

Abstract
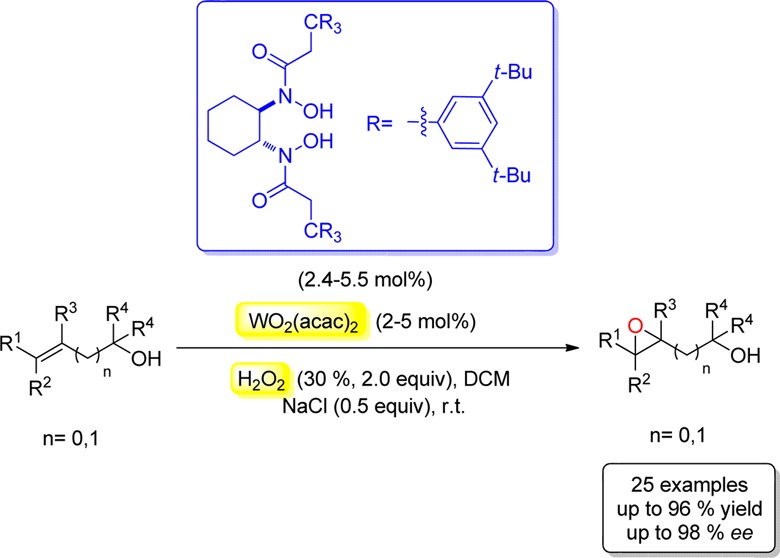
A simple, efficient, and environmentally friendly asymmetric epoxidation of primary, secondary, tertiary allylic, and homoallylic alcohols has been accomplished. This process was promoted by a tungsten–bishydroxamic acid complex at room temperature with the use of aqueous 30% H2O2 as oxidant, yielding the products in 84–98% ee.
Asymmetric epoxidation is one of the most important transformations in organic synthesis, since it provides a straightforward access to various optically active epoxides, which are highly useful chiral building blocks for the synthesis of natural products and synthetic analogues with biological activities.1 Much progress has been achieved in this field since Sharpless et al. reported the discovery of titanium-catalyzed asymmetric epoxidation of allylic alcohols in the early 1980s.2 In recent years our group developed a series of unique bishydroxamic acids (BHA) and applied them successfully as ligands for highly enantioselective epoxidation of allylic, homoallylic, and bishomoallylic alcohols as well as N-alkenyl sulfonamides and N-tosyl imines.3 However, all these reactions employ toxic alkyl peroxide as the oxidant, functioning with low atom economy. Therefore, from both economical and ecological viewpoints, it is desirable to develop a catalytic epoxidation with broad substrate scope using aqueous hydrogen peroxide (H2O2) as the oxidant, which is safe, cheap, easy to handle and generates water as the sole byproduct.4,5 Recently, Katsuki et al. described H2O2-mediated epoxidation of allylic alcohols in highly enantioselective manner using niobium–salem complexes as catalysts.6 Nonetheless, the substrate scope of this protocol is limited to primary allylic alcohols. Over the last decades the use of peroxotungstates as catalysts for the epoxidation with H2O2 has attracted much attention as a result of their high capability for oxygen transfers and low activity for disproportion of H2O2.7 Despite intensive research in this area, highly enantioselective tungsten-catalyzed epoxidation remains still elusive. Recently, Mizuno et al. developed dinuclear peroxotungstates, which could catalyze the H2O2-mediated epoxidation of various allylic and homoallylic alcohols efficiently;7g,7k but no asymmetric induction was reported in these two contexts. Herein, we report a tungsten-catalyzed asymmetric epoxidation of both primary, secondary, and tertiary allylic as well as homoallylic alcohols with aqueous H2O2 as oxidant by using our W-BHA catalyst system.
Since BHA-1 and BHA-2 showed high catalytic activity and selectivity in the vanadium- and hafnium-catalyzed epoxidation of allylic and homoallylic alcohols,3 we initially investigated them as ligands in the epoxidation reaction of cis-2-hexen-1-ol (1a) using WO2(acac)2 as the tungsten source. Unfortunately, both reactions gave the product 2a in moderate yields and low enantioselectivities, while higher enantioselectivity was obtained in the case of BHA-2 (Table 1, entries 1 and 2). For this reaction, three other ligands, BHA-3–5, with modified ligand arms were synthesized and studied on the basis of the scaffold of BHA-2 (Table 1, entries 3–5). The best results with respect to both yield and enantioselectivity were achieved in the case of the BHA-5 (Table 1, entry 5). Furthermore, (−)-TADDOL was also investigated as ligand, but the reaction gave only traces of product after 8 h, indicating that the bishydroxamic acids were essential for the outcome of this process (Table 1, entry 6). In all the cases using BHA as ligand it was observed that the epoxide 2a underwent a ring-opening reaction with H2O2 as nucleophile. In order to halt this undesired reaction, we screened five different alkali salts as additives.7m In the case of LiCl the side reaction was totally blocked, but the epoxidation was also slowed down (Table 1, entry 7). When LiBr was employed, no reaction occurred, and gas evolution was observed, suggesting the disproportion of H2O2 (Table 1, entry 8). The use of LiF, Na2SO4, or NaCl as additive afforded similar results, and all inhibited the ring-opening reaction effectively, while the asymmetric induction of epoxidation remained excellent (Table 1, entries 9–11). Considering that Na2SO4 and toxic LiF are more expensive than NaCl, we chose NaCl for further optimization. The reactions were then conducted in toluene and THF, but there were no better results (Table 1, entries 12, 13). Furthermore, the commercially available WO2Cl2 was also tested and proved to be less reactive (Table 1, entry 14). Reducing the catalyst loading to 2 mol % led to longer reaction time and lower yield (Table 1, entry 15). Then we attempted to improve the catalytic activity by adjusting the amount of additive used and substrate concentration. By lowering the amount of NaCl to 0.5 equiv the reaction time could be shortened to 8 h, and the yield improved to 87% (Table 1, entry 16). When the concentration of the olefin 1a in DCM was increased to 0.1 M, the reaction was completed within 3 h, affording the product in 92% yield and 95% ee (Table 1, entry 17). Reducing the catalyst to 1 mol % resulted in a decrease of the yield to 63% (Table 1, entry 18). Performing the reaction with 0.25 equiv NaCl could improve the yield to 86%, but the ee diminished to 79% (Table 1, entry 19). Finally, the reaction was carried out on a scale of 10.0 mmol 1a, furnishing the product in 89% yield and 96% ee (Table 1, entry 20). In this case BHA-5 could be recycled through column chromatography with 92% yield in analytically pure form. Importantly, all the reactions were conducted under air and needed no anhydrous solvents or additional preparation of the BHA-W complexes prior to the epoxidation reaction. The olefin 1a and H2O2 could be added to the mixture of BHA-5 and WO2(acac)2 immediately, giving the product 2a with excellent ee. In addition, conducting the reaction in the absence of BHA gave only traces of epoxide after 3 h, indicating that the BHA ligand is crucial not only for the stereoselectivity but also for the catalytic activity.
Table 1. Ligand, Additive, and Solvent Screening for the Asymmetric Epoxidationa.
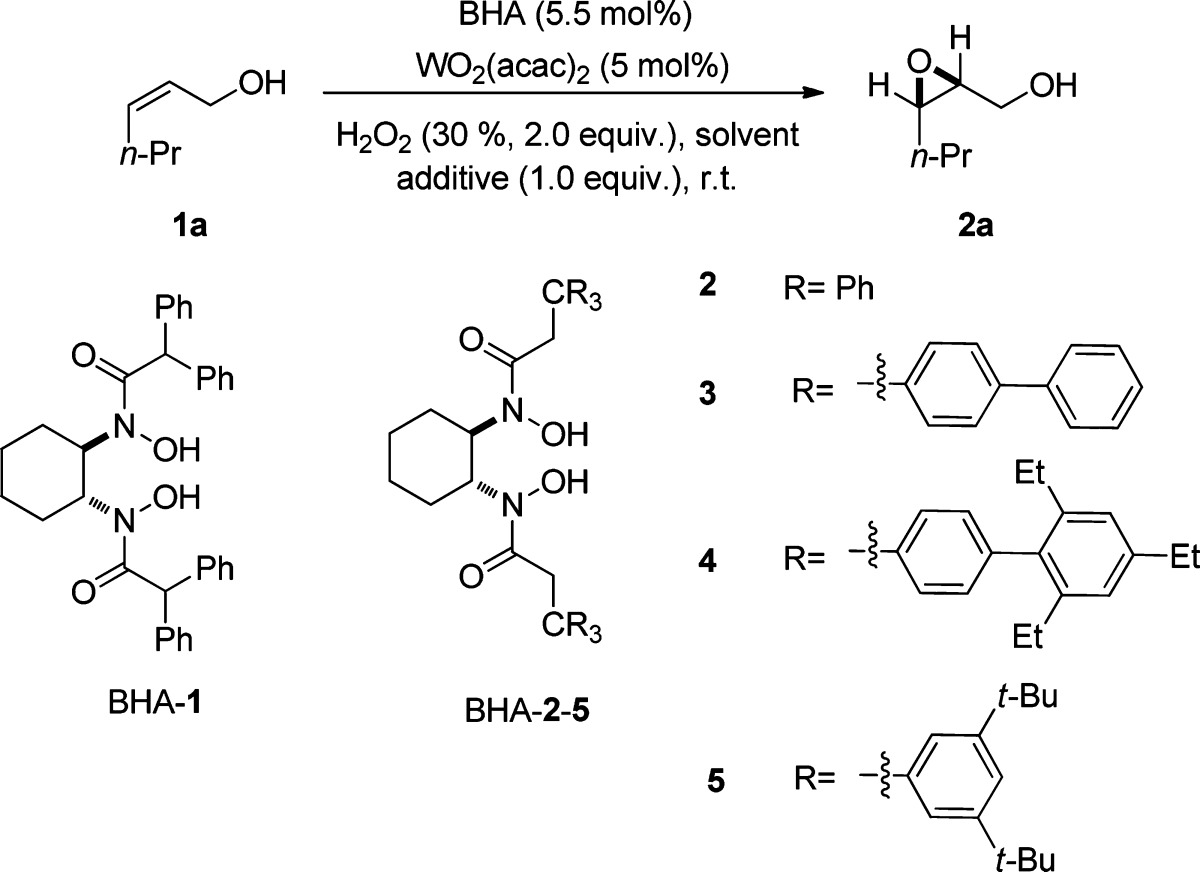
| entry | ligand | solvent | additive | t (h) | yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|
| 1 | 1 | DCM | – | 2.5 | 38 | –12 |
| 2 | 2 | DCM | – | 2.5 | 54 | 37 |
| 3 | 3 | DCM | – | 2.5 | 69 | 49 |
| 4 | 4 | DCM | – | 2.5 | 71 | 41 |
| 5 | 5 | DCM | – | 2.5 | 84 | 94 |
| 6 | d | DCM | – | 8 | traces | n.d.e |
| 7 | 5 | DCM | LiCl | 24 | 81 | 93 |
| 8 | 5 | DCM | LiBr | 24 | 0 | – |
| 9 | 5 | DCM | NaCl | 2.5 | 91 | 96 |
| 10 | 5 | DCM | LiF | 2.5 | 92 | 95 |
| 11 | 5 | DCM | Na2SO4 | 2.5 | 92 | 95 |
| 12 | 5 | toluene | NaCl | 24 | 83 | 93 |
| 13 | 5 | THF | NaCl | 24 | 49 | 55 |
| 14f | 5 | DCM | NaCl | 24 | 51 | 92 |
| 15g | 5 | DCM | NaCl | 24 | 73 | 94 |
| 16g,h | 5 | DCM | NaCl | 8 | 87 | 95 |
| 17g,h,i | 5 | DCM | NaCl | 3 | 92 | 95 |
| 18h,j | 5 | DCM | NaCl | 24 | 62 | n.d.e |
| 19j,k | 5 | DCM | NaCl | 24 | 86 | 79 |
| 20g,h,i,l | 5 | DCM | NaCl | 3 | 89 | 96 |
Unless otherwise stated, reactions were performed on a 0.25 mmol scale of cis-2-hexen-1-ol (1a) using 2.0 equiv; 30% aqueous H2O2, 5 mol % WO2(acac)2, 5.5 mol % BHA ligand, and 1.0 equiv additive at rt in 5.0 mL solvent.
Yields of isolated products.
Determined by HPLC on a chiral stationary phase on the corresponding benzoate.
(−)-TADDOL was used as ligand.
Not determined.
WO2Cl2 was used instead of WO2(acac)2.
2.0 mol % WO2(acac)2 and 2.4 mol % BHA-5 were used.
0.5 equiv NaCl was used.
Reaction was performed in 2.5 mL DCM.
Reactions were performed on 1.0 mol % WO2(acac)2, 1.2 mol % BHA-5 in 0.5 mL DCM.
0.25 equiv NaCl was used.
The reaction was performed on a scale of 10.0 mmol 1a.
After optimizing the reaction conditions we started to evaluate the substrate scope of this reaction. We first investigated a variety of allylic alcohols with different substituted patterns; see Chart 1 for summarized results. Excellent results in terms of both yield and enantioselectivity were obtained in the case of aliphatic cis and trans disubstituted allylic alcohols 1a–1e. Cinnamyl alcohol 1f and its fluorinated analogue 1g turned out to be less reactive. In both cases the reactions were carried out with 5 mol % catalyst furnishing the products 2f and 2g in 87 and 72% yields and 94 and 86% ee, respectively. Two α-substituted cinnamyl alcohols 1h and 1i were employed as substrates for the epoxidation reaction, giving the products 2h and 2i in good yields (87 and 79%) and 84 and 86% ee. Symmetric β,β-disubstituted allylic alcohol 1j was also a suitable substrate for this method, giving the product 2j in 86% yield and 92% ee. The reaction using nerol, 1k, farnesol, 1l, and its derivative, 1m, as precursors proceeded smoothly with high regioselectivities under the optimized conditions, giving the products 2k–m in 84–96% yields and 84–90% ee. A tertiary allylic alcohol 1n was also subjected to the epoxidation reaction, requiring longer reaction time and higher catalyst loading (5 mol %), and the product 2n was obtained in a 86% yield and 90% ee.
Chart 1. Asymmetric Epoxidation of Allylic Alcoholsa,b,c,d,e,f,8.

a Unless otherwise stated, reactions were performed on a 0.50 mmol scale of allylic alcohols 1 using 2.0 equiv; 30% aqueous H2O2, 2 mol % WO2(acac)2, 2.4 mol % BHA ligand, and 0.5 equiv NaCl at rt in 5 mL DCM.
b Yields of the isolated products.
c Determined by HPLC on a chiral stationary phase on the corresponding benzoates.
d Determined by HPLC on a chiral stationary phase.
e 5 mol % WO2(acac)2 and 5.5 mol % BHA-5 were used. Reaction time: 24 h
f Determined by GC on a chiral stationary phase.
Chart 2. Asymmetric Epoxidation of Homoallylic Alcoholsa,b,c,d,e,f,g,8.

a Unless otherwise stated, reactions were performed on a 0.50 mmol scale of homoallylic alcohols 3 using 2.0 equiv; 30 % aqueous H2O2, 2 mol % WO2(acac)2, 2.4 mol % BHA-5, and 0.5 equiv NaCl at rt in 5 mL DCM.
b Yields of the isolated products.
c Determined by HPLC on a chiral stationary phase on the corresponding benzoates.
d Determined by HPLC on a chiral stationary phase.
e Reaction was performed at 0 °C using 1.0 equiv NaCl; reaction time 8 h.
f Determined by GC on a chiral stationary phase.
g 5 mol % WO2(acac)2 and 5.5 mol % BHA-5 were used; reaction time: 24 h.
Being similar to their allylic analogues, primary cis and trans disubstituted homoallylic alcohols 3a–f were excellent substrates for this epoxidation method, and the corresponding products 4a–f were obtained in high yields (80–92%) with good to excellent asymmetric induction (88–97% ee). In the case of cyclic olefin 3g the reaction was conducted at 0 °C using 1.0 equiv NaCl as additive to prevent the undesired side reaction. Under this condition the product 4g was afforded in 72% yield and 96% ee. In contrast, the tertiary homoallylic alcohol 3h underwent an epoxidation/intramolecular ring-opening cascade reaction giving a tetrasubstituted tetrahydrofuran 4h as product in 75% yield, >98% de, and 90% ee.
The method was also applied to the kinetic resolution of racemic α-vinylbenzyl alcohol (rac-5) furnishing both the allylic alcohol 5 and the epoxide 6 with high ee’s (Chart 3).
Chart 3. Kinetic Resolution of α-Vinylbenzyl Alcohol.

Since our W-catalyst exhibits not only excellent stereoselectivity but also distinct reactivity for various types of alcohols, our method could be used not only for the synthesis of a simple chiral pool but also as a late-stage oxidation for the synthesis of complex molecules. However, we have to provide more information about the influence of the anchor groups as well as that of the geometry of the olefins on the reactivity of the substrates. Hopefully, Tables 2 and 3 will provide at least some basic information. The results obtained show the following trend of the reactivity of different substrates: (a) primary alcohols ≫ tertiary alcohols and phenyl-substituted secondary alcohols; (b) cis- or trans-disubstituted olefins ≈ trisubstituted olefins > geminal disubstituted olfefins.
Table 2. Investigation of the Chemoselectivity of Various Allylic Alcoholsa,9.
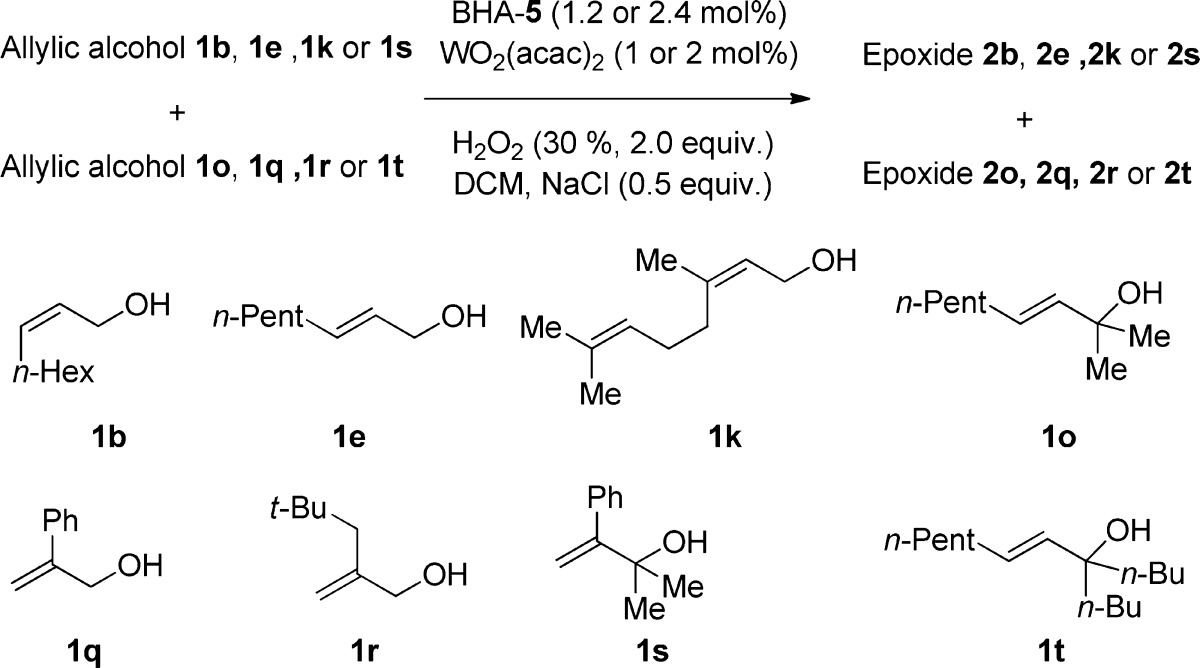
| 1o | 1q | 1r | 1t | |
|---|---|---|---|---|
| 1b | – | 82/7b | – | – |
| 1e | 84/25 | 79/7 | 90/35 | 86/10 |
| 1k | – | 71/5 | – | – |
| 1s | 9/69 | – | – | – |
Reactions were performed with a 1:1 mixture of two allylic alcohols using 2.0 equiv; 30% aqueous H2O2, 1 or 2 mol % WO2(acac)2, 1.2 or 2.4 mol % BHA-5 and 0.5 equiv NaCl in DCM.
Conversions were determined by 1H NMR spectroscopy of the reaction mixtures. The conversions were given in form of a/b with a as conversion of the compound shown in the first column and b as conversion of the compound shown in the top of the respective row.
Table 3. Investigation of the Chemoselectivity of Various Homoallylic Alcoholsa,9.
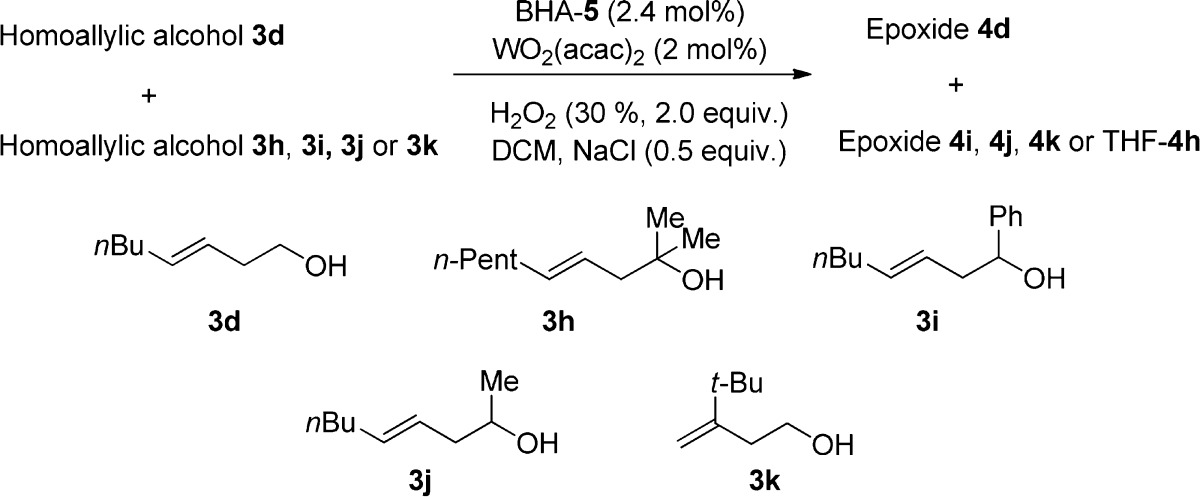
| 3h | 3i | 3j | 3k | |
|---|---|---|---|---|
| 3d | 92/3b | 89/5 | 81/27 | 64/21 |
Reactions were performed with a 1:1 mixture of two homoallylic alcohols using 2.0 equiv; 30% aqueous H2O2, 2 mol % WO2(acac)2, 2.4 mol % BHA-5, and 0.5 equiv NaCl in DCM.
Conversions were determined by 1H NMR spectroscopy of the reaction mixtures. The conversions are given in form of a/b: where a = conversion of 3d and b = conversion of 3h–k.
Last, two farnesol derivatives 1u and 1v bearing three olefins and two alcohol moieties were employed as precursors of the epoxidation reaction. To our delight, the corresponding products 2u and 2v were furnished in almost complete regioselectivities, 85 and 74% yields, and 88–92% ee (Chart 4).
Chart 4. Regioselective Epoxidation of the Farnesol Derivatives 1u and 1v.

To conclude, we have developed a tungsten-catalyzed asymmetric epoxidation with the following advantages and breakthroughs: (1) the first highly enantioselective epoxidation using tungsten catalyst; (2) the use of environmentally benign aqueous H2O2 as oxidant instead of toxic organic alkyl peroxides; (3) simple conditions: reactions are performed under air and in most cases at RT requiring no anhydrous solvent or preparation of metal–catalyst complex prior to the catalytic process; (4) broad substrate scope, i.e. both primary, secondary, and tertiary allylic as well as homoallylic alcohols are successfully employed as precursors for this epoxidation reaction furnishing the products in high ee’s; (5) good chemoselectivities for primary alcohols over secondary and tertiary alcohols, promising the use of this method in late-stage, complex molecule synthesis.
Acknowledgments
We thank the National Institutes of Health (NIH) for financial support (2R01GM068433). C.W. thanks the Alexander von Humboldt Foundation for his postdoctoral fellowship. We also thank Dr. Antoni Jurkiewicz and Dr. Jin Qin for their respective expertise in NMR spectroscopy and mass spectrometry.
Supporting Information Available
Experimental details; characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For general reviews on asymmetric epoxidation:; a Katsuki T.; Martin V. S. Org. React. 1996, 48, 1. [Google Scholar]; b Katsuki T. In Comprehensive Asymmetric Catalysis; Jacobsen E. N., Pfaltz A., Yamamoto H., Eds.; Springer: Berlin, 1999; Vol. 2, p 621. [Google Scholar]; c Adam W.; Zhang A. Synlett 2005, 37, 1047. [Google Scholar]; d McGarrigle E. M.; Gilheany D. G. Chem. Rev. 2005, 105, 1563. [DOI] [PubMed] [Google Scholar]; e Xia Q.-H.; Ge H.-Q.; Ye C.-P.; Liu Z.-M.; Su K.-X. Chem. Rev. 2005, 105, 1603. [DOI] [PubMed] [Google Scholar]; f Wong O. A.; Shi Y. Chem. Rev. 2008, 108, 3958. [DOI] [PubMed] [Google Scholar]; g Matsumoto K.; Sawada Y.; Katsuki T. Pure Appl. Chem. 2008, 80, 1071. [Google Scholar]
- Katsuki T.; Sharpless K. B. J. Am. Chem. Soc. 1980, 102, 5974. [Google Scholar]
- a Murase N.; Hoshino Y.; Oishi M.; Yamamoto H. J. Org. Chem. 1999, 64, 338. [Google Scholar]; b Hoshino Y.; Murase N.; Oishi M.; Yamamoto H. Bull. Chem. Soc. Jpn. 2000, 73, 1653. [Google Scholar]; c Hoshino Y.; Yamamoto H. J. Am. Chem. Soc. 2000, 122, 10452. [Google Scholar]; d Makita N.; Hoshino Y.; Yamamoto H. Angew. Chem., Int. Ed. 2003, 42, 941. [DOI] [PubMed] [Google Scholar]; e Zhang W.; Basak A.; Kosugi Y.; Hoshino Y.; Yamamoto H. Angew. Chem., Int. Ed. 2005, 44, 4389. [DOI] [PubMed] [Google Scholar]; f Zhang W.; Yamamoto H. J. Am. Chem. Soc. 2007, 129, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Barlan A. U.; Zhang W.; Yamamoto H. Tetrahedron 2007, 63, 6075. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Li Z.; Zhang W.; Yamamoto H. Angew. Chem., Int. Ed. 2008, 47, 7520. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Li Z.; Yamamoto H. J. Am. Chem. Soc. 2010, 132, 7878. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Olivares-Romero J. L.; Li Z.; Yamamoto H. J. Am. Chem. Soc. 2012, 134, 5440. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Olivares-Romero J. L.; Li Z.; Yamamoto H. J. Am. Chem. Soc. 2013, 135, 3411. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Li Z.; Yamamoto H. Acc. Chem. Res. 2013, 46, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For epoxidation of olefins with H2O2:; a Arends I. W. C. E.; Sheldon R. A. Top. Catal. 2002, 19, 133. [Google Scholar]; b Grigoropoulou G.; Clark J. H.; Elings J. A. Green Chem. 2003, 5, 1. [Google Scholar]; c Benjamin S. L.; Burges K. Chem. Rev. 2003, 103, 2457. [DOI] [PubMed] [Google Scholar]; d De Faveri G.; Ilyashenko G.; Watkinson M. Chem. Soc. Rev. 2011, 40, 1722. [DOI] [PubMed] [Google Scholar]; e Russo A.; De Fusco C.; Lattanzi A. ChemCatChem 2012, 4, 901. [Google Scholar]
- For examples not in ref (4) on asymmetric epoxidation of olefins with H2O2:; a Romney D. K.; Miller S. J. Org. Lett. 2012, 14, 1138. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chu Y.; Liu X.; Li W.; Hu X.; Lin L.; Feng X. Chem. Sci. 2012, 3, 1996. [Google Scholar]; c Lifchits O.; Mahlau M.; Reisinger C. M.; Lee A.; Farès C.; Polyk I.; Gopakumar G.; Thiel W.; List B. J. Am. Chem. Soc. 2013, 135, 6677. [DOI] [PubMed] [Google Scholar]; d Berkessel A.; Günther T.; Wang Q.; Neudörfl J.-M. Angew. Chem., Int. Ed. 2013, 52, 8467. [DOI] [PubMed] [Google Scholar]; e Cussó O.; Garcia-Bosch I.; Ribas X.; Lloret J.; Costas M. J. Am. Chem. Soc. 2013, 135, 14871. [DOI] [PubMed] [Google Scholar]; f Dai W.; Li J.; Li G.; Yang H.; Wang L.; Gao S. Org. Lett. 2013, 15, 4138. [DOI] [PubMed] [Google Scholar]
- Egami H.; Ogama T.; Katsuki T. J. Am. Chem. Soc. 2010, 132, 5886. [DOI] [PubMed] [Google Scholar]
- For examples of tungsten-catalyzed epoxidation of olefins:; a Herrmann W. A.; Haider J. J.; Fridgen J.; Lobmaier G. M.; Spiegler M. J. Organomet. Chem. 2000, 603, 69. [Google Scholar]; b Xi Z.; Zhou N.; Sun Y.; Li K. Science 2001, 292, 1139.11349143 [Google Scholar]; c Denis C.; Misbahi K.; Kerbal A.; Ferrières V.; Plusquellec D. Chem. Commun. 2001, 37, 2460. [DOI] [PubMed] [Google Scholar]; d Adam W.; Alsters P. L.; Neumann R.; Saha-Möller C. R.; Slobada-Rozner D.; Zhang R. Synlett 2002, 34, 2011. [Google Scholar]; e Adam W.; Alsters P. L.; Neumann R.; Saha-Möller C. R.; Slobada-Rozner D.; Zhang R. J. Org. Chem. 2003, 68, 1721. [DOI] [PubMed] [Google Scholar]; f Wang X.-Y.; Shi H.-C.; Sun C.; Zhang Z.-G. Tetrahedron 2004, 60, 10993. [Google Scholar]; g Kamata K.; Yamaguchi K.; Mizuno N. Chem.—Eur. J. 2004, 10, 4728. [DOI] [PubMed] [Google Scholar]; h Maheswari P. U.; de Hoog P.; Hage R.; Gamez P.; Reedijk J. Adv. Synth. Catal. 2005, 347, 1759. [Google Scholar]; i Sartorel A.; Carraro M.; Bagno A.; Scorrano G.; Bonchio M. Angew. Chem., Int. Ed. 2007, 46, 3255. [DOI] [PubMed] [Google Scholar]; j Kamata K.; Kotani M.; Yamaguchi K.; Hikichi S.; Mizuno N. Chem.—Eur. J. 2007, 13, 639. [DOI] [PubMed] [Google Scholar]; k Kamata K.; Hirano T.; Kuzuya S.; Mizuno N. J. Am. Chem. Soc. 2009, 131, 6997. [DOI] [PubMed] [Google Scholar]; l Kamata K.; Yonehara K.; Sumida Y.; Hirata K.; Nijima S.; Mizuno N. Angew. Chem., Int. Ed. 2011, 50, 12062. [DOI] [PubMed] [Google Scholar]; m Hachiya H.; Kon Y.; Ono Y.; Takumi K.; Sasagawa N.; Ezaki Y.; Sato K. Synlett 2012, 44, 1672. [Google Scholar]
- Absolute stereochemistries of 2a, 2c, 2f, 2h, 2i, 2k, 2l, 4c, 4e, 5, and 6 were assigned by comparison with known compounds (see the Supporting Information). Hence, 2b, 2d, 2e, 2g, 2j, 2m, 2n, 2u, 2v, 4a, 4b, 4d, 4f, 4g, and the epoxide precursor of 4h were assigned by analogy, assuming a common reaction pathway. The absolute configuration of the tetrahydrofuran 4h was assigned by assuming that the ring-opening reaction proceeds in an SN-2-type reaction.
- For detailed reaction conditions, see the SI.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.