflect the coordination geometry around the metal39 and are similar to those found in pseudo-octahedral model complexes, although the 1s → 3d peak area is larger than what is typically found (∼1.0–4.0 × 10–2 eV). This indicates a geometry that is more distorted from centrosymmetric in the β2m–Ni(II) complex than in the models. The M–L distances are consistent with a high-spin, S = 1, electronic configuration for Ni(II), and thus consistent with a six-coordinate pseudo-octahedral geometry.39 Previous measurements of the β2m–Ni(II) dissociation constant (Kd) found that this metal had a much lower affinity for the protein than Cu(II) does (400 μM vs 2.5 μM).22 The lower affinity for Ni(II) might suggest that some of the ligands identified as N/O donors might not arise from the protein but instead from water molecules.
The XAS data indicate that the Ni(II) coordination environment is different from that of Cu(II), but it does not identify the specific amino acids bound to Ni(II). To determine the Ni(II) binding residues, we used metal-catalyzed oxidation (MCO) reactions along with mass spectrometry (MS) detection. In the MCO–MS method, an oxidizing agent and a reducing agent are added to allow the site-selective oxidation of metal-bound amino acid residues, and MS is then used to sequence and identify the modified amino acids. This method has been used successfully to identify Cu(II) binding sites in proteins,38,40−45 and we have also shown that it can be used to identify metal binding sites in Mn(II), Fe(II), Co(II), and Ni(II) binding peptides and proteins.45 After a 20 min MCO reaction in the presence of ascorbate as the reducing agent and persulfate as the oxidizing agent, the addition of up to two oxygen atoms to β2m is reproducibly observed with a modification percent of 45–50% (Figure S2a of the Supporting Information). To pinpoint the oxidation sites, β2m was subjected to proteolysis followed by LC–MS/MS analyses. An example of extracted ion chromatograms of modified and unmodified forms of a histidine-containing peptide is shown in Figure S2b of the Supporting Information. The oxidized peptide fragments were sequenced by tandem MS to identify the modified amino acids. An example of how tandem MS was used to identify oxidation sites is illustrated in Figure S2c of the Supporting Information.
The results from the MCO reactions of the β2m–Ni(II) complex indicate that the N-terminus and His31 are part of the Ni(II) binding site (Table 2), as only these two residues show a significant increase in their level of oxidation compared to that from the control experiment. Oxidation of His51, Trp60, and Trp95 is measured, but their extent of oxidation is similar to that from the control experiment. Moreover, increases in the oxidant concentration indicate that the extents of oxidation of the N-terminus and His31 further increase whereas those of His51, W60, and W95 do not (Figure S3 of the Supporting Information). Unfortunately, no amino acids other than the N-terminus and His31 are significantly oxidized, making it difficult to identify any other protein-based ligands.
Table 2. Percentages of Modified Residues Observed after a 20 min MCO Reaction of the β2m–Ni(II) Complex.
| MCOa | controlb | |
|---|---|---|
| N-terminus | 27.9 ± 3.6% | 0 ± 0% |
| His31 | 14.5 ± 1.9% | 0.1 ± 0.2% |
| His51 | 0.4 ± 0.2% | 0.2 ± 0.2% |
| Trp60 | 4.4 ± 2.0% | 8.4 ± 3.0% |
| Trp95 | 1.6 ± 1.2% | 1.7 ± 1.5% |
β2m (200 μM) was reacted with 250 μM NiSO4, 5 mM ascorbate, 0.5 mM persulfate in 150 mM potassium acetate, and 25 mM MOPS (pH 7.4).
In the control experiment, 200 μM β2m was reacted with 250 μM NiSO4 and 0.5 mM persulfate in 150 mM potassium acetate and 25 mM MOPS (pH 7.4).
Because Cu(II) and Ni(II) have at least two β2m ligands in common, one might predict that high concentrations of Ni(II) might displace Cu(II) and influence the rate of Cu(II)-induced oligomer formation. Even though Ni(II) itself does not cause the protein to form oligomers, if it displaces Cu(II) at higher concentrations, it will decrease the population of Cu(II)−β2m species in solution, thereby slowing Cu(II)-induced oligomer formation. To test this idea, we incubated 100 μM β2m and 150 μM Cu(II) in the absence and presence of 3 mM Ni(II). On the basis of previous dissociation constant (Kd) measurements of binding of β2m to Cu(II) and Ni(II) (KdCu = 2.5 μM; KdNi = 400 μM),22 the addition of 3 mM Ni(II) should result in a decrease in β2m–Cu(II) loading from 96 to 77% and a corresponding decrease in the extent of oligomerization (see the Supporting Information for details). According to SEC measurements, the amounts of dimer and tetramer formed after 2 and 5 days decrease (Figure S4 of the Supporting Information). These results confirm our expectation, indicating that Ni(II) competes with Cu(II) for the same binding locus.
Binding of Zn(II) to β2m
The effect of Zn(II) on β2m oligomerization is rather intriguing because the metal induces the formation of dimers and hexamers but no amyloid fibrils. Previous studies had found a β2m–Zn(II) Kd of 1.5 mM using a metal competition assay with a fluorescence readout;22 however, our ThT and SEC results are not consistent with this previously measured Kd. Considering a β2m–Zn(II) Kd of 1.5 mM and the Zn(II) and β2m concentrations used to give the results in Figures 1 and 2, it is unlikely that Zn(II) would be able to have such a significant effect on β2m oligomer formation with only ∼8% of the protein bound to Zn(II). One possible explanation is that the previously measured Kd value is incorrect. The previous measurements relied on fluorescence quenching, presumably of Trp60, which is solvent-exposed and near the identified Cu(II) binding site. It is possible that Zn(II) binds very distant from Trp60 and therefore does not quench the fluorescence of this residue at concentrations that otherwise lead to β2m–Zn(II) binding. If so, then Zn(II) may bind the protein at a site very different from that to which Cu(II) binds.
To test the idea that Cu(II) and Zn(II) bind at different sites, we monitored the effect of added Zn(II) on the Cu(II)-induced oligomerization of β2m. We incubated 100 μM β2m with 150 μM Cu(II) in the absence and presence of 150 μM Zn(II). Unlike the comparable experiment with Ni(II), the addition of Zn(II) suppresses the formation of the tetramer but does not significantly change the extent of β2m dimer formation (compare the black and blue traces in Figure 5a). The absence of any significant change in the concentration of the dimer implies that Zn(II) does not displace Cu(II). Instead, Zn(II) suppresses the formation of the tetramer, which implies its binding occurs at a site that interferes with tetramer formation. Also, the presence of Cu(II) completely prevents Zn(II)-induced hexamer formation (compare the red and blue traces in Figure 5a). Adding a similar amount of Ni(II) (i.e., 200 μM) with Zn(II) does not disrupt Zn(II)-induced oligomer formation (compare the black and red traces in Figure 5b), but an excess of Ni(II) (i.e., 3 mM) does suppress oligomerization to some extent (compare the black and blue traces in Figure 5b). The effect of Ni(II) on Zn(II)-induced oligomerization is not the same as the effect of Cu(II), indicating some subtle differences between how Ni(II) and Cu(II) bind β2m. Collectively, these data imply that Zn(II) binds β2m at a site different from those at which Cu(II) and Ni(II) bind.
Figure 5.
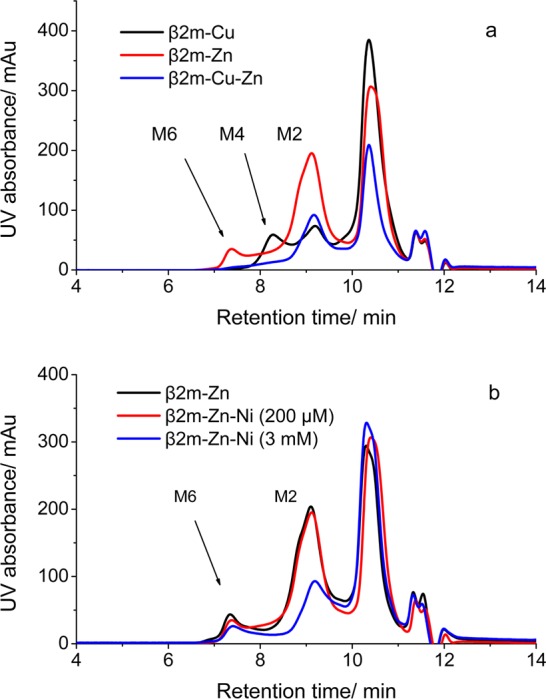
SEC analyses of (a) 100 μM β2m incubated with 150 μM Cu(II) (black), 150 μM Zn(II) (blue), and both 150 μM Cu(II) and 150 μM Zn(II) (red) and (b) 100 μM β2m incubated with 150 μM Zn(II) without (black) and with (red) 200 μM Ni(II) or (blue) with 3 mM Ni(II) in 500 mM urea, 150 mM potassium acetate, and 25 mM MOPS (pH 7.4) at 37 °C for 4 days.
Characterization of the coordination structure of β2m with Zn(II) is more challenging than characterization of that with Cu(II) or Ni(II) because of Zn(II)’s electronic structure. Zn(II) does not undergo a one-electron redox process, so the MCO–MS method cannot be directly applied. However, the MCO reactions of the β2m–Cu(II) complex can be performed in the presence of increasing concentrations of Zn(II) to test if Zn(II) competes with Cu(II) for the same binding site. If Zn(II) does bind to β2m at a site with ligands in common with the Cu(II) complex, the expectation would be that the level of site specific oxidation of the Cu(II)-bound residues would decrease as Zn(II) concentrations are increased. From Figure S5 of the Supporting Information, it is clear that Zn(II) does not displace Cu(II) as the oxidation levels of the N-terminus and His31, which are two of the Cu(II) binding residues, remain unchanged. These two residues maintain approximately the same level of oxidation at Zn(II) concentrations as high as 3 mM, a result that is consistent with the SEC data in Figure 5.
Because Zn(II) appears to bind to β2m at a site different from those at which Ni(II) and Cu(II) bind, we set out to identify the Zn(II) binding site. Given that His residues are the second most common Zn(II) ligands in proteins,46 we assumed that one or more His residues would be part of the Zn(II) binding site, especially because the protein’s two Cys residues, Cys being the most common Zn ligand, form a buried disulfide bond. To find the bound His residue(s), we measured the hydrogen–deuterium exchange of the hydrogen on the C2 atom of the imidazole ring on each of the four histidines (His13, His31, His51, and His84) in β2m. This hydrogen is known to exchange with a half-life of ∼2 days, and when His is bound to a metal, the exchange rate is even slower,47−49 allowing Zn binding His residues to be identified.25 Using the protocol described in Materials and Methods, but without added urea so that oligomer formation is slowed, β2m was mixed in D2O with and without Zn(II), and aliquots of the solution were analyzed over the course of 4 days. The data in Table 3 indicate that only His51 undergoes a statistically significant change in the extent of deuterium incorporation over the entire course of the experiment, suggesting that this His residue is bound to Zn(II). His31 does show a slight decrease in the level of exchange in the presence of Zn after 4 days, but this result can be rationalized by the formation of oligomers after 1 day under these conditions (Figure S6 of the Supporting Information). It is conceivable that His31 becomes less solvent accessible as more oligomers are formed, thereby decreasing its extent of H–D exchange.
Table 3. Hydrogen–Deuterium Exchange Analyses of 100 μM β2m Incubated in the Presence or Absence of 100 μM Zn(II) in D2O Containing 150 mM Potassium Acetate and 25 mM MOPS (pH 7.4) at 37 °C.
| no. of deuteriums
incorporated |
||||
|---|---|---|---|---|
| residue | Zn(II) | 1 day | 2 days | 4 days |
| His13 | – | 0.171 ± 0.004 | 0.32 ± 0.02 | 0.568 ± 0.005 |
| + | 0.161 ± 0.009 | 0.31 ± 0.03 | 0.53 ± 0.03 | |
| His31 | – | 0.104 ± 0.008 | 0.25 ± 0.01 | 0.46 ± 0.02a |
| + | 0.10 ± 0.01 | 0.241 ± 0.001 | 0.41 ± 0.03a | |
| His51 | – | 0.195 ± 0.007a | 0.33 ± 0.01a | 0.52 ± 0.02a |
| + | 0.14 ± 0.02a | 0.24 ± 0.01a | 0.44 ± 0.03a | |
| His84 | – | <0.05b | <0.05b | 0.051 ± 0.001 |
| + | <0.05b | <0.05b | 0.054 ± 0.006 | |
Deuterium levels in the Zn-bound protein that are statistically different from the deuterium levels in the Zn-free protein based on a t test (p < 0.05).
Number of deuteriums incorporated into His84 at 1 and 2 days were too low to be confidently measured.
XAS was further used to probe the coordination structure of Zn(II)-bound β2m (Figure 4 and Table 1). Being a d10 metal, Zn(II) shows no possible pre-edge transitions. One can, however, make qualitative determinations of the coordination number based on the intensity of the XANES spectra, where the normalized intensity increases with coordination number: four coordinate has a normalized intensity of ∼1.3, and five- and six-coordinate have normalized intensities between 1.3 and 2.50 Because the intensity of the white line for the β2m–Zn(II) samples is ∼1.5, a five-coordinate arrangement would be consistent with this qualitative analysis and the EXFAS data. It is interesting that two imidazoles (i.e., two His residues) are found to bind Zn(II) from the best fit of the EXAFS data. These data seem to contradict the hydrogen–deuterium exchange data in Table 3, but it should be pointed out that the EXAFS experiments were performed on solutions that contained much higher concentrations of Zn(II) (1.2 mM) and β2m (2 mM) to obtain a sufficient XAS signal. Thus, it is possible that Zn(II) binding facilitates the bridging of two β2m molecules at these high concentrations via His51, which sits on the surface of the protein. Indeed, SEC of the sample used for the XAS experiment shows extensive dimer formation (Figure S7 of the Supporting Information). An alternate explanation is that two His residues from the same protein molecule bind Zn(II) simultaneously, and the slight decrease in the level of exchange seen for His31 reflects binding of this residue. However, if this were to occur, β2m would have to undergo a major structural change to allow these two His residues to bind Zn(II) because these residues are ∼22 Å from one another in the native structure. Far-UV circular dichroism (CD) measurements, however, indicate that the β2m–Zn(II) complex has a global structure that is very similar to the protein with no metal bound or even with Cu(II) or Ni(II) bound (Figure S8 of the Supporting Information), ruling out such a major conformational change. Moreover, backbone amide hydrogen–deuterium exchange experiments further indicate that the Zn(II)-bound protein does not undergo any major structural reorganization (Table S11 of the Supporting Information), as the increase in the level of exchange in the presence of Zn or the other metals is modest compared to that without metal.
As a final set of experiments, we attempted to explain the fact that Zn(II) causes β2m oligomerization at concentrations much lower than expected on the basis of the previously measured Kd value of 1.5 mM.22 The extent of hydrogen–deuterium exchange of the hydrogen at C2 of His51 was monitored at increasing Zn(II) concentrations and compared to the extent of exchange in the absence of Zn. The resulting decrease in the rate of deuterium uptake as a function of Zn(II) concentration was then fit using eq 1 to obtain an apparent Kd of 24 ± 11 μM (Figure 6). This measured Kd value for the β2m–Zn(II) complex is more consistent with the ThT and SEC results (Figures 1 and 2) than the previously reported Kd value of 1.5 mM.22
| 1 |
Figure 6.
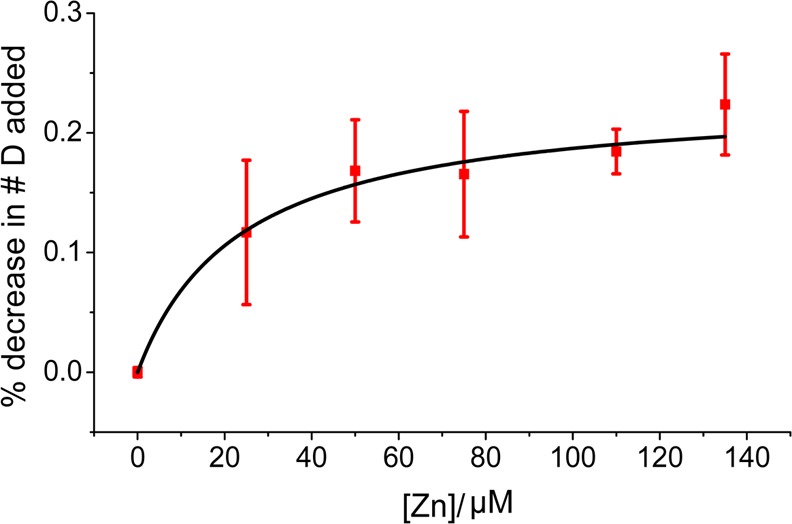
Percent decrease in the level of deuterium incorporation, as compared to that of the Zn(II)-free protein, fit to eq 1 to obtain a Kd value (error bars represent standard errors of the mean and have considered propagations of error in the calculated values).
Discussion
A comparison of the binding properties and effects of Ni(II) and Zn(II) on β2m oligomerization and amyloid formation give us additional insight into the unique nature of Cu(II) in allowing β2m amyloid fibril formation. We previously established that Cu(II) binds β2m via the N-terminus, a backbone amide between IIe1 and Gln2, His31, and Asp59.38 Evidence indicates that Cu(II) binding causes the repositioning of Asp59 and Arg3 to allow two pairs of dimer-stabilizing salt bridges to be formed between Asp59 and Lys19 and between Arg3 and Glu16 (Figure 7a).51 Cu(II) also facilitates the cis–trans isomerization of Pro32, which in turn exposes Phe30 to the solvent and creates a hydrophobic patch that drives dimer assembly.52,53
Figure 7.
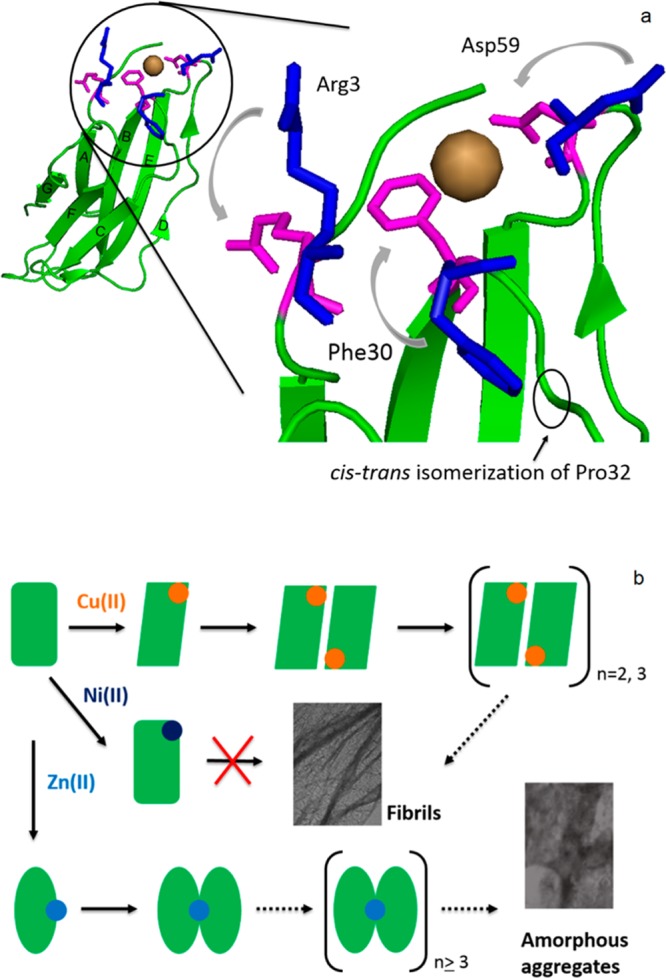
(a) Structural changes of β2m upon Cu(II) binding, including repositioning of Arg3 and Asp59 and exposure of Phe30 to the solvent followed by cis–trans isomerization of Pro32 (the β2m backbone is colored green; repositioning of the residues is indicated from blue to magenta). (b) Proposed model for the different effects of Cu(II), Ni(II), and Zn(II) on β2m oligomerization and fibril formation.
The effect (or lack thereof) of Ni(II) on β2m aggregation provides insight into the metal–protein interactions that are essential for allowing dimer formation. In contrast to Cu(II), no oligomers are formed with Ni(II) present, even though the MCO–MS and SEC data (Table 2 and Figure 5) imply that Ni(II) and Cu(II) bind to the same region of the protein. A similar binding site was suggested previously on the basis of intrinsic fluorescence and native MS experiments,22 and the data here provide stronger support for this conclusion. Moreover, the CD and amide hydrogen–deuterium exchange data indicate that the Cu(II)- and Ni(II)-bound forms of the protein have similar overall global structures. The XAS and MCO–MS data, though, do indicate some key differences between binding of Cu(II) and Ni(II) to β2m. First, the β2m–Cu(II) and β2m–Ni(II) complexes have different coordination numbers and geometries. Ni(II) is hexacoordinate and presumably adopts a pseudo-octahedral ligand arrangement, whereas Cu(II) is five-coordinate. For Cu(II), all four protein-based ligands are known from the MCO–MS experiments, and the fifth ligand is an anion. The MCO–MS experiments with Ni(II) indicate only two protein-based ligands, the N-terminus and His31. Given the higher Kd value for Ni(II) [400 μM vs 2.5 μM for Cu(II)], it makes sense that Ni(II) has a smaller number of protein-based ligands than Cu(II) and has more non-protein-based ligands in its coordination sphere. Indeed, the MCO–MS and XAS results are truly complementary in this regard as they provide this insight in a way that would not be possible by either method alone. A key difference would appear to be that Ni(II) does not bind to the backbone amide between Ile1 and Gln2, whereas Cu(II) does. Interestingly, binding of Cu(II) to amide nitrogens is a common feature of several amyloid-forming systems such as the prion protein and α-synuclein as probed by circular dichroism and electron paramagnetic resonance titrations.54,55 The apparent requirement of metal–amide binding in the amyloid formation of β2m suggests the importance of this interaction for inducing amyloid formation, which was suggested previously.56 Another difference between Cu(II) and Ni(II) binding is the absence of Asp59 as a ligand for Ni(II). The unique participation of Asp59 in Cu(II) coordination and the resulting amyloidogenicity of Cu(II) are consistent with previous work that indicates the importance of the loop between strands D and E that contains Asp59. Heegaard et al. found that β2m has an increased level of amyloid formation upon being cleaved at Lys58.57 Presumably, cleavage of this loop at Lys58 and binding of Cu(II) at Asp59 both reposition Asp59 in a way that allows amyloid formation. In contrast, Ni(II) probably does not reposition this residue to form a dimer-stabilizing salt bridge.51 On the basis of a comparison between Cu(II) and Ni(II) binding, it appears that the essential features of the β2m–Cu(II) interaction that allow dimer formation, and eventually amyloid formation, are Ile1-Gln2 amide and/or Asp59 binding (Figure 7b). It seems likely, although no evidence is provided here, that Ni(II) also does not induce the cis–trans isomerization of Pro32, which is thought to be another important conformational change allowing dimer assembly.52,53
Like the presence of Cu(II), the presence of Zn(II) causes β2m to form oligomers. These oligomers cause changes in ThT fluorescence and appear as dimers and hexamers according to SEC, but the resulting aggregates are not amyloid-like morphologically, as indicated by TEM, or chemically, as indicated by SDS dissolution experiments. The difference in aggregation between the two metals appears to be caused by differences in metal–protein binding sites. Substitution MCO–MS reaction and hydrogen–deuterium exchange of the C2 hydrogens of imidazoles indicate that the two metals bind β2m quite differently, with His51 serving as the main Zn(II) binding residue. Because it is located in a region of the protein completely different than the Cu(II) binding site, Zn(II) does not promote the repositioning of Arg3 or Asp59, nor does it likely promote the cis–trans isomerization of Pro32 like Cu(II) does. Instead, dimer formation by Zn(II) may be induced by the bridging of His51 residues from two different monomers as suggested by the XAS data and consistent with SEC showing the formation of dimers. The ability of Zn(II) to inhibit Cu(II)-induced β2m tetramer formation (Figure 5a) is consistent with this binding mode based upon the structure of the Cu(II)-induced β2m tetramer.58 The Cu(II)-induced tetramer is mediated by interactions between strand D (residues 50–56) of one dimer unit and strand G (residues 91–95) of another dimer unit. Binding of Zn(II) to His51 along strand D would be expected to inhibit the interactions necessary to stabilize the Cu(II)-induced tetramer (Figure 7b).
Interestingly, the dimer formed in the presence of Zn(II) causes the fluorescence maximum of ThT to shift in a manner similar to those of other amyloid-forming proteins, even though the eventual aggregates are not amyloid-like. While speculative, this change in fluorescence might occur as Zn(II) bridges two monomers via His51, allowing the formation of an extended β sheet structure mediated at the D strands and extending over the E, B, and A strands of β2m (Figure 8). This proposed dimer differs from the crystallographic dimer of the P32A mutant of β2m,53 which is also mediated by the D strands, in that it has a parallel rather than an antiparallel arrangement of the monomeric units. This parallel arrangement would seem to be important for orienting the two His51 in the proximity of each other. The Zn(II)-induced dimer could eventually proceed to a hexamer and larger aggregates, but our current data provide no insight into how this occurs. Even so, the proposed Zn(II) dimer has an interface very different from that of the Cu(II)-induced dimer, highlighting the importance of the correct dimer structure in allowing eventual amyloid formation. Again, the Cu(II) coordination site and geometry are essential for assembling an amyloid-competent dimer, and Zn(II) appears to be unable to adopt the correct binding motif.
Figure 8.
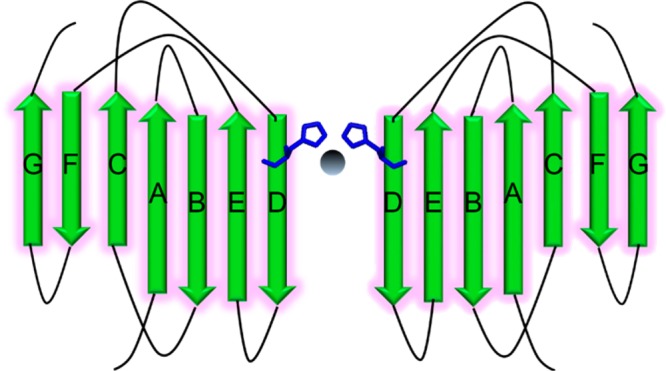
Proposed Zn(II)-bridged β2m dimer via His51.
The full β2m–Zn(II) coordination structure is difficult to obtain from our current data set, but our measured Kd value of ∼30 μM indicates a fairly robust binding site. As an aside, our Kd value is much lower than one previously reported (1.5 mM);22 however, we think that the competitive intrinsic fluorescence assay used in the prior study is probably misleading given that Cu(II) and Zn(II) bind at very different sites and the Trp60 residue that is monitored in the fluorescence assay is >20 Å from the likely His51 binding site. Other than His51, the likely Zn(II) binding sites could be Glu50 and/or Asp53 as Zn(II) is reasonably capable of binding acidic residues.46 Glu50 and Asp53 are solvent accessible and close to His51, and their binding along with His51 could facilitate an intermolecular complex between two β2m monomers.
Conclusions
When compared to Ni(II) and Zn(II), we find that Cu(II) is unique in its ability to induce the formation of β2m oligomers that can eventually form amyloid fibrils. Cu(II)’s ability to induce amyloid formation arises from the nature of its coordination structure with the protein. Specifically, cooperative binding to the N-terminal amine, His31, the backbone amide between Ile1 and Gln2, and Asp59 appear to be essential for allowing the formation of dimers that eventually progress to form amyloid fibrils. The essentiality of the backbone amide is evident from a comparison of Cu(II) and Ni(II) binding. Ni(II) appears to be unable to bind this backbone amide, and as a result, this metal is unable to induce the formation of β2m oligomers or amyloids. As is evident from β2m’s interactions with Zn(II), the binding of a metal to other sites on the protein can allow oligomers and aggregates to be formed. Unless the binding site involves the aforementioned residues, however, the structural changes necessary for the formation of amyloid-competent oligomers (e.g., cis–trans isomerization of Pro32 and repositioning of Arg3 and Asp59) do not occur. In a broader context, our work helps identify some of the key important interactions that make Cu(II) a general motif responsible for protein amyloid formation. Future work to delineate the detailed structural differences caused by the binding of Cu(II), Ni(II), and Zn(II) should reveal other essential structural changes necessary for β2m to progress to amyloid.
Acknowledgments
We thank Prof. Lila Gierasch for access to the CD spectrometer. We also thank Dr. Louis Raboin for help with the TEM experiments.
Supporting Information Available
A figure showing how the oxidation site is identified after MCO reactions of β2m and Ni(II); a figure showing the modification percentages of oxidized residues found in the MCO reaction of β2m and Ni(II) as a function of persulfate concentration; calculations estimating the percent decrease in β2m–Cu(II) loading in the presence and absence of Ni(II); SEC analyses illustrating the effect of the addition of Ni(II) on Cu(II)-induced β2m oligomerization for up to 5 days; a figure showing the modification percentages of Ni(II) binding residues in the substitution MCO reaction as a function of Zn(II) concentration; SEC analysis of the sample used in the hydrogen–deuterium exchange of the C2 hydrogens of histidine incubated for 1 day; SEC analysis of the Zn(II) sample used in XAS experiments; CD spectra of β2m in the absence and presence of Cu(II), Ni(II), and Zn(II); a table containing global hydrogen–deuterium exchange rates of the amide hydrogens for β2m incubated in the absence and presence of Cu(II), Ni(II), and Zn(II); and EXAFS fitting tables for Ni(II)–, Cu(II)–, and Zn(II)−β2m complexes. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by National Institutes of Health Grants R01 GM 075092 (R.W.V.) and R01 GM 069696 (M.J.M.).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wearsch P. A.; Cresswell P. (2008) The quality control of MHC class I peptide loading. Curr. Opin. Cell Biol. 20, 624–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menaa C.; Eseer E.; Sprague S. M. (2008) β-2-microglobulin stimulates osteoclast formation. Kidney Int. 73, 1275–1281. [DOI] [PubMed] [Google Scholar]
- Keating M. J. (1999) Chronic lymphocytic leukemia. Semin. Oncol. 26, 107–114. [PubMed] [Google Scholar]
- Malaguarnera M.; Restuccia S.; Di Fazio I.; Zoccolo A. M.; Trovato B. A.; Pistone G. (1997) Serum β-2-microglobulin in chronic hepatitis C. Dig. Dis. Sci. 42, 762–766. [DOI] [PubMed] [Google Scholar]
- McParland V. J.; Kad N. M.; Kalverda A. P.; Brown A.; Kirwin-Jones P.; Hunter M. G.; Sunde M.; Radford S. E. (2000) Partially unfolded states of β2-microglobulin and amyloid formation in vitro. Biochemistry 39, 8735–8746. [DOI] [PubMed] [Google Scholar]
- Esposito G.; Michelutti R.; Verdone G.; Viglino P.; Hernanadez H.; Robinson C. V.; Amoreseno A.; Dal Piaz F.; Monti M.; Pucci P.; Mangione P.; Stoppini M.; Merlini G.; Ferri G.; Bellotti V. (2000) Removal of the N-terminal hexapeptide from human β2-microglobulin facillitates protein aggragation and fibril formaiton. Protein Sci. 9, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relini A.; Canale C.; De Stefano S.; Rolandi R.; Giorgetti S.; Stoppini M.; Rossi A.; Fogolari F.; Corazza A.; Esposito G.; Gliozzi A.; Belloti V. (2006) Collogan plays an active role in the aggregation of β2-microglobulin under physiopathological conditions of dialysis-related amyloidosis. J. Biol. Chem. 281, 16521–16529. [DOI] [PubMed] [Google Scholar]
- Ohhashi Y.; Kihara M.; Naiki H.; Goto Y. (2005) Ultrasonication-induced amyloid fibril formation of β2-microglobulin. J. Biol. Chem. 280, 32843–32848. [DOI] [PubMed] [Google Scholar]
- Morgan C. J.; Gelfans M.; Atreya C.; Miranker A. D. (2001) Kidney dialysis-associated amyloidosis: A molecular role for copper in fiber formation. J. Mol. Biol. 309, 339–345. [DOI] [PubMed] [Google Scholar]
- Villanueva J.; Hoshino M.; Katou H.; Kardos J.; Hasegawa K.; Naiki H.; Goto Y. (2004) Increase in the confirmational flexibility of β2-microglobulin upon copper binding: A possbile role for copper in dialysis-related amyloidosis. Protein Sci. 13, 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ypersele de Strihou C.; Jadoul M.; Malghem J.; Maldague B.; Jamart J. (1991) Effect of dialysis membrane and patient’s age on signs of dialysis-related amyloidosis. The Working Party on Dialysis Amyloidosis. Kidney Int. 39, 1012–1019. [DOI] [PubMed] [Google Scholar]
- Miura Y.; Ishiyama T.; Inomata A.; Takeda T.; Senma S.; Okuyama K.; Suzuki Y. (1992) Radiolucent bonecysts and the type of dialysis membrane used in patients undergoing long-term hemodialysis. Nephron 60, 268–273. [DOI] [PubMed] [Google Scholar]
- Viles J. H. (2012) Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer’s, Parkinson’s and prion diseases. Coord. Chem. Rev. 256, 2271–2284. [Google Scholar]
- Rasia R. M.; Bertoncini C. W.; Marsh D.; Hoyer W.; Cherny D.; Zweckstetter M.; Griesinger C.; Jovin T. M.; Fernández C. O. (2005) Structural characterization of copper(II) binding to α-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 102, 4294–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Li J.; Fink A. L. (2001) Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein. A possible molecular link between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 276, 44284–44296. [DOI] [PubMed] [Google Scholar]
- Sarell C. J.; Wilkinson S. R.; Viles J. H. (2010) Substoichiometric levels of Cu(II) ions accelerate the kinetics of fiber formation and promote cell toxicity of amyloid-β from Alzheimer disease. J. Biol. Chem. 285, 41533–41540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen J. T.; Østergaard J.; Rozlosnik N.; Gammelgaard B.; Heegaard N. H. (2011) Cu(II) mediates kinetically distinct, non-amyloidogenic aggregation of amyloid-β peptides. J. Biol. Chem. 286, 26952–26963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viles J. H.; Klewpatinond M.; Nadal R. C. (2008) Copper and the structural biology of the prion protein. Biochem. Soc. Trans. 36, 1288–1292. [DOI] [PubMed] [Google Scholar]
- Davis D. P.; Gallo G.; Vogen S. M.; Dul J. L.; Sciarretta K. L.; Kumar A.; Raffen R.; Stevens F. J.; Argon Y. (2001) Both the environment and somatic mutations govern the aggregation pathway of pathogenic immunoglobulin light chain. J. Mol. Biol. 313, 1021–1034. [DOI] [PubMed] [Google Scholar]
- Fox J. H.; Kama J. A.; Lieberman G.; Chopra R.; Dorsey K.; Chopra V.; Volitakis I.; Cherny R. A.; Bush A. I.; Hersch S. (2007) Mechanisms of copper ion mediated Huntington’s disease progression. PLoS One 2, e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha C.; Ryu J.; Park C. B. (2007) Metal ions differentially influence the aggregation and deposition of alzheimer’s β-amyloid on a solid template. Biochemistry 46, 6118–6125. [DOI] [PubMed] [Google Scholar]
- Eakin C. M.; Knight J. D.; Morgan C. J.; Gelfand M. A.; Miranker A. D. (2002) Formation of a copper specific binding site in non-native states of β-2-microglobulin. Biochemistry 41, 19646–10656. [DOI] [PubMed] [Google Scholar]
- Eakin C. M.; Attenello F. J.; Morgan C. J.; Miranker A. D. (2004) Oligomeric assembly of native-like precursors precedes amyloid formation by β-2-microglobulin. Biochemistry 43, 7808–7815. [DOI] [PubMed] [Google Scholar]
- Kassirer J. P. (1971) Clinical evaluation of kidney function: Glomerular function. N. Engl. J. Med. 285, 385–389. [DOI] [PubMed] [Google Scholar]
- Dong J.; Borotto N. B.; Callahan K. L.; Vachet R. W. (2014) Locating Zn-bound histidine residues in metalloprotein using hydrogen deuterium exchange mass spectrometry. Anal. Chem. 86, 766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins K. A.; Chivers P. T.; Maroney M. J. (2012) The role of the N-terminus in determining metal-specific responses in the E. coli Ni- and Co-responsive metallogegulator, RcnR. J. Am. Chem. Soc. 134, 7081–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins K. A.; Hu H. Q.; Chivers P. T.; Maroney M. J. (2012) The Effects of Select Histidine to Cysteine Mutations onTranscriptional Regulation by E. coli RcnR. Biochemistry 51, 7816–7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch S.; Bradley M. J.; Rowe J. L.; Chivers P. T.; Maroney M. J. (2007) Nickel-specific response in the transcriptional regulator, Escherichia coli NikR. J. Am. Chem. Soc. 129, 5085–5095. [DOI] [PubMed] [Google Scholar]
- a Banaszak K.; Martin-Diaconescu V.; Bellucci M.; Zambelli B.; Rypniewski W.; Maroney M. J.; Ciurli S. (2012) Crystallographic and X-ray absorption spectroscopic characterization of Helicobacter pylori UreE bound to Ni2+ and Zn2+ reveals a role for the disordered C-terminal arm in metal trafficking. Biochem. J. 441, 1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ryan K.; Johnson O.; Cabelli D.; Brunold T.; Maroney M. (2010) Nickel superoxide dismutase: Structural and functional roles of Cys2 and Cys6. JBIC, J. Biol. Inorg. Chem. 15, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb S. M. (2005) SIXpack: A graphical user interface for XAS analysis using IFEFFIT. Phys. Scr. T115, 1011. [Google Scholar]
- Ravel B.; Newville M. (2005) ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541. [DOI] [PubMed] [Google Scholar]
- Newville M. (2001) IFEFFIT: Interactive XAFS analysis and FEFF fitting. J. Synchrotron Radiat. 8, 322–324. [DOI] [PubMed] [Google Scholar]
- Engh R. A.; Huber R. (1991) Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. A47, 392–400. [Google Scholar]
- a Blackburn N. J.; Hasnain S. S.; Pettingill T. M.; Strange R. W. (1991) Copper K-extended X-ray absorption fine structure studies of oxidized and reduced dopamine β-hydroxylase. Confirmation of a sulfur ligand to copper(I) in the reduced enzyme. J. Biol. Chem. 266, 23120–23127. [PubMed] [Google Scholar]; b Ferreira G. C.; Franco R.; Mangravita A.; George G. N. (2002) Unraveling the substrate-metal binding site of ferrochelatase: An X-ray absorption spectroscopic study. Biochemistry 41, 4809–4818. [DOI] [PubMed] [Google Scholar]
- Herbst R. W.; Perovic I.; Martin-Diaconescu V.; O’Brien K.; Chivers P. T.; Pochapsky S. S.; Pochapsky T. C.; Maroney M. J. (2010) Communication between the zinc and nickel sites in dimeric HypA: Metal recognition and pH sensing. J. Am. Chem. Soc. 132, 10338–10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana R.; Coleman C.; Ionescu-Zanetti C.; Carter S. A.; Krishna V.; Grover R. K.; Roy R.; Singh S. (2005) Mechanism of thioflavin T binding to amyloid fibrils. J. Struct. Biol. 151, 229–238. [DOI] [PubMed] [Google Scholar]
- Antwi K.; Mahar M.; Srikanth R.; Olbris M. R.; Tyson J. F.; Vachet R. W. (2008) Cu(II) organizes β-2-microglobulin oligomers but is released upon amyloid formation. Protein Sci. 17, 748–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth R.; Mendoza V. L.; Bridgewater J. D.; Zhang G. S.; Vachet R. W. (2009) Copper binding to β-2-microglobulin and its pre-amyloid oligomer. Biochemistry 48, 9871–9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colpas G. J.; Maroney M. J.; Bagyinka C.; Kumar M.; Willis W. S.; Suib S. L.; Baidya N.; Mascharak P. K. (1991) X-ray spectroscopy studies of nickel-complexes, with application to the structure to the structure of nickel sites in hydrogenases. Inorg. Chem. 30, 920–928. [Google Scholar]
- Lim J.; Vachet R. W. (2003) Development of a methodology based on metal-catalyzed oxidation reactions and mass spectrometry to determine the metal binding sites in copper metalloproteins. Anal. Chem. 75, 1164–1173. [DOI] [PubMed] [Google Scholar]
- Bridgewater J. D.; Vachet R. W. (2005) Using microwave-assisted metal-catalyzed oxidation reactions and mass spectrometry to increase the rate at which the copper-binding sites of a protein are determined. Anal. Chem. 77, 4649–4653. [DOI] [PubMed] [Google Scholar]
- Bridgewater J. D.; Vachet R. W. (2005) Metal-catalyzed oxidation reactions and mass spectrometry: The roles of ascorbate and different oxidizing agents in determining Cu-protein-binding sites. Anal. Biochem. 341, 122–130. [DOI] [PubMed] [Google Scholar]
- Bridgewater J. D.; Lim J.; Vachet R. W. (2006) Using metal-catalyzed oxidation reactions and mass spectrometry to identify amino acid residues within 10 angstrom of the metal in Cu-binding proteins. J. Am. Soc. Mass Spectrom. 17, 1552–1559. [DOI] [PubMed] [Google Scholar]
- Srikanth R.; Wilson J.; Burns C. S.; Vachet R. W. (2008) Identification of the copper(II) coordinating residues in the prion protein by metal-catalyzed oxidation mass spectrometry: Evidence for multiple isomers at low copper(II) loadings. Biochemistry 47, 9258–9268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgewater J. D.; Lim J.; Vachet R. W. (2006) Transition metal-peptide binding studied by metal-catalyzed oxidation reactions and mass spectrometry. Anal. Chem. 78, 2432–2438. [DOI] [PubMed] [Google Scholar]
- Zheng H. P.; Chruszcz M.; Lasota P.; Lebioda L.; Minor W. (2008) Data mining of metal ion environments present in protein structures. J. Inorg. Biochem. 102, 1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi M.; Nakazawa T. (2008) Determination of pKa values of individual histidine residues in proteins using mass spectrometry. Anal. Chem. 80, 6481–6487. [DOI] [PubMed] [Google Scholar]
- Buisson D. H.; Jones J. R.; Taylor S. E. (1975) Effects of metal ions on rates of detritiation-new probe in the study of metal-substrate interactions. J. Chem. Soc., Chem. Commun. 20, 856–856. [Google Scholar]
- Buncel E.; Clement O.; Onyido I. (2000) Metal ion effects in isotopic hydrogen exchange in biologically important heterocycles. Acc. Chem. Res. 33, 672–678. [DOI] [PubMed] [Google Scholar]
- Jacquamet L.; Aberdam D.; Adrait A.; Hazemann J.-L.; Latour J.-M.; Michaud-Soret I. (1998) X-ray absorption spectroscopy of a new zinc site in the Fur protein from Escherichia coli. Biochemistry 37, 2564–2571. [DOI] [PubMed] [Google Scholar]
- Mendoza V. L.; Antwi K.; Baron-Rodriguez M. A.; Blanco C.; Vachet R. W. (2010) Structure of the preamyloid dimer of β-2-microglobulin from covalent labeling and mass spectrometry. Biochemistry 49, 1522–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaho D. V.; Miranker A. D. (2009) Delineating the conformational elements responsible for Cu2+-induced oligomerization of β-2-microglobulin. Biochemistry 48, 6610–6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eakin C. M.; Berman A. J.; Miranker A. D. (2006) A native to amyloidogenic transition regulated by a backbone trigger. Nat. Struct. Mol. Biol. 13, 202–208. [DOI] [PubMed] [Google Scholar]
- Klewpatinond M.; Davies P.; Bowen S.; Brown D. R.; Viles J. H. (2008) Deconvoluting the Cu2+ binding modes of full-length prion protein. J. Mol. Biol. 283, 1870–1881. [DOI] [PubMed] [Google Scholar]
- Dudzik C. G.; Walter E. D.; Millhauser G. L. (2011) Coordination features and affinity of the Cu2+ site in the α-synuclein protein of Parkinson’s disease. Biochemistry 50, 1771–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese M. F.; Miranker A. D. (2009) Metal binding sheds light on mechanisms of amyloid assembly. Prion 3, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heegaard N. H.; Jørgensen T. J.; Rozlosnik N.; Corlin D. B.; Pedersen J. S.; Tempesta A. G.; Roepstorff P.; Bauer R.; Nissen M. H. (2005) Unfolding, aggregation, and seeded amyloid formation of lysine-58-cleaved β-2-microglobulin. Biochemistry 44, 4397–4407. [DOI] [PubMed] [Google Scholar]
- Mendoza V. L.; Baron-Rodriguez M. A.; Blanco C.; Vachet R. W. (2011) Structural insights into the Pre-amyloid tetramer of β-2-microglobulin from covalent labeling and mass spectrometry. Biochemistry 50, 6711–6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.