

Abstract
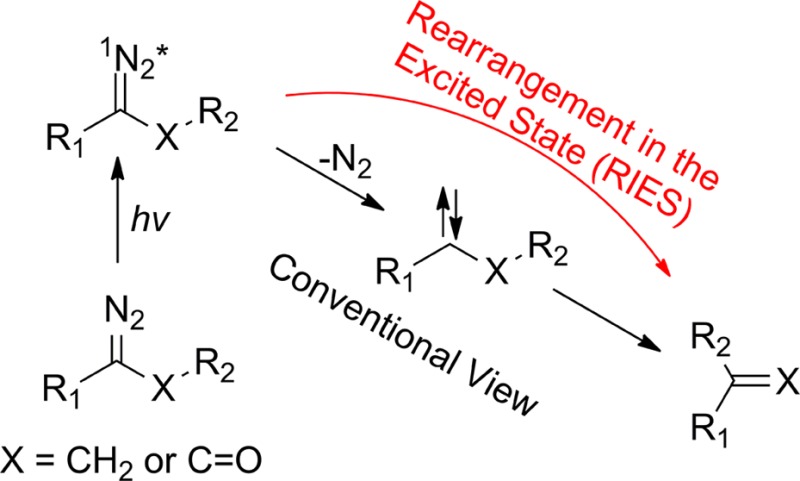
A perspective on the development of mechanistic carbene chemistry is presented. The author will point out questions that have been answered, and a next generation of questions will be proposed.
I. Introductory Remarks
I feel incredibly honored to be named the recipient of the 2014 James Flack Norris Award in physical organic chemistry. I also feel very much humbled to see my name associated with previous award winners, particularly my mentors Jerome Berson and Gerhard Closs and P.D. Bartlett, the mentor of Shelton Bank, who more than anyone else has been my role model as an educator.
I feel tremendously indebted to my wonderful students whose efforts are actually being recognized by this award. It is also a pleasure to gratefully acknowledge the National Science Foundation for support of my undergraduate and graduate research, for a postdoctoral fellowship, and continuous support of my laboratory in Columbus since 1979. As NSF Division Director, I was incredibly impressed by the work ethic and professionalism of the Program Officers, the men and women who are the unsung heroes of the US research enterprise. Finally, I want to thank special collaborators, friends, and unofficial teachers over the years, especially Thomas Bally, Barry Carpenter, Wes Borden, Nina Gritsan, Christopher Hadad, Maitland Jones, Eva Migirdicyan, Josef Michl, Robert Moss, Tito Scaiano, and Jakob Wirz, all of whom pushed me to be the best chemist I could be. I also want to thank The Journal of Organic Chemistry for giving me the opportunity to publish this Perspective. I will try to point out some contributions from our joint efforts that I am particularly proud of contributing to the field of carbene chemistry. This is not a comprehensive review but is rather a personal memoir describing the influences behind the experiments we performed and their historical context. I also want to point out how our knowledge of carbenes has grown since I started paying attention, how the everyday tools used by reactive intermediate chemists have changed, and mostly note the questions that have been answered and point out some new questions of interest.
II. Student Days
Of course, the field of carbene chemistry predates my own awareness of it by at least a century. By the time I was a freshman in college, the classic kinetic work of Hine1 on the hydrolysis of chloroform had been published and had made its way into the introductory organic textbooks of my youth. This work led to the postulation of dichlorocarbene as a reactive intermediate. Doering and Hoffmann2 would soon intercept this transient species with cyclohexene to form a geminal dichlorocyclopropane. This discovery led to a new synthetic method that has been refined and brought to widespread use around the world. I still find it noteworthy that a curiosity inspired study of the kinetics of hydrolysis of chloroform would lead directly to a now standard synthetic method. Additional mechanistic studies by Doering,3 Skell,4 Closs,5 Moss,6 and Jones7 provided a sturdy mechanistic foundation for solution-phase carbene chemistry which has stood the test of time and which has suffered no serious challenge after decades of study by physical and theoretical methods.
My first formal exposure
to chemistry was in a high school course.
My teacher, Alexander Goros, was a stern and exceptionally demanding
and effective individual. I still do stoichiometric calculations the
way he taught the method. My first exposure to carbene chemistry came
during my freshman year (1969–1970) at the State University
of New York at Albany (now the University
at Albany). I was a chemistry major matriculating through the experimental
George Hammond curriculum developed at Caltech. Henry Kuivila (a Mel
Newman mentored graduate of Ohio State!) taught the class about hybridization.
Based on the best spectroscopy of the time,8 Professor Kuivila informed us that triplet methylene
was linear and was an exemplar of sp hybridization, with two singly
occupied p orbitals. This example also illustrated the extension of
Hund’s Rule to molecules and introduced the class to the Pauli
Principle and its underlying principles based on electron–electron
repulsion.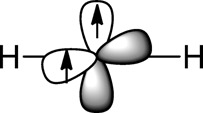
In my sophomore year, I came under the influence of a charismatic Professor, Shelton Bank, who in the language of the day “turned me on” to the study of organic reaction mechanisms. He gave me the opportunity to work in his laboratory, and I became fascinated by the thought that one could actually understand and control the reactions of organic molecules swirling in a round-bottom flask. I learned that I enjoyed lab work and that I could go to graduate school tuition free and receive a $300/month stipend as well. It turned out to be the best deal I would ever be offered and led me to New Haven, CT, to further my education in physical organic chemistry.
As a graduate student at Yale, I heard Ed Wasserman lecture on exciting new ESR experiments that demonstrated that triplet methylene has a bond angle of 136°, a result in line with qualitative VSEPR theory.9 I learned to love simple qualitative arguments like VSEPR and, more importantly, that disagreements between simple arguments and sophisticated calculations almost always resolve in favor of the former. This lesson also applies to university administration but, sadly, not to government service.
In my doctoral work, I studied trimethylenemethane biradicals under the tutelage of Jerome Berson and was greatly influenced by the work of the Bell Laboratories team10 (Wasserman, Trozzolo, Roth, and others) and the Closs11 group at Chicago. Their pioneering use of physical methods to study carbenes had a profound influence on my development as a scientist.
At one of the legendary Berson Monday night group meetings, I became
aware of the work of the Lineberger group12 and the controversy surrounding the singlet–triplet splitting
of methylene. This had great relevance to my thesis work,
and at one point, I felt that I had “disproved” some
calculations of Wes Borden. Further experimental work demonstrated
that the original results, although correct, had been misinterpreted
by me. This became clear in the hindsight of later experimental work,
which produced completely unexpected findings. As I look back forty
years later, it is clear that a simple density functional theory (DFT)
calculation, a now standard tool used by advanced undergraduates,
would have prevented my misinterpretation of the data.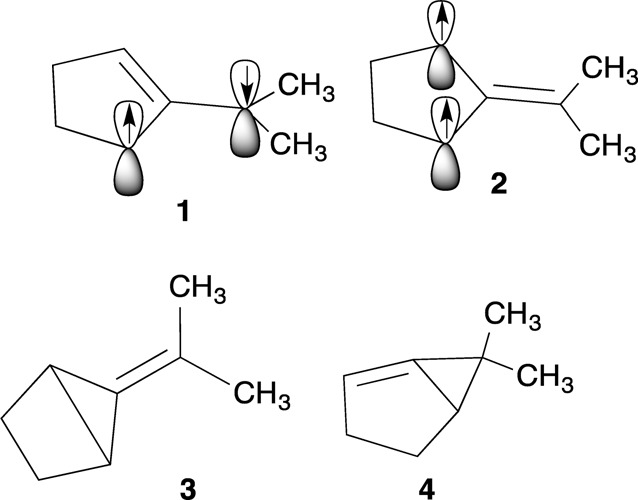
Forty years ago, theory could not accurately predict the energies of 1–4 and point out then (as opposed to now) that 3 and 4 could serve as a reservoir for 1 under certain conditions.13 With the benefit of hindsight, then as now, it is very easy to uncritically love ones own ideas and to be wishful.
Today, calculations are a necessary and required part of presenting a mechanism, not unlike the need to obtain a melting point or NMR spectrum of a new compound. Theory does not prove a mechanism any more than does experimentation, but like experimental work, it can now disprove many mechanistic hypotheses by revealing that the thermodynamics and/or kinetics of a proposed mechanism are nonsensical. Calculations should also be performed before conducting many experiments by demonstrating that the desired results have plausible kinetics and/or thermodynamics.
One last remembrance of graduate school days was my sense of frustration that there were no experimental tools available to directly observe singlet biradicals such as 1. In addition, although it was possible to detect triplet biradicals such as 2 by low-temperature EPR spectroscopy, this tool could do little more than confirm ground-state multiplicity and provide some limited structural insights. It could not, for example, give information on the rate of intersystem crossing (ISC) or of bimolecular reactions of ground-state triplet species. The tools needed to answer those questions would eventually be found in picosecond time-resolved optical methods.
Following a year and a half of postdoctoral work with Gerhard Closs, I joined the faculty of The Ohio State University in 1978. I learned later that my chief attribute as a job candidate was that I wanted to make heavy use of an idle ESR spectrometer! I was far from being considered the best and brightest job candidate in the class of 1978 and will ever be grateful to OSU for giving me an opportunity.
In the Closs Laboratory, I studied the triplet states of chlorophylls by NMR line broadening techniques. As a newly minted Assistant Professor, I hurried back to my first love, the study of triplet biradicals by ESR spectroscopy.
III. Carbenes as a Source of Biradicals?
While my group was still small I had the time
to synthesize 5 hoping that it would form carbene 6 and ultimately
biradical 7. At that time I viewed triplet carbenes as
a novel source of more interesting biradical species.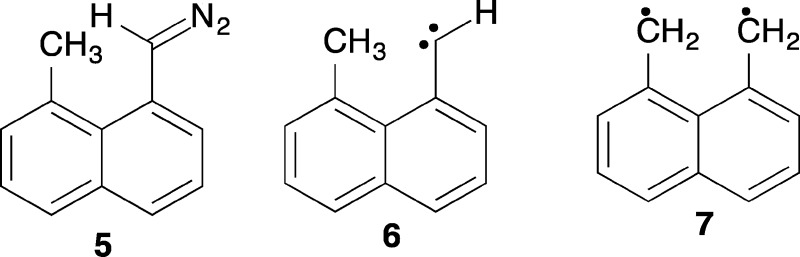
Photolysis of 5 at 77 K did indeed produce the EPR spectrum of 7. Triplet carbene 6 was not observed, even at 4 K, and the formation of 7 was “instantaneous” on human time scales at all temperatures.14 I found the fast rate of this hydrogen transfer intriguing, and a review of the literature revealed that the absolute rates of triplet carbene H-atom transfer reaction rates were essentially unknown in the condensed phase. I then undertook such studies in frozen solids, believing it to be a temporary detour from the study of biradicals.
We found that we could use EPR spectroscopy to follow the rate of H-atom transfer processes in frozen matrices such as that shown in eq 1.15a
 |
1 |
The temperature dependence of the kinetics, the H/D isotope effects, and prior studies of Ffrancon Williams and co-workers15b convinced us that the H atom transfer process was an example of hydrogen atom tunneling. In collaboration with Bill McCurdy, we modeled Brad Wright’s kinetic data and predicted that the classical activation energy of the process would be on the order of 6.9 kcal/mol in the example shown above.16 To test that prediction, we would need to study the same reaction in fluid solution, where presumably, quantum mechanical tunneling (QMT) effects would be less important.
About that time, Tito Scaiano and Dave Griller17 at the NRCC in Ottawa and Bob Moss (Rutgers) and Nick Turro (Columbia)18 were pioneering the use of nanosecond (ns) time-resolved laser flash photolysis methods for the study of bimolecular reactions of carbenes in their ground states (ground triplet states for aryl carbenes, ground singlet states for arylhalo carbenes). These two teams had clearly ushered in a new era in the study of carbenes, an exclusive club that I very much wanted to join.
Having spent a bit of time with Tito Scaiano in his Notre Dame days (when I was close by in Chicago), it was quite natural for me to reach out to him in Ottawa for the purpose of collaboration. It was also convenient to drive to the home of my in-laws in upstate New York, kiss my wife and children a goodbye, and continue on to Ottawa for hands on research. Linda Hadel and other students would later visit Ottawa for longer periods of time to complete a number of projects. I am extremely indebted to Tito Scaiano, who taught me everything I know about ns time-resolved spectroscopy. His patience and expertise allowed me to establish this technology in my laboratory.
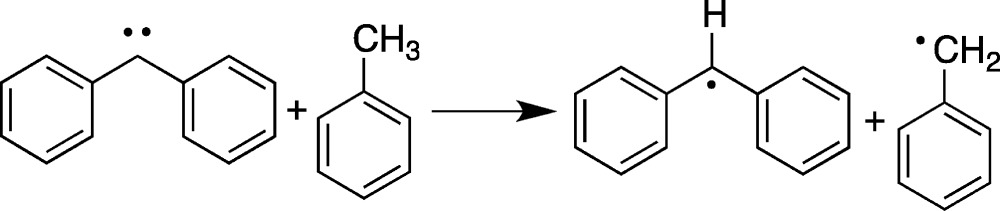
The activation energy for the reaction of triplet diphenylcarbene and toluene in solution at temperatures near ambient was found to be 3.2 kcal/mol, in only fair agreement with McCurdy’s QMT calculations.19 Later work would show that QMT also contributed to this reaction, even in fluid solutions at ambient temperature, depressing the experimental result relative to a QMT free process.20
For over two decades, the Moss laboratory21 and my own22 have used ns time-resolved spectroscopy to mainly study the reactivity of carbenes in their ground states. Although fascinating, I had the same frustrations as a graduate student. The ns time-resolved spectroscopic methods were still “too slow” to allow direct observation of the singlet states of carbenes, where the triplet is the ground state. It was therefore impossible to systematically study singlet to triplet intersystem crossing (ISC) or the wonderful rearrangements of simple singlet carbenes (eq 2).
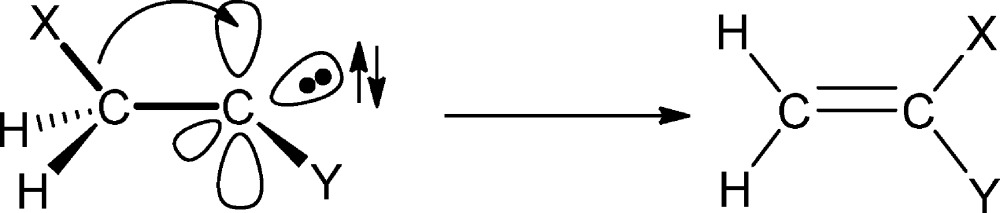 |
2 |
In the mid twentieth century, chemists discovered that the mode of generation of “carbenes” has a large impact on the mixture of persistent products that are formed, as illustrated in Table 1.23
Table 1. Experimental Product Distribution for the Decomposition of Nitrogenous Precursors of Ethylmethylcarbene.
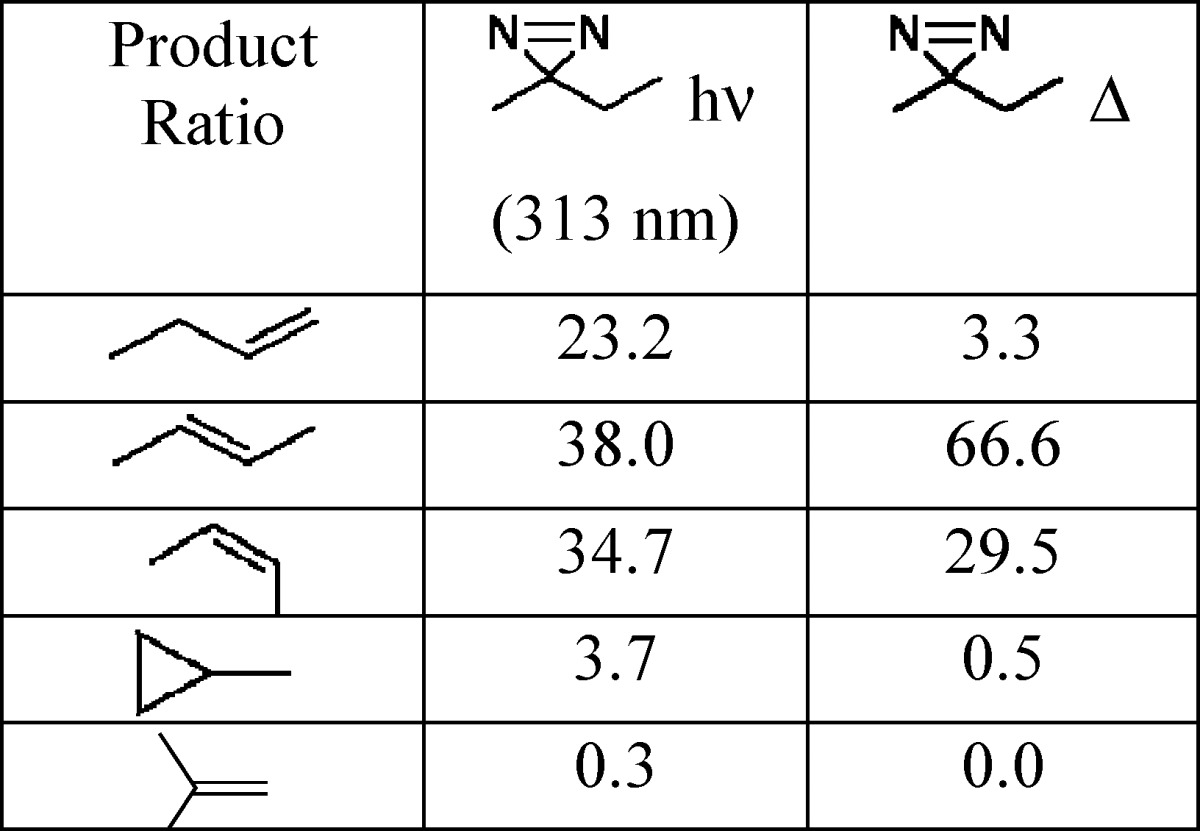
A quick inspection of Table 1 reveals that different “ethylmethylcarbenes” (EMCs) are produced (as measured by their chemistry) depending on whether they are generated thermally or photochemically. DFT calculations are in excellent agreement with the finding that thermolysis of precursors produces the thermodynamic mixture of products. Clearly, something remarkable is happening upon photolysis of the precursors, but ns spectroscopy was again too slow to contribute to the resolution of this mystery. Progress in understanding this dichotomy would require pico (ps) and femtosecond (fs) time-resolved methods, along with the development of new theoretical methods
Terry Gustafson, Bern Kohler, Christopher Hadad, and Claudio Turro led the effort to establish the Center for Chemical and Biophysical Dynamics (CCBD) at The Ohio State University. I thank these colleagues and many students, especially Dr. Gotard Burdzinski, for building the CCBD. I am particularly grateful to Gotard, Dr. Jin Wang, Dr. Yunlong Zhang, and Dr. Jacek Kubicki for the experimental work discussed in the remainder of this perspective, which finally allowed our investigation of ISC and singlet carbene rearrangements. Of course, I also want to thank the NSF and The Ohio State University for the financial support needed to establish the center. In the interest of full disclosure, I must confess that portions of our ultrafast work have been previously reviewed by the author and passages in this memoir will necessarily be very similar to those in previous reviews of the same topics.
IV. Influence of Solvent on Carbene Intersystem Crossing Rates
Ironically, I joined the Closs group as a postdoctoral student (1977–1978) after the peak of his research activity in carbene chemistry. Closs was a gifted teacher, and he used the singlet/triplet interconversion and spin specific reaction products model, developed for carbenes, to teach the basic principles of chemically induced dynamic nuclear polarization (CIDNP) in his group meetings. Later, in many late night beer-fueled conversations in smoked-filled bars, in the 1980s, Closs would often tell me how mysterious he found “spin flips” in general and intersystem crossing in particular. I have felt the same sense of mystery throughout my career as well.
The first systematic study of carbene singlet to triplet ISC absolute rates was reported by Eisenthal and co-workers for diphenylcarbene, a molecule which has a triplet ground state.24 In this pioneering work, a linear dependence was found between the log of the first-order rate constant of ISC versus the Dimroth ET(30)25 parameter. This study utilized a variety of nonhalogenated solvents. The fastest ISC rates were observed in nonpolar solvents with correspondingly small ET(30) values. In nonpolar solvents, the singlet–triplet (S–T) energy gap is relatively large because the zwitterionic closed-shell singlet carbene is poorly solvated relative to the biradical-like triplet spin isomer. Thus, the fast ISC rate in nonpolar solvents, where the singlet is preferentially stabilized and the S–T gap is small, apparently contradicts the “Golden Rule”26−28 of radiationless transitions, which states that smaller energy separations promote faster radiationless transitions. Eisenthal and co-workers accepted that polar solvents preferentially stabilize the singlet state relative to the triplet state of the arylcarbene, as confirmed later by theoretical29 and experimental studies.30 To explain the slower than expected ISC rates in polar solvents, the Eisenthal group concluded that the diphenylcarbene (DPC) system fortuitously suffered from poor S–T vibronic coupling.
Thus, once we had ready access to ultrafast time-resolved spectroscopy in Columbus, the Eisenthal experiments were among the first I wanted to revisit and expand upon. In addition to diphenylcarbene (DPC), we also studied its close relative fluorenylidene (FL), which also has a triplet ground state.10,11 In the 1970s. it was thought that singlet to triplet ISC in DPC should be faster than in FL because the latter molecule is rigidly planar. There was speculation that bond angle deformation or ring rotation in DPC might couple to ISC and accelerate the radiationless transition.31 This speculation was fueled by classic experiments of Closs and Closs5 which indicated that in DPC, spin interconversion was faster than bimolecular chemistry. It was also stimulated by the classic work of Jones and Rettig7 with FL, in which they showed that dilution with an “inert” solvent, hexafluorobenzene, allowed singlet FL (unlike DPC where there was no dilution effect) time to relax to the lower energy triplet.
Our work has demonstrated that rates of ISC do not always correlate with bulk solvent polarity. Rather, ISC rates are impacted by specific carbene-solvent interactions. We presented two hypotheses32−35 to explain the observed deceleration of the rate of ISC for FL (and DPC) in their lowest, closed-shell singlet configurations in solvents containing atoms with nonbonding pairs of electrons (Figure 1).
Figure 1.

Intersystem crossing rate of a closed-shell singlet carbene is retarded by (top) solute–solvent interactions producing a Franck–Condon-like factor or (bottom) surmounting of an energy barrier to achieve desolvation to release “free” singlet carbene prior to ISC (see ref (32)).
In the case of a coordinating solvent such as acetonitrile, as shown in Figure 1, the solvation of the singlet and triplet carbenes is significantly different. We posited that this could produce a Franck–Condon-like factor (Figure 1 top). Alternatively (Figure 1 bottom), we speculated that the origin of the effect might resemble the well-known ability of protic solvents to depress SN2 reaction rates relative to dipolar aprotic solvents.25 This may be another example where the rate-limiting step might be the surmounting of a small energy barrier to release “free” carbene, prior to the ISC process. Table 2 reveals that the ISC rates in cyclohexane or benzene, two nonpolar, zero dipole moment solvents are larger than those measured in acetonitrile. Much to our surprise, another slow ISC rate, comparable to acetonitrile, was found7,11 in a halogenated solvent (hexafluorobenzene) which has zero dipole moment. To explain this result, we posited the formation of an ylide-like complex formed by a bonding interaction between the empty p orbital of the singlet carbene with a nonbonding pair of electrons of the halogen. Turro and co-workers first proposed this type of solvation and ISC rate retardation for the simplest singlet carbene, methylene.36,37 A different but related type of interaction is possible in aromatic solvents: the formation of a π complex stabilizing the singlet carbene.
Table 2. Solvent-Dependent Singlet Carbene Lifetimes (Inverse Intersystem Crossing Rates)32−34 and Associated ET(30) Parameters25,35.
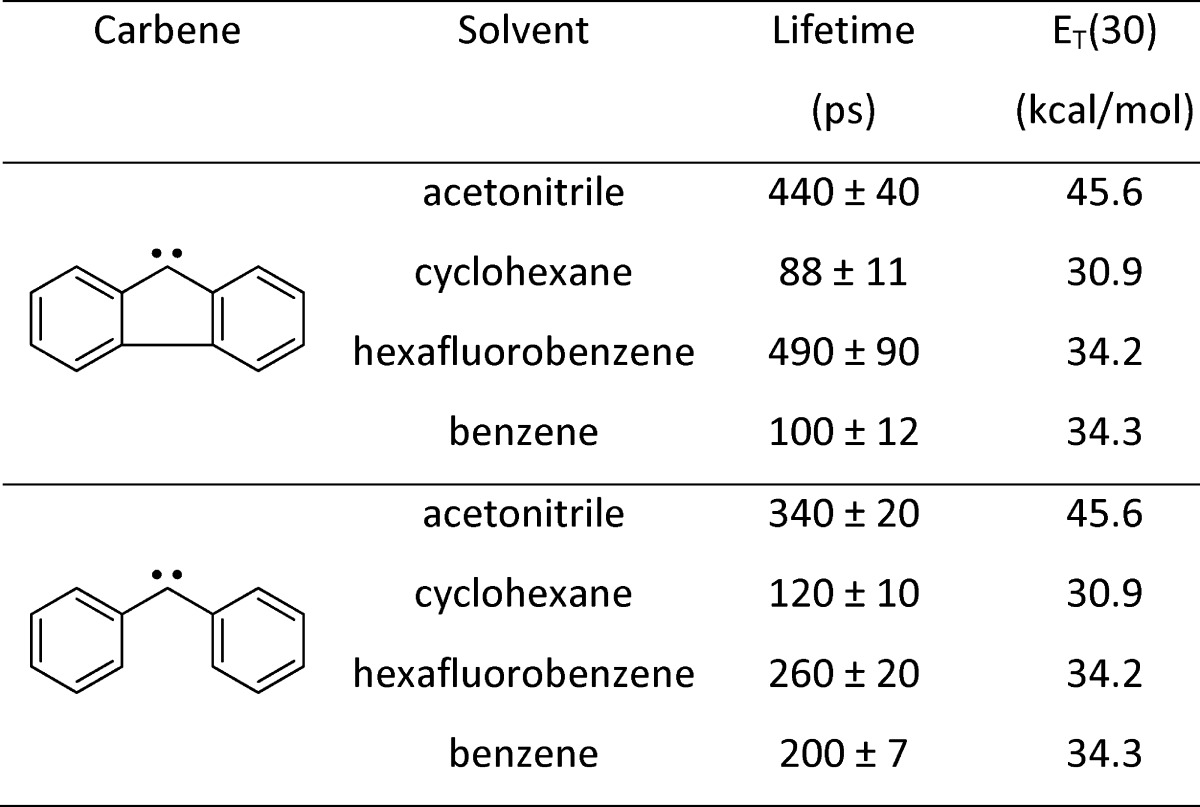
We wish to note that in the cases of singlet diphenylcarbene and fluorenylidene, ISC to the ground triplet state is the main deactivation pathway in these solvents. Thus, we are confident that the decay rates are indeed the ISC rates.7,11
Our ultrafast studies have led to a more sophisticated view of ISC in aryl carbenes. The flexibility of the carbene is not a factor in controlling the rate as posited by Salem and Rowland.31 The difference in the Closs and Closs5 and Jones and Rettig7 dilution experiments is based on the difference in bimolecular reactivities of DPC (slow-sterically encumbered) and fluorenylidene (fast, unencumbered) rather than ISC rate differences. It is also clear that there is no violation of the “Golden Rule” of radiationless transitions. The data show that weak solvent–solute interactions impact carbene ISC rates. To my knowledge, theory has not been successfully applied to describe these interactions. In fact, attempts to do so usually show no enthalpic barrier between solvent–solute interaction and exothermic product formation. My intuition tells me that these interactions have many practical consequences for carbene chemistry and are much in need of more attention. As an aside, Bill Doering once told me that in the early days of modern carbene chemistry, he felt that the product distribution obtained by reaction of an alkene with a carbene was “steered” by weak interactions between the reactants. Our data suggest that weak solvation has an impact as well. Support for this idea has been provided recently by Moss and co-workers in their studies of carbene complexations with arenes.6
Thirty years ago, the assignment of transient spectra to certain carbenes required either courage or wishfulness, depending on your point of view. Today, transient spectra can be assigned with considerable confidence to the lowest energy states of singlet and triplet carbenes thanks to improvements in computational chemistry. Time-dependent density functional theory (TD–DFT) calculations allow the assignment of transient absorption spectra to singlet or triplet carbenes in a reliable manner.
During my Assistant Professor days, the assignment of spectra to ground-state triplet carbenes was confirmed using cryogenic matrix isolation experiments or by the shortening of transient lifetimes in the presence of oxygen in nanosecond laser flash photolysis experiments. Today, the shortening of singlet carbene lifetimes in neat alcohols is the “test”, along with TD DFT calculations, that confirms the assignment of transient absorption bands to the singlet state of a carbene. Assigning transient bands in the 21st century can be done with more success then was possible in the 1970s.
V. Dynamics of Carbene Vibrational Cooling and Solvation
Diazo and diazirine compounds are commonly employed photochemical precursors of carbenes. In these cases, the resultant carbenes are often born with an excess of vibrational energy (#). The “hot” carbenes undergo subsequent cooling upon collision with nearby solvent molecules. This is termed “vibrational cooling” and proceeds with a time constant of 5–30 ps (ps).35 The observation of carbene absorption band narrowing (in both the UV–vis and IR transient absorption experiments) and a blue shift of the absorption maximum announces the vibrational cooling of vibrationally excited intermediates.35
The photochemistry of p-biphenylyltrifluoromethyldiazomethane BpCN2CF3 was studied in our laboratory because it was not expected to undergo 1,2 F shift reactions. These studies were motivated by a desire to further understand the photochemistry of BpCN2CH3, to be described later. Serendipitously, these experiments led us to what we felt was a more interesting effect (eq 3).38
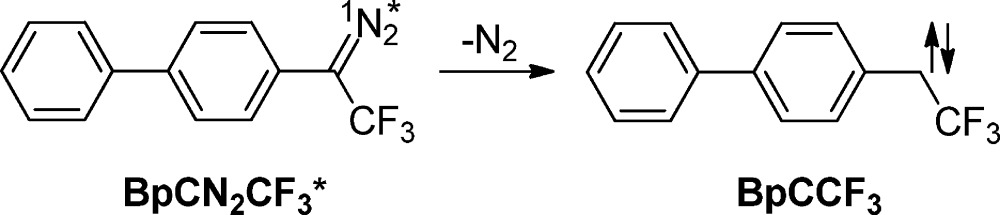 |
3 |
A pattern typical of vibrational cooling was observed in nonpolar solvents such as cyclohexane (Figure 2A). A very different pattern was observed in the polar solvent methanol (Figure 2B), which we attributed to the dynamics of solvation. The observation of a red shift of the carbene absorption band, typically over 1–15 ps in UV–vis transient absorption experiments, was assigned as the time required to solvate the carbene formed within the 300 fs laser pulse.
Figure 2.
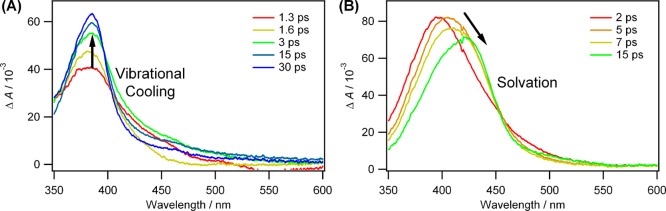
Transient absorption spectra recoded in (A) nonpolar cyclohexane and in (B) polar and coordinating methanol solvent. The spectra were generated by photolysis (λexc = 308 nm) of p-biphenylyltrifluoromethyldiazomethane (see ref (38)).
Carbene solvation rates were similarly studied in solvents of differing polarities, including hydrogen-bonding solvents,38 using p-biphenylyltrifluoromethylcarbene (BpCCF3) and ultrafast time-resolved techniques. The initial carbene absorption band in acetonitrile undergoes a red shift from 400 to 410 nm with a time constant of 0.4 ps. In methanol, which can participate in hydrogen bonding, the red shift is more dramatic (from 394 to 427 nm). The dynamics of solvation are slower in the hydrogen-bonding solvent, with a time constant ∼10 ps (Figure 2B). The singlet carbene BpCCF3 has a closed-shell electronic structure with a filled and an empty nonbonding orbital on the carbene carbon. The trifluoromethyl group makes the carbene center even more electron deficient. This increases the strength of the interaction between the empty p-orbital of the carbene and the nonbonding electrons on the nitrogen atom of acetonitrile. The red shift is more dramatic in methanol than in acetonitrile, because of the greater polarity in the former solvent. However, we believe that this is mostly because the alcohol solvent will have bonding interactions with both the filled and the empty nonbonding orbitals on the carbene carbon (eq 4).
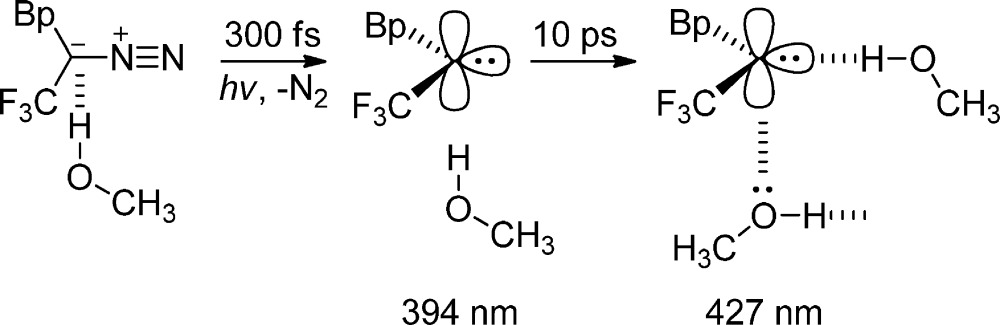 |
4 |
The nonbonding electrons of the oxygen atom of the alcohol will have a bonding interaction with the empty orbital of the singlet carbene. The acidic alcohol proton (of the same or yet another alcohol molecule) will form a hydrogen bond with the filled in-plane nonbonding orbital of the carbene. The kinetic O–H/O–D isotope effect on the red shift in alcohol solvents demonstrates the formation of a new hydrogen bond, in our opinion. There is a linear dependence between solvent viscosity and the time constant of the solvation shift (from ten to hundreds of ps, Figure 3) over a homologous series of alcohols.
Figure 3.

Dependence of solvation shift time constant of p-biphenylyltrifluoromethylcarbene on alcohol solvent viscosity. (Reprinted with permission from ref (38). Copyright 2008 American Chemical Society).
Intuition suggests that solvent viscosity plays
an important role
in the rate of solvation in the manner observed, the more viscous
the solvent, the longer the time constant of solvation. A similar
pattern was realized for 2-trifluoromethylfluorenyl (FlCCF3), which is a rigid analogue of BpCCF3. This demonstrates
that torsional motions of the two phenyl rings of the latter carbene
do not explain the observations we prefer to assign to solvation dynamics.38
This is one of the many unexpected results obtained in the ultrafast time-resolved experiments we encountered. To be honest, over the course of this research, I often felt more like an explorer then a scientist. It was great fun to make new precursors and simply see what we would discover upon their study by ultrafast techniques, rather than to synthesize a compound to precisely answer a specific question.
VI. Carbonyl Carbenes
VI.1. Concerted or Stepwise Wolff Rearrangement (WR)?
Aryl carbonyl carbenes have been a rich area of study for ultrafast time-resolved spectroscopy because both UV–vis and IR spectroscopies can be usefully employed. In these studies, we found that solvation influences the rate of a chemical process (WR) in a manner reminiscent of its influence on ISC rates. This will be discussed in the next section, after a quick recap of ultrafast time-resolved observations and quantum yields of two carbonyl carbenes.
In the early years of the 20th century, it was discovered that diazo carbonyl compounds will extrude nitrogen upon exposure to heat or light. Ketenes are commonly formed upon decomposition of diazo carbonyl compounds. This reaction is known, of course, as the Wolff rearrangement (WR).39 Kirmse has relatively recently published a comprehensive review of the WR process. We have more recently reviewed the ultrafast time-resolved studies of diazo carbonyl photochemistry.40
α-Diazo carbonyl compounds usually have a planar configuration of the O=C–C=N2 group.40 There are two conformers, syn and anti, as shown in Scheme 1.
Scheme 1. Stepwise and Concerted Photoinduced Wolff Rearrangement Processes.

Stepwise and concerted mechanisms have been proposed for photoinduced WR in α-diazo carbonyl compounds (Scheme 1).8 In 1966, Kaplan and Meloy proposed that ketene formation, in concert with nitrogen extrusion, is favored upon decomposition of the syn conformers of α-carbonyl compounds.9 The anti conformers, however, prefer to decompose to form trappable carbenes. These carbonyl carbenes may subsequently isomerize to ketenes or can be consumed by other decay routes. The Kaplan–-Meloy rule has been well supported since its inception (mainly based on chemical analyses of stable photoproducts).
The photochemistry of acyclic aryl diazo ketone 8 and ester 9 (eq 5)40−42 were studied by time-resolved UV–vis and IR spectroscopies. The expected ketene (2020 cm–1) was formed in both a fast (τ < 0.4 ps) process from the diazo ketone excited state 8 (concerted WR) and in a slower process (700 ps) from the relaxed singlet keto carbene.
Vibrational cooling of the ketene was evident by band narrowing and a blue shift to 2110 cm–1 over a 50 ps time window. The precursor of the hot ketene was assigned to the singlet diazo excited state 18*based on the following logic. The 18* transient species is observed at 450 nm and decays with a time constant shorter than 300 fs. In addition, the decay of 18* is accompanied by the growth of the singlet carbene, observed at 380 nm. The carbene band decays with an 800 ± 100 ps time constant. This fact is in excellent agreement with the value of the “long” time constant (700 ps) of ketene formation. The observations confirm that the quantum yield of the stepwise WR process is about the same as the faster concerted WR process.40−42
A different story is told by acyclic aryl diazo ester 9. Photolysis of this precursor only produced the carbene. In fact, we found no evidence of fast ketene formation from the diazo ester excited state (eq 5.40−42
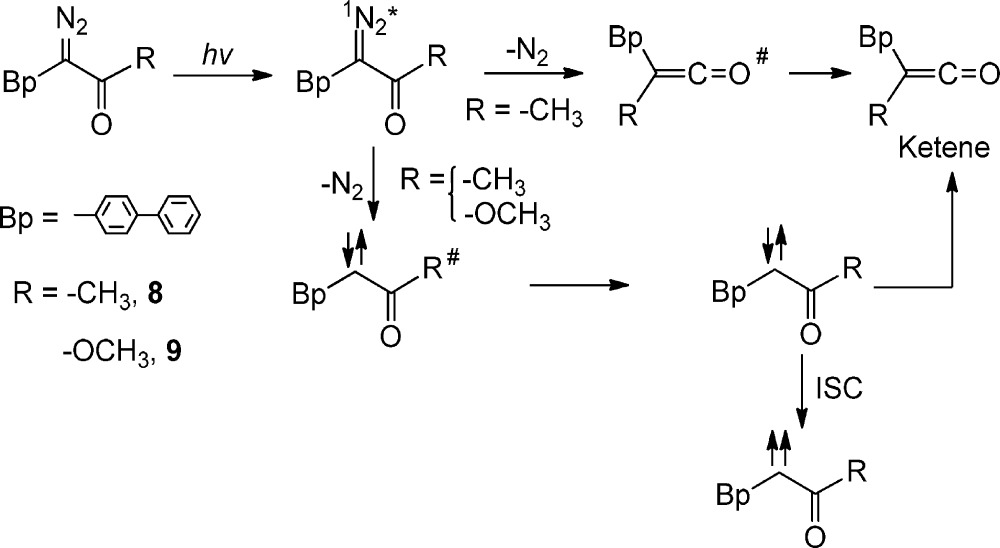 |
5 |
Ultrafast time-resolved photolysis of p-biphenyl diazo ketone 8 and ester 9 produces the readily observable transient absorption of their respective diazo excited states. In one aspect, the photochemistry is similar: the optical yield of the excited diazo ester and ketones are about the same. However, the transient absorption of the singlet carbene ester is about twice that of the singlet keto carbene.40−42 It seems unlikely that this is due to large differences in carbene extinction coefficients as TD-DFT calculations predict that the molar absorptivities of the two carbenes are similar. It seems reasonable then that the quantum yield of carbene ester formation is twice that of the related keto carbene. We explain the lower yield of singlet keto carbene relative to carbene ester as a consequence of more efficient WR in the diazo ketone excited state. This is completely consistent with the previously described IR experiment (eq 6).40−42
 |
6 |
Tomioka and co-workers had previously come to similar conclusions in their studies43 of the parent phenyl system. Our chemical analysis of reaction products derived from the biphenyl diazo carbonyl compounds told the same story as once again there was no evidence of ketene formation from photolysis of the diazo ester.
To explain the observations, we speculated that diazo ester excited states and singlet carbene esters are less prone to WR rearrangement, relative to ketone analogues, because of ester resonance. DFT calculations predict that the barrier to WR of the relaxed singlet biphenyl ketocarbene is 4.4 kcal/mol while that of the corresponding ester is 8.9 kcal/mol. The loss of ester resonance makes the rearrangement of an ester less exothermic than the rearrangement of the corresponding ketone. Classical physical organic reasoning predicts that the ester rearrangement will therefore be somewhat slower than the more exothermic ketone analogue.40−42 Analogous calculations for the diazo carbonyl excited-state concerted WR processes have not been reported and would certainly be most welcome.
VI.2. Solvent Effects on a Carbene S–T Gap and ISC Rates
The lifetime of p-biphenyl keto carbene (Ar-C-COCH3) is 180 ± 20 ps in cyclohexane and is 700 ± 30 ps in acetonitrile. In both solvents, the dynamics are controlled largely by the WR process.40−42 A traditional explanation immediately suggests itself; the polar solvent better stabilizes the reactant than the transition state. Nevertheless, the time constant of WR in dichloromethane, which has a much lower dielectric constant than acetonitrile, is also long: 770 ± 40 ps.
The impact of a solvent atom bearing a nonbonding pair of electrons on the intersystem crossing (ISC) rates of an aryl carbene was discussed earlier and followed exactly the same pattern as the WR solvent dependence data. Thus, we again propose that a desolvation effect precedes and effectively retards the rate of Wolff rearrangement (eq 7).
 |
7 |
Systematic studies of solvent effects on the ISC rates of carbonyl carbenes reminiscent of fluorenylidene immediately suggested themselves. Unfortunately, they could not be performed because the nature of the carbonyl carbene ground state is solvent dependent and also because of a geometry change involved in the ISC of carbonyl carbenes. It is clear from experiment and DFT calculations that keto carbene (Ar–C–COCH3) has a singlet ground state in cyclohexane, dichloromethane, and acetonitrile, thereby preventing measurement of the time constant of singlet to triplet relaxation.40−42 On the other hand, the carbene ester (Ar–C–CO2CH3), is predicted by theory to have a triplet ground state in cyclohexane but a singlet ground state in dichloromethane and acetonitrile. Indeed, these predictions were confirmed by time-resolved spectroscopy.
The “orthogonal” structure of the
singlet carbene
allows conjugation between the filled nonbonding orbital of the carbene
with the π system of the carbonyl group.40
In the case of the ester carbene, the conjugation is weaker due to the internal resonance of the ester moiety. Students of sophomore organic chemistry are taught this effect in a different context. Thus, the same structural factor that makes methyl ketones more acidic than methyl esters stabilizes and favors singlet keto carbenes relative to carbene esters in their ground states.
The orthogonal structure of the singlet carbene also impacts ISC rates. The singlet to triplet ISC rate of (noncarbonyl) carbenes in cyclohexane40−42 is twice that of the p-biphenyl carbene ester (Ar–C–CO2CH3). The orthogonal singlet carbene ester is lower in energy than the “orthogonal” triplet. ISC of the relaxed singlet to the orthogonal triplet is actually endothermic. In order to relax to the lower energy triplet carbene, the singlet carbene must rotate to a higher energy structure. Theory predicts that the singlet and triplet surfaces (of the parent phenyl analogue) become degenerate when the plane of the ester is 40° relative to the plane defined by the carbonyl carbon, the carbene carbon and the carbon of the phenyl ring. The surfaces cross 6.2 kcal/mol above the orthogonal singlet. The energy cost to rotation of the singlet carbene, to achieve a geometry isoenergetic with a triplet carbene, effectively provides a barrier to ISC.
Thus, this is an example of carbene motion controlling an ISC rate, as posited by Salem and Rowland,31 but it is not due to coupling a radiationless process to a physical motion. It is a physical motion that changes the relative energies of singlet and triplet to access a geometry where the two spin states are energetically degenerate. This provides a pathway to the lowest energy state and offers a relaxation pathway out of the lowest energy geometry of an excited state.
I must confess that none of the questions posed and answered in this section are original. The experimental technology needed to do these experiments has only recently become available to the nonphysicist, and it was my good fortune to have early (for an organic chemist) access to this instrumentation. But the interpretation of the data was critically dependent on advances in theory. In the last century, it was just a dream to be able to accurately predict absorption maxima, molar absorptivities, and singlet–triplet gaps in the gas phase and as a function of solvent.
VI.3. 2-Naphthylcarbomethoxycarbene
Aryl diazo excited states decompose within 300 fs. Thus, it seems likely that the corresponding singlet carbonyl carbenes are born in the planar geometry of their diazo precursors (eq 8.44a
 |
8 |
The IR spectrum of the singlet carbonyl vibration undergoes a red shift, whereas the C=C vibration blue shifts, as is typical of vibrational cooling. We propose that the initially populated planar diazo-excited state fragments in 300 fs to form the planar singlet carbene, which subsequently relaxes over 3 ps to form the orthogonal singlet. Simply put, the diazo excited state does not undergo geometric change during its short (<300 fs lifetime) and the initially formed planar singlet requires 3 ps to rotate the ester group by 90°. The relaxed, orthogonal singlet undergoes slow (ns) ISC in cyclohexane because of the mismatch of the singlet and triplet geometries, necessitating a rotation of the ester plane of ∼80° to become energetically degenerate with the triplet carbene (eq 9).
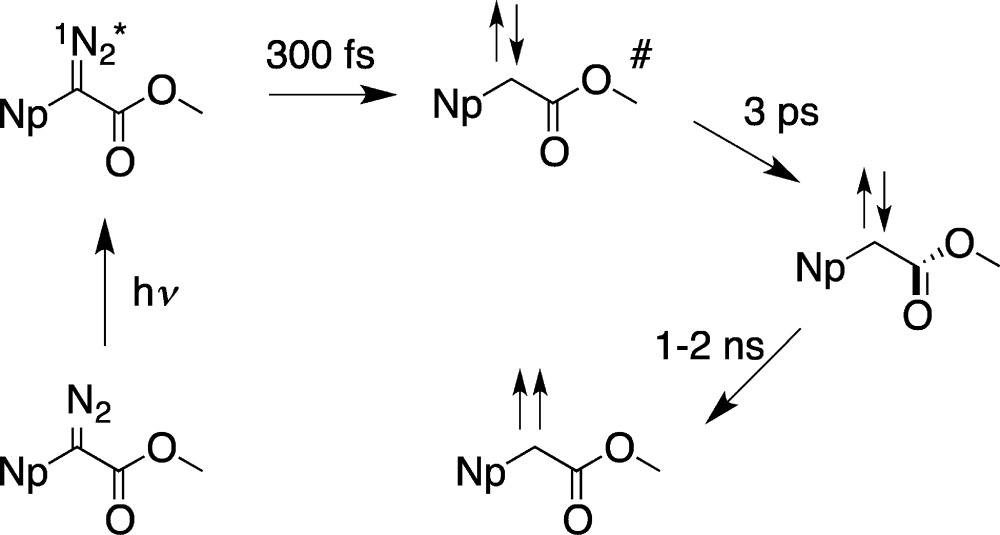 |
9 |
VI.4. Carbonyl Carbene–Carbene Isomerization
Oxirenes have intrigued chemists for decades. Calculations suggest that oxirenes mediate the isomerization of carbonyl carbenes.40 We demonstrated this isomerization process with photoexcited 18* and 110* using femtosecond time-resolved UV–vis and IR transient absorption spectroscopy (eq 10).45
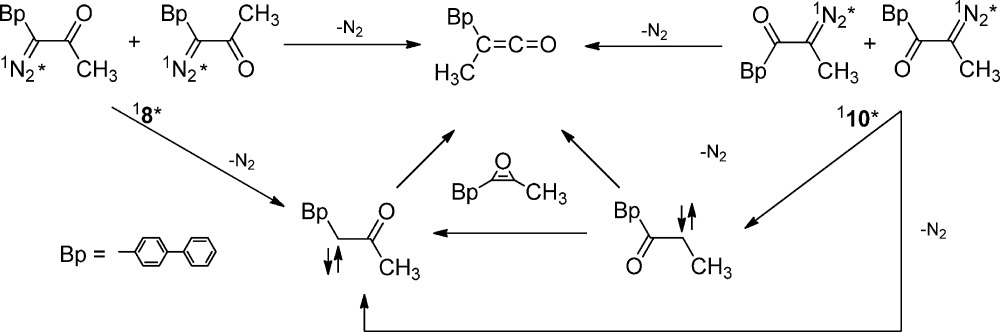 |
10 |
Ultrafast time-resolved photolysis of 8 and 10 produced the transient absorption band of 1BpCCOMe which was detected near 380 nm, when either compound was used as the precursor. Interestingly, the dynamics of carbene growth is precursor dependent. The excited diazo carbonyl 110*can decay by three mechanisms. The excited state can extrude nitrogen to form 1BpCOCMe, which subsequently isomerizes to the more stable carbene 1BpCCOMe with a time constant of 5 ps in methanol. Alternatively, excited-state extrusion of nitrogen and formation of carbene 1BpCCOMe can proceed in concert, without the intermediacy of 1BpCOCMe. Finally, nitrogen extrusion and ketene formation (confirmed by fast growth τ < 0.4 ps of IR ketene band) can proceed in concert in the diazo excited state 110*.
Theory predicts that 1BpCCOMe is lower in energy than 1BpCOCMe by 7.8 kcal/mol in the gas phase at 0 K. The oxirene minimum is predicted to be 0.7 kcal/mol higher in energy than 1BpCOCMe. The potential energy surface connecting the oxirene and carbene is clearly very flat. Consequently, the activation barrier for carbene interconversion must be very small, in agreement with the fast time constant (5 ps) measured in the experiment. The oxirene intermediate was not detected in our experiments despite its strong oscillator strength (f = 0.6307) at 375 nm as predicted by a TD-DFT calculation. We suspect that the oxirene is formed vibrationally excited and has a very short (sub ps) lifetime. We cannot exclude the possibility that the oxirene is actually a transition state, rather than an intermediate. What is certain is that photolysis of diazo precursor 10 forms the isomerized carbene “instantaneously” (less than 5 ps). The initially formed carbene isomerizes through an oxirene as rapidly as one can observe, even on ultrafast time scales. The initially formed hot carbene flies over the oxirene barrier to form the lower energy carbene isomer and one cannot determine experimentally if either species is an intermediate or a nonstationary point on the potential energy surface.
VII. Rearrangements in the Excited State of the Carbene Precursor
Numerous chemists have proposed that formal carbene rearrangement products can be formed by at least two pathways.40−42 Carbene mimetic products can be formed directly from the excited state of the precursor (rearrangement in the excited state, termed RIES by Liu46) as exemplified, but not limited to the Wolff rearrangement. Alternatively, products can be formed “classically” from a free carbene species.22b Historically, mechanistic studies of carbenes relied on chemical analysis of persistent photolysis products. The observation of a “non trappable carbene” route to rearranged products, by analysis of persistent products, was taken as support of the RIES mechanism.22b,40−42
We have reported studies that are consistent with a RIES mechanism (concerted molecular nitrogen extrusion and rearrangement) in alkyl diazo compounds47,48 and diazirines49(Scheme 2).
Scheme 2.

Ultrafast time-resolved photolysis of p-biphenylyldiazoethane (BpCN2CH3) and p-biphenylyldiazomethane (BpCN2H) were performed under identical conditions to allow meaningful comparison of the data.47 The absorption bands detected at 360 nm were assigned to the corresponding singlet carbenes. The carbene lifetimes varied between a few hundred picoseconds in acetonitrile to ∼10 ps in a carbene scavenging solvent such as methanol. In acetonitrile, in cyclohexane, and in methanol, the observed quantum yield of 1BpCCH3 formation was 30–40% lower than that of 1BpCH. TD-DFT calculations predict that that the two carbenes have similar extinction coefficients. This justified in our minds equating relative optical yields and relative quantum yields. This is the same type of comparison made previously in a study of the photochemistry of a diazo ketone and a diazo ester. The observation was again taken as an example of an RIES mechanism. The simplest explanation of the data is that the diazo excited state can form either p-vinylbiphenyl directly (Scheme 3) or the carbene. The competitive RIES process decreases the yield of carbene produced from BpCN2CH3, relative to BpCN2H, where an RIES pathway does not exist.
Scheme 3.

Thus far, we have described the use of ultrafast time-resolved methods to demonstrate that “instantaneous” yields of carbenes can provide indirect evidence of RIES. A subsequent study provided evidence of “instantaneous” alkene formation, as predicted by the RIES mechanism. In these experiments, the photochemistry of alkyl diazo esters were studied using ultrafast time-resolved IR spectroscopy. This was accomplished by monitoring the C=O vibration of the vinyl esters produced from alkyl diazo esters.48 Diazo esters rather than diazo ketones were used in these studies because the ester group is less prone to Wolff rearrangement and ketene formation. Ultrafast photolysis of methyl 2-diazopropionate (CH3CN2CO2CH3) produced two distinctive spectral features (see Figure 4). A bleaching band was observed at 1693 cm–1 corresponding to the diazo consumption. There was no further signal evolution, due to the lack of diazo ground state recovery (ΦIC = 0). In addition, we observed a positive, broad vinyl ester (C=O stretching) band. This spectral feature was initially observed with a maxima at 1720 cm–1. The absorption maximum blue-shifted to 1733 cm–1 as a result of vibrational cooling. This transpired over a 70 ps time window. Thus alkenes, and not just carbenes, are produced from diazo excited states on ultrafast time scales.
Figure 4.
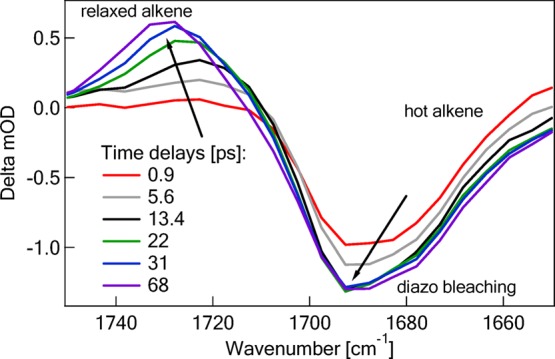
Ultrafast formation of a vinyl ester species (<0.4 ps) produced upon photoexcitation of methyl 2-diazopropionate at 266 nm. (Reprinted with permission from ref (48). Copyright 2010 American Chemical Society).
We will conclude this perspective by stating that 1,2-hydrogen RIES can explain an over 50-year-old mechanistic question that has intrigued carbene chemists these many years,23 but first we must revisit an aspect of the ultrafast time-resolved spectroscopy of fluorenylidene (FL).
VII. Electronically Excited (Open-Shell) Singlet Carbenes
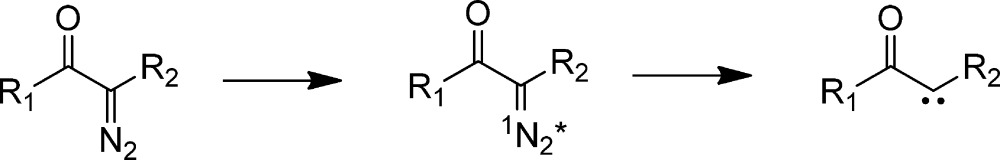
Closed-shell singlet carbenes are typically “born” in a vibrationally excited state (#) upon ultrafast photolysis of diazo compounds or diazirines. But is the singlet carbene formed in closed-shell, zwitterionic-like singlet electronic state or in a higher energy (open-shell) biradical-like excited state (*)? Theory predicts that both decay channels are operative but that the S1 state of diazomethane will predominantly produce the open-shell singlet state of methylene!50
The C=N double bond in a diazo compound is rather weak (e.g., 30.6 kcal/mol in diazomethane).51 UV excitation of ∼310 nm corresponds to 92 kcal/mol. As a result, there is sufficient energy to form the open shell singlet 1carbene* species upon photolysis of diazo precursors with UV light.
Our ultrafast studies of diazofluorene persuaded us that 1fluorenylidene* (1Fl*) is produced upon UV photolysis of 9-diazofluorene (Scheme 4, Figure 5).
Scheme 4.

Figure 5.
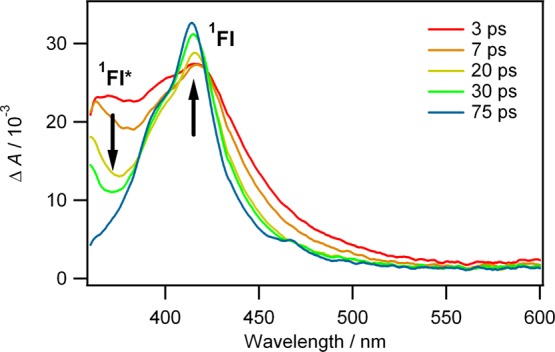
Proposed ultrafast photophysics and photochemistry of 9-diazofluorene*. The time-resolved transient UV–vis absorption spectra were recorded over a 3–75 ps time window. (Reprinted with permission from ref (32). Copyright 2007 American Chemical Society).
The chemical species responsible for the decaying band at 370 nm was assigned to the excited open-shell singlet 1FL* (see Figure 5). The rising band at 420 nm was assigned to the 1carbene in the lowest (closed-shell) singlet state. The estimated lifetime of the open-shell singlet carbene was 20 ps. There is clearly an intermediate between the excited state of diazofluorene and the closed-shell singlet state of fluorenylidene. We believe it is the open-shell, biradical like, singlet carbene, as predicted by theory.50
VIII. Mechanistic Aspects of Singlet Carbene Formation from Phenyldiazirine
The decay of the S1 state of a diazo compound can proceed by at least three pathways: (1) by S1→S0 internal conversion; (2) by extrusion of nitrogen and the formation of closed-shell singlet carbene and nitrogen; and (3) by extrusion of nitrogen and formation of the open-shell singlet carbene, as described previously for fluorenylidene.
The decay of the S1 state of a diazirine compound can proceed by the same three pathways described above, but there is in addition a fourth major pathway; isomerization to the thermodynamically more stable diazo isomer. The photoisomerization of phenyldiazirine to diazo compound in its ground state (S0) was studied using ultrafast time-resolved IR spectroscopy (N=N stretching at 2064 cm–1). The 270 nm light induced formation of closed-shell singlet phenylcarbene was demonstrated by monitoring its characteristic vibrational band (C=C at 1582 cm–1).52−54
Phenyldiazirine and phenyldiazomethane were studied computationally at the B3LYP/6-31+G(d) and RI-CC2/TZVP levels of theory. In each case, the three lowest singlet excited states were optimized at the RI-CC2/TZVP level. Theory predicts that the S1 state of phenyldiazirine is σ → π*. This state has a quinoidal structure. Interestingly, the C–N bonds of the diazirine group are slightly deformed from the CS symmetry of the geometry of the ground state of the diazirine. The S2 and S3 states are both predicted to be π→ π* in nature with the excitation energy localized largely on the aromatic ring. The calculations predict that the S1 state has a very large dipole moment and consequently an active aromatic C=C vibrational mode around ∼1600 cm–1. This IR band is not predicted in the other electronic states mentioned previously.
An economical use of theory is to assign the polar intermediate observed by ultrafast time-resolved UV–vis and IR spectroscopic studies of arylhalo- and arylalkyldiazirines to the calculated S1 state of phenyldiazirine.53,55
Unsurprisingly, theory predicts that the ground (S0) and S1 excited state of phenyldiazirine will have different chemistry. Thermolysis of phenyldiazirine is predicted to form singlet phenylcarbene, whereas excitation to the S1 excited state leads to isomerization to the first excited state of phenyldiazomethane. This excited state is predicted to rapidly extrude nitrogen and form carbene (eq 11).
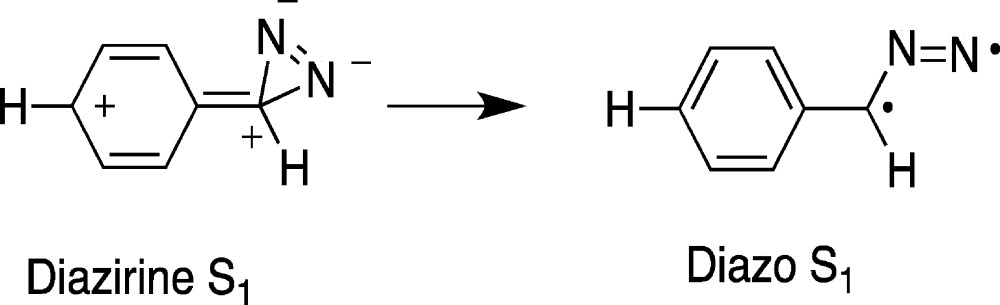 |
11 |
The calculations indicate that the S1-diazo compound will be born with a large excess of vibrational energy (over 50 kcal/mol). This results from the highly exothermic S1–S1 diazirine–diazo isomerization50,51 and this in turn will lead to ultrafast carbene formation from the hot, diazo excited state. Upon excitation of phenyl diazirine, an intense singlet carbene IR band is observed within a few picoseconds of the 270 nm laser pulse.24 The excited state, nitrogenous precursors of singlet phenylcarbene are interesting short-lived reactive intermediates in their own right!
IX. An Explanation for the Different Thermo- and Photochemistry of Diazirines
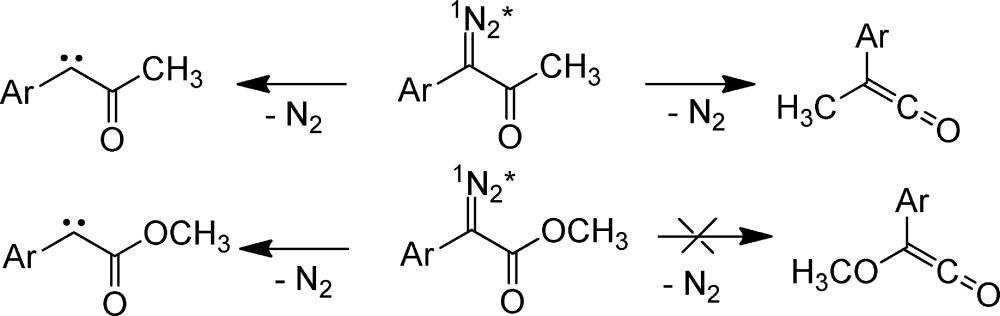
Photolysis of ethylmethyldiazirine gives a very random mixture of alkene products as mentioned earlier (Table 1, Scheme 5).23 Thermolysis of the same diazirine generates a thermodynamic mixture of products, in excellent agreement with DFT calculations.
Scheme 5.

For over fifty years, carbene chemists have speculated about the origin of the different results of thermal and photochemical activation. Chemists have proposed electronic and vibrationally excited states of the nitrogenous precursor and of the singlet carbene. Our ultrafast time-resolved experiments, and modern theory, do not exclude any possibilities but do provide experimental support for a specific sequence of events.
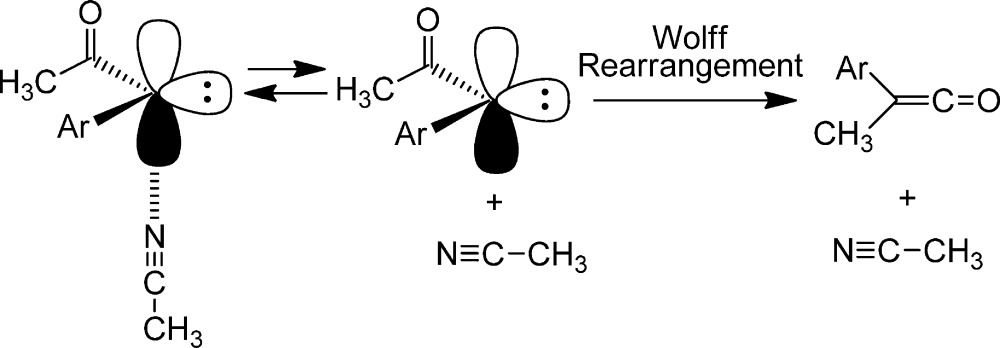
Our current working hypothesis (see Scheme 6) is that promotion of ethylmethydiazirine to the S1 state leads to the S1 state of the isomeric diazo compound, as predicted by calculations on phenyl diazirine.52−54 The diazo S1 excited state then fragments to form the vibrationally hot open-shell singlet (OSS) carbene of ethylmethylcarbene; the analogue of the biradical-like singlet fluorenylidene-detected previously.
Scheme 6.

Unpublished calculations by Hoi-Ling Luk and Christopher Hadad56 find a conical intersection (CI) between the vibrationally excited open-shell singlet (OSS) carbene and alkene products. Perhaps this is the reverse of the long-known photoisomerization of alkene to carbene reaction.57 We posit that in less than one ps, the hot OSS carbene isomerizes to a nonthermodynamic mixture of alkenes (alkenes′, see below) via the aforementioned CIs. Hot OSS carbene isomerization is also in competition with internal conversion to ultimately form the closed-shell singlet carbene. The closed-shell singlet (CSS) carbene forms the thermodynamic mixture of alkenes, exactly as in the pyrolysis of the diazirine. If this mechanistic hypothesis is correct, then the dynamics of alkene′ formation, via hot OSS carbene, and RIES in a diazo excited state, are kinetically equivalent (sub ps). The dynamics will be much faster than that of thermodynamic alkene mixture formation, via thermally relaxed OSS (10–20 ps) and CSS (1 ns) ethylmethylcarbene (Scheme 6). (Note that the vertical axis below (energy) is not drawn to scale.)
X. The Next Generation of Mechanistic Questions
Forty years ago, determining the bond angle of triplet methylene and the singlet triplet energy separation of this compound were at the forefront of research of both theory and experiment. Studies of the kinetics of bimolecular reactions of carbenes were in their infancy. Intramolecular reactions of carbenes were thought to be too fast to study because simple dialkylcarbenes could not be trapped with alkenes. Intersystem crossing rates in solution were unknown and structural and solvent effects on ISC were areas of pure speculation.
Today we know that dialkylcarbenes have singlet ground states and that their rearrangements are not too fast to study, either experimentally or computationally. Past failures to trap these intermediates stem from their poor yields of formation, as excited state (vibrational and electronic of nitrogenous precursor and carbene) siphon off the yields of relaxed singlet carbenes. The same issues permeate carbonyl carbene chemistry.
In my opinion, the most interesting mechanistic questions these days concern the excited state surfaces of diazirines, diazo compounds and carbenes. How do these surfaces connect, and what carbene mimetic rearrangements do these excited states undergo? Another problem is the solvation of carbenes. It seems clear that there are intimate interactions between solvent and carbene that have implications for intersystem crossing and chemical reactivity. These interactions require better modeling and better understanding.
I think that there are many similarities between 2013 and 1973. Once again, theory and experiment must work together to understand excited state surfaces, and solvation, to achieve the level of insight we have obtained with relaxed carbenes. I look forward to reading about it!58
Acknowledgments
I gratefully acknowledge the support of the NSF for over 40 years: first as a graduate student, then with a postdoctoral fellowship, and for over 35 continuous years as a faculty member at The Ohio State University. I will always be grateful to OSU for giving me an opportunity when frankly no other institution would do so and for access to so many resources. I also thank Bob Moss and Wes Borden for critical readings of the manuscript. Finally, words cannot express my appreciation to my former students and collaborators who made the research so much fun and for the patience and support of my wife and best friend Joan, every step of the way. The Platz laboratory has also been supported by the NIH, the NSF, and the biomedical industry.
Biography

Matthew S. Platz became Vice Chancellor for Academic Affairs at the University of Hawaii-Hilo, on January 2, 2013. Platz is a native of New York City, and he obtained a B.Sc. degree from the State University of New York at Albany in 1973 and the Ph.D. from Yale University in 1977. His studies were guided by Professors Shelton Bank and Jerome Berson, respectively. Following a post doctoral year at the University of Chicago with Gerhard Closs, he joined the faculty of The Ohio State University as an assistant professor of chemistry in 1978. Platz was promoted to associate professor in 1984 and to full professor in 1990 and served as department chair from 1994–1999. He won Ohio State University awards for distinguished teaching and research, and in 2001 Platz was named Distinguished University Professor of Chemistry. He also served Ohio State as a Vice Provost and Dean. From September 2010 through the end of 2012 Platz was on leave from Ohio State serving as Director of the Division of Chemistry of the National Science Foundation. At NSF, he led the development of a new program in Sustainable Chemistry, Engineering and Materials (SusChEM) and a new NSF–EPA joint program in Green Chemistry. Platz studies the photochemistry of small organic molecules in media as diverse as simple organic solutions and in blood products used in transfusion medicine. He has mentored well over 100 undergraduate, graduate, and post doctoral research students, published over 300 peer reviewed papers, edited or coedited three books, delivered over 200 invited lectures, and holds over a dozen patents.
Author Present Address
† Administration Building, 200 West Kawili St, The University of Hawai’i, Hilo Hilo, HI 96720
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Hine J.Divalent Carbon; Ronald Press Co.: New York, 1964. [Google Scholar]
- Doering W. v. E.; Hoffmann A. K. J. Am. Chem. Soc. 1954, 76, 6162–6165. [Google Scholar]
- Doering W. v. E.; Buttery R. G.; Laughlin R. G.; Chuadhuri N. J. Am. Chem. Soc. 1956, 78, 3224–3224. [Google Scholar]
- Skell P. S.; Woodworth R. C. J. Am. Chem. Soc. 1956, 78, 4496–4497. [Google Scholar]
- Closs G. L. Top. Stereochem. 1968, 3, 193–235. [Google Scholar]
- Jones M. Jr., Moss R. A.. Singlet Carbenes. In Reactive Intermediate Chemistry; Moss R. A., Platz M. S., Jones M. Jr., Eds.; John Wiley & Sons: Hoboken, NJ, 2004; pp 273–328. [Google Scholar]
- Jones M. Jr.; Rettig K. R. J. Am. Chem. Soc. 1965, 87, 4013–4015. [Google Scholar]; See also:; Baron W. J.; DeCamp M. R.; Hendrick M. E.; Jones M. Jr.; Levin R. H.; Sohn M. B.. Carbenes form diazo compounds. In Carbenes; Jones M. Jr., Moss R. A., Eds.; John Wiley & Sons: New York, 1973; Vol. 1, pp 79–84. [Google Scholar]
- a Herzberg G. Proc. R. Soc. London Ser. A 1961, 262, 291–317. [Google Scholar]; b Bernheim R. A.; Bernard H. W.; Wang P. S.; Wood L. S.; Skell P. S. J. Chem. Phys. 1970, 53, 1280–1281. [Google Scholar]
- Wasserman E.; Yager W. A.; Kuck V. J. Chem. Phys. Lett. 1970, 7, 409–413. [Google Scholar]
- Trozzolo A. M.; Wasserman E. In Carbenes; Moss R. A., Jones M. Jr., Eds.; John Wiley & Sons, New York, 1975; Vol. 2, pp 185–206. [Google Scholar]
- Closs G. L. In Carbenes; Moss R. A., Jones M. Jr., Eds.; John Wiley & Sons: New York, 1975; Vol. 2, p 159. [Google Scholar]
- a Zittel P. F.; Ellison G. B.; O’Neil S. V.; Herbst E.; Lineberger W. C.; Reinhardt W. P. J. Am. Chem. Soc. 1976, 98, 3731–3732. [Google Scholar]; b Leopold D. G.; Murray K. K.; Miller A. E. S.; Lineberger W. C. J. Chem. Phys. 1985, 83, 4849–4865. [Google Scholar]; c Shavitt I. Tetrahedron 1985, 41, 1531–1542. [Google Scholar]; d Schaefer H. F. Science 1986, 231, 1100–1107. [DOI] [PubMed] [Google Scholar]
- Berson J. A.Non-Kekulé Molecules as Reactive Intermediates. In Reactive Intermediate Chemistry; Moss R. A., Platz M. S., Jones M. Jr., Eds.; John Wiley & Sons: Hoboken, NJ, 2004; pp 165–203. [Google Scholar]
- Platz M. S. J. Am. Chem. Soc. 1979, 101, 3398–3399. [Google Scholar]
- a Senthilnathan V. P.; Platz M. S. J. Am. Chem. Soc. 1980, 102, 7637–7643. [Google Scholar]; b LeRoy R. J.; Murai H.; Williams F. J. Am. Chem. Soc. 1980, 102, 2325–2334. [Google Scholar]
- Platz M. S.; Senthilnathan V. P.; Wright B. B.; McCurdy C. W. Jr. J. Am. Chem. Soc. 1982, 104, 6494–6501. [Google Scholar]
- Scaiano J. C.Solution Photochemistry of Carbenes and Biradicals. In Kinetics and Spectroscopy of Carbenes and Biradicals; Platz M. S., Ed.; Plenum: New York, 1990; pp 353–368. [Google Scholar]
- Moss R. A., Turro N. J.. Laser Flash Photolytic Studies of Arylhalocarbenes. In Kinetics and Spectroscopy of Carbenes and Biradicals; Platz M. S., Ed.; Plenum: New York, 1990; pp 213–238. [Google Scholar]
- Hadel L. M.; Platz M. S.; Scaiano J. C. J. Am. Chem. Soc. 1984, 106, 283–287. [Google Scholar]
- Shaffer M. W.; Leyva E.; Soundararajan N.; Chang E.; Chang D. H. S.; Capuano V.; Platz M. S. J. Phys. Chem. 1991, 95, 7273–7277. [Google Scholar]
- Moss R. A.Advances in Carbene Chemistry; Brinker U. H., Ed.; JAI Press: Greenwich, CT, 1994; Vol. 1, pp 59–88. [Google Scholar]
- a Jackson J. E.; Platz M. S.. Advances in Carbene Chemistry; Brinker U. H., Ed.; JAI Press: Greenwich, CT, 1994; Vol. 1, pp 89–160. [Google Scholar]; b Platz M. S.Advances in Carbene Chemistry; Brinker U. H., Ed.; JAI Press: Stamford, CT, 1998; Vol. 2, pp 133–174. [Google Scholar]
- a Sulzbach H. M.; Platz M. S.; Schaefer H. F. III; Hadad C. M. J. Am. Chem. Soc. 1997, 119, 5682–5689. [Google Scholar]; b Mansoor A. M.; Stevens I. D. R. Tetrahedron Lett. 1966, 16, 1733–1737. [Google Scholar]; c Chang K. T.; Shechter H. J. Am. Chem. Soc. 1979, 101, 5082–5084. [Google Scholar]; d Frey H. M.Photolysis of the Diazirines. In Advances in Photochemistry; John Wiley & Sons: New York, 1966; Vol. 4, pp 225–256. [Google Scholar]; e Frey H. M.; Stevens I. D. R. J. Chem. Soc. 1965, 3101–3108. [Google Scholar]
- a Langan J. G.; Sitzmann E. V.; Eisenthal K. B. Chem. Phys. Lett. 1984, 110, 521–527. [Google Scholar]; b Langan J. G.; Sitzmann E. V.; Eisenthal K. B. Chem. Phys. Lett. 1986, 124, 59–62. [Google Scholar]; c Sitzmann E. V.; Langan J. G.; Griller D.; Eisenthal K. B. Chem. Phys. Lett. 1989, 161, 353–360. [Google Scholar]; d Sitzmann E. V.; Langan J. G.; Eisenthal K. B. J. Am. Chem. Soc. 1984, 106, 1868–1869. [Google Scholar]
- Reichardt C. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar]
- Jortner J.; Rice S. A.; Hochstrasser R. M.. Radiationless Transitions in Photochemistry. In Advances in Photochemistry; John Wiley & Sons: New York, 1969; Vol. 7, pp 149–310. [Google Scholar]
- Robinson G. W.; Frosch R. P. J. Chem. Phys. 1962, 37, 1962–1973. [Google Scholar]
- Robinson G. W.; Frosch R. P. J. Chem. Phys. 1963, 38, 1187–1203. [Google Scholar]
- Geise C. M.; Wang Y.; Mykhaylova O.; Frink B. T.; Toscano J. P.; Hadad C. M. J. Org. Chem. 2002, 67, 3079–3088. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Hadad C. M.; Toscano J. P. J. Am. Chem. Soc. 2002, 124, 1761–1767. [DOI] [PubMed] [Google Scholar]
- Salem L.; Rowland C. Angew. Chem., Int. Ed. Engl. 1972, 11, 92–111. [Google Scholar]
- Wang J.; Kubicki J.; Hilinski E. F.; Mecklenburg S. L.; Gustafson T. L.; Platz M. S. J. Am. Chem. Soc. 2007, 129, 13683–13690. [DOI] [PubMed] [Google Scholar]
- Wang J.; Kubicki J.; Peng H. L.; Platz M. S. J. Am. Chem. Soc. 2008, 130, 6604–6609. [DOI] [PubMed] [Google Scholar]
- Wang J.; Zhang Y.; Kubicki J.; Platz M. S. Photochem. Photobiol. Sci. 2008, 7, 552–557. [DOI] [PubMed] [Google Scholar]
- Burdzinski G.; Platz M. S.. Ultrafast Kinetics of Carbene Reactions. In Contemporary Carbene Chemistry; Moss R. A., Doyle M. P., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2014; pp 166–192. [Google Scholar]
- Turro N. J.; Cha Y.; Gould I. R. J. Am. Chem. Soc. 1987, 109, 2101–2107. [Google Scholar]
- Eisenthal K. B.; Moss R. A.; Turro N. J. Science 1984, 225, 1439–1445. [DOI] [PubMed] [Google Scholar]
- Wang J.; Kubicki J.; Gustafson T. L.; Platz M. S. J. Am. Chem. Soc. 2008, 130, 2304–2313. [DOI] [PubMed] [Google Scholar]
- Wolff L. Justus Liebigs Ann. Chem. 1902, 325, 129–195. [Google Scholar]
- a Kirmse W. Eur. J. Org. Chem. 2002, 2193–2313. [Google Scholar]; b Kaplan F.; Meloy G. K. J. Am. Chem. Soc. 1966, 88, 950–956. [Google Scholar]; c Burdzinski G.; Platz M. S. J. Phys. Org. Chem. 2010, 23, 308–314. [Google Scholar]
- Burdzinski G.; Wang J.; Gustafson T. L.; Platz M. S. J. Am. Chem. Soc. 2008, 130, 3746–3747. [DOI] [PubMed] [Google Scholar]
- Wang J.; Burdzinski G.; Kubicki J.; Platz M. S. J. Am. Chem. Soc. 2008, 130, 11195–11209. [DOI] [PubMed] [Google Scholar]
- Tomioka H.; Okuno H.; Kondo S.; Izawa Y. J. Am. Chem. Soc 1980, 102, 7123–7125. [Google Scholar]
- a Zhang Y.; Kubicki J.; Wang J.; Platz M. S. J. Phys. Chem. A 2008, 112, 11093–11098. [DOI] [PubMed] [Google Scholar]; b Wang Y.; Hadad C. M.; Toscano J. P. J. Am. Chem. Soc. 2002, 124, 1761–1767. [DOI] [PubMed] [Google Scholar]
- Wang J.; Burdzinski G.; Kubicki J.; Gustafson T. L.; Platz M. S. J. Am. Chem. Soc. 2008, 130, 5418–5419. [DOI] [PubMed] [Google Scholar]
- Bonneau R.; Liu M. T. H.; Kim K. C.; Goodman J. L. J. Am. Chem. Soc. 1996, 118, 3829–3837. [Google Scholar]
- Wang J.; Burdzinski G.; Gustafson T. L.; Platz M. S. J. Am. Chem. Soc. 2007, 129, 2597–2606. [DOI] [PubMed] [Google Scholar]
- Burdzinski G.; Zhang Y.; Selvaraj P.; Sliwa M.; Platz M. S. J. Am. Chem. Soc. 2010, 132, 2126–2127. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Kubicki J.; Platz M. S. Org. Lett. 2010, 12, 3182–3184. [DOI] [PubMed] [Google Scholar]
- Lee H.; Miyamoto Y.; Tateyama Y. J. Org. Chem. 2009, 74, 562–7. [DOI] [PubMed] [Google Scholar]
- Zimmerman P. M.; Toulouse J.; Zhang Z.; Musgrave C. B.; Umrigar C. J. J. Chem. Phys. 2009, 131, 124103. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Burdzinski G.; Kubicki J.; Platz M. S. J. Am. Chem. Soc. 2008, 130, 16134–16135. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Burdzinski G.; Kubicki J.; Vyas S.; Hadad C. M.; Sliwa M.; Poizat O.; Buntinx G.; Platz M. S. J. Am. Chem. Soc. 2009, 131, 13784–13790. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Vyas S.; Hadad C. M.; Platz M. S. J. Phys. Chem. A 2010, 114, 5902–5912. [DOI] [PubMed] [Google Scholar]
- a Wang J.; Burdzinski G.; Kubicki J.; Platz M. S.; Moss R. A.; Fu X.; Piotrowiak P.; Myahkostupov M. J. Am. Chem. Soc. 2006, 128, 16446–16447. [DOI] [PubMed] [Google Scholar]; b Zhang Y.; Wang L.; Moss R. A.; Platz M. S. J. Am. Chem. Soc. 2009, 131, 16652–16653. [DOI] [PubMed] [Google Scholar]
- Unpublished calculations of Luk H. L. and Hadad C. M..
- Kropp P.Photorearrangement and Fragmentation of Alkenes. In CRC Handbook of Organic Photochemistry and Photobiology; Horspool W., Lenci F., Eds.; CRC Press: Boca Raton, 2004; pp 13.1–13.15. [Google Scholar]
- Space did not permit a discussion of my other interest of decades, nitrenes, but I would hate for my students and collaborators to feel this was not a passion of mine equal to carbenes. It certainly is not, and their interactions are just as precious; see:Platz M. S. In Nitrenes; Moss R. A., Platz M. S., Jones M. Jr., Eds.; John Wiley & Sons, Hoboken, NJ, 2004; pp 501–560. [Google Scholar]