

Abstract
Redox reactions have been established as major biological players in many cellular signaling pathways. Here we review mechanisms of redox signaling with an emphasis on redox-active signaling endosomes. Signals are transduced by relatively few reactive oxygen species (ROS), through very specific redox modifications of numerous proteins and enzymes. Although ROS signals are typically associated with cellular injury, these signaling pathways are also critical for maintaining cellular health at homeostasis. An important component of ROS signaling pertains to localization and tightly regulated signal transduction events within discrete microenvironments of the cell. One major aspect of this specificity is ROS compartmentalization within membrane-enclosed organelles such as redoxosomes (redox-active endosomes) and the nuclear envelope. Among the cellular proteins that produce superoxide are the NADPH oxidases (NOXes), transmembrane proteins that are implicated in many types of redox signaling. NOXes produce superoxide on only one side of a lipid bilayer; as such, their orientation dictates the compartmentalization of ROS and the local control of signaling events limited by ROS diffusion and/or movement through channels associated with the signaling membrane. NOX-dependent ROS signaling pathways can also be self-regulating, with molecular redox sensors that limit the local production of ROS required for effective signaling. ROS regulation of the Rac-GTPase, a required co-activator of many NOXes, is an example of this type of sensor. A deeper understanding of redox signaling pathways and the mechanisms that control their specificity will provide unique therapeutic opportunities for aging, cancer, ischemia-reperfusion injury, and neurodegenerative diseases.
I. Cellular Redox Signaling and Its Impact on Health
The cell signaling that drives all homeostatic and disease processes involves specific interactions between proteins and low-molecular weight compounds at precise intra- and extracellular locations. Cell signaling is dysregulated under numerous pathological conditions, both genetic and acquired. Because all aspects of healthy cell signaling are highly specific and precise, researchers were surprised to discover, over the past few decades, that many essential cellular signaling processes involve reactive oxygen species (ROS), which had initially been characterized as agents of rapid, damaging redox chemistry. Progress toward redox-related therapies is now coming from studies ranging from analyses of the inheritance of high levels of redox-active compounds to clinical trials of the effectiveness of applying such compounds to combat cancer.1,2 However, the mechanisms that control the production and function of cellular ROS remain subjects of much intense study. Compartmentalization of ROS in the redoxosome (i.e., the redox-active endosome) is one important means of controlling cellular ROS signals and is a main focus of this review. Redoxosomes are signaling endosomes that form within cells in response to specific extracellular stimuli. Their formation is closely linked with endocytic machinery and the coordinated endocytosis of ROS-producing enzymes and signal-specific proteins from the cell surface. Specific to redoxosomes is the requirement for ROS production in the interior of the endosome that induces redox-dependent changes in protein structure on or near the cytoplasmic surface of the redoxosome. These redox-dependent signals trigger translocation of cytoplasmic proteins to endosomal receptor complexes that are required for activation and signal transduction. These pathways are covered in detail in this review. Newly discovered and less studied areas of ROS production within the nuclear envelope will also be briefly reviewed.
What Are ROS?
In the field of redox signaling biology, and in particular in the field of redoxosomal signaling, only hydrogen peroxide (H2O2) and superoxide (O2•–, or more simply designated as O2–) are commonly considered to be the signaling ROS (Table 1). In broader terms, ROS can include ordinary molecular triplet-state oxygen (O2), hydroxyl radical (OH•), singlet oxygen (1O2), and ozone (O3).3 In addition, the term “ROS” has often been extended to include several nitrogen-containing compounds, which are also known as reactive nitrogen species, such as nitric oxide (NO•), nitroxyl anion (NO–), and peroxynitrite (ONOO–); these nitrogen-containing compounds function in many cell signaling pathways but have not been demonstrated to participate in redoxosomal signaling.
Table 1. Redoxosome Signaling Components.
| protein or molecule | function(s) in redoxosome-mediated signaling | redox-modified? |
|---|---|---|
| NOX1 or -2 | transfers electrons from NADPH onto O2, producing O2– inside the redoxosomal lumen | unknown |
| p67phox (NOXA1?) | binds and thereby activates NOX | unknown |
| p47phox (NOXO1, Tks4, Tks5?) | “organizes” NOX (necessary for NOX activity) | unknown |
| p22phox | binds and thereby activates NOX | unknown |
| superoxide (O2–) | anion, free radical ROS; may exit some redoxosomes via anion channels; dismutates, spontaneously or with the help of SOD, to form H2O2; may be involved in several redox reactions that trigger signaling | N/A |
| hydrogen peroxide (H2O2) | neutral (uncharged) ROS, not a free radical; might diffuse through redoxosomal membranes or be transported by aquaporins; oxidizing agent, can oxidize specific redox-active cysteine residues to form sulfenamides, sulfenic acids, or disulfides | N/A |
| ClC-3 and IClswell | anion channels, may allow controlled diffusion of O2– out of the redoxosomal lumen in specific signal transduction pathways; may control charge and pH gradients at redoxosomal membranes | unknown |
| interleukin 1-β (IL-1β) | cytokine, initiates redoxosomal signaling, eventually activates NF-κB signaling pathway | unknown |
| tumor necrosis factor α (TNFα) | cytokine, initiates redoxosomal signaling, eventually activates the NF-κB and/or ATF-1 signaling pathway | unknown |
| interferon γ (IFNγ) | cytokine, might activate some redoxosomal signaling pathways | unknown |
| cholesterol | structure of lipid raft; essential for NOX2 activation in phagosomes and likely essential in redoxosomes; may bind acylated tails of Src family proteins | unknown |
| sphingolipids | structure of lipid raft; may hold membrane leaflets together | unknown |
| caveolin | coats the cytoplasmic face of a lipid raft prior to endocytosis | unknown |
| dynamin | pinches vesicle neck to form nascent endosome; usually but perhaps not always involved in all redoxosome formation | unknown |
| early endosome antigen 1 (EEA1) | marker of early endosomes; function in redoxosome signaling unclear | unknown |
| Rab5 | biogenesis and trafficking of endosomes; as a GTPase, might be involved in the regulation of Rac activity | unknown |
| IL-1R | dimer of IL-1R1 and IL-1RAcP; serves as a receptor for IL-1β | unknown |
| rho guanine nucleotide dissociation inhibitor (RhoGDI) | sequesters the prenylated tail of Rac, thereby preventing Rac from translocating to membranes; keeps Rac in the cytosol; after IL-1R binds IL-1β, RhoGDI becomes phosphorylated by unknown means, releasing Rac | unknown |
| Rac1 or -2 | as a GTPase, dephosphorylates GTP to GDP, stopping NOX activity; when bound to GTP, actively turns on NOX activity | yes |
| Vav1 (or Vav2?) | guanine exchange factor (GEF), exchanges GTP for GDP on Rac; is recruited to lipid rafts, function at rafts unclear | unknown |
| c-Src | activates redoxosomal signaling, mechanism(s) unknown; may phosphorylate Vav1 or -2 | probably |
| MyD88 | adaptor, links IL-1R to downstream IKK kinases | unknown |
| protein tyrosine phosphatases (PTPs) | can activate c-Src; direct evidence of the participation of PTP in redoxosomal signaling remains elusive | yes |
| superoxide dismutase 1 (SOD1; Cu,Zn SOD) | dismutates O2– to H2O2; binds Rac, preventing conversion of Rac from the GTP- to GDP-bound form; by binding Rac, keeps NOX active | unknown |
| TRAF6 | recruited to IL-1R, activates downstream IKK kinase proteins | probably |
| IRAK | recruited to IL-1R, activates downstream IKK kinase proteins | unknown |
| TNFR1 | receptor for TNFα | unknown |
| TRAF2 | recruited to TNFR1, activating downstream IKK kinase proteins | probably |
| TRADD/RIP | recruited to TNFR1, activating downstream IKK kinase proteins | unknown |
| alsin | GEF activity, might act as a GEF on Rac1 or Rab5; implicated in ALS; affects ROS levels at the redoxosomal level in glia | unknown |
| angiotensin II | activates Rac1/NOX2-active redoxosomes in cardiac myocytes | unlikely |
Signal Transduction by ROS
Homeostatic signal transduction by ROS requires that a physiological trigger induce the production of a particular ROS molecule, in the right quantities and at the right cellular location, and that this ROS react with a very specific target. Furthermore, the signal must be terminated or reversed at the appropriate time and location. Such control is achieved by the interactions of many activator and inhibitor proteins, often within membrane-bound organelles. The reaction of one O2– molecule with another (dismutation) to form H2O2 is vital in most of the well-described signaling pathways, because it is often H2O2 (rather than O2–) that transduces the signal, by oxidizing a specific amino acid (usually cysteine) at a certain site within a specific protein.
Specific Signaling Functions of ROS: Many Signaling Proteins Are ROS Targets
Among the many redox-modifiable proteins that contribute to cellular signaling are receptors, kinases, phosphatases, transcription factors, and peroxiredoxins.4−7 These proteins contain redox-active cysteines or methionines whose oxidation by ROS affects their signaling activity either positively or negatively. Two of the major families of redox-modifiable proteins are the kinases of the Src family and the protein tyrosine phosphatases (PTPs); both are discussed below (see also Table 1). Although a number of specific cysteine and methionine residues on various proteins have been identified as participants in redox signaling, their counterparts on many other proteins known to be vital to redox signaling remain unidentified.
PTP Redox Modifications
One notable example of redox signaling is the inhibition of PTPs by H2O2. The mammalian genome encodes approximately 100 PTPs.8 These proteins contain a catalytic site that features a redox-active cysteine with an unusually low pKa (<6.0), whose oxidation by H2O2 produces the sulfenic acid (RS-OH) form of cysteine. In the presence of either a low-molecular weight thiol such as cysteine or glutathione or a neighboring cysteine residue within the same PTP protein, this sulfenic acid can be further oxidized to the disulfide (RS-SR′) form.9,10 Once the PTP active site contains a disulfide, it can be reduced back to the original free thiol form by a specific thiol-reducing protein, such as peroxiredoxin, thioredoxin, or glutaredoxin (all of which also have redox-active cysteines that interact with additional redox-active, cysteine-containing enzymes). Another oxidative reaction in which the active-site cysteine residue of PTP participates is the formation of a sulfenamide bond, between the sulfur atom of the catalytic cysteine and the amide nitrogen atom of a neighboring serine residue.11 Like sulfenic acid and disulfide modifications, sulfenamide bonds in PTPs are reversible. However, reversal requires a reduction reaction, and until this occurs, the oxidized (sulfenamide-, sulfenic acid-, or disulfide-containing) PTP is inactive as a phosphatase. This redox-mediated inactivation is crucial in signaling because each of the numerous PTPs has very specific targets,8 and when any one PTP is inactivated, every one of its downstream signaling molecules is affected. As one specific example, analysis of insulin signaling has shown that in a cell that is not being stimulated by insulin PTP1B actively blocks intracellular insulin signaling pathways, whereas when insulin binds at the cell surface, H2O2 is immediately produced, PTP1B is oxidatively inactivated, and insulin signaling is initiated.8,12 Similarly, a study of growth factor signaling (which also is associated with elevations in levels of ROS and inactivation of PTPs) demonstrated that PTP cysteines were oxidized by H2O2 (and not O2–).13
c-Src Redox Modifications
In addition to the PTPs, c-Src and the Src family of protein tyrosine kinases are redox-sensitive and important members of redox signaling pathways.14 Src-regulated redox pathways include those that are active in endosomes, and, in particular, redoxosomes, as described below. The mechanism whereby Src contributes to redox signaling is not entirely understood but appears to involve multiple layers of redox control (see Table 1). Oxidation of certain cysteines within c-Src itself may have distinct results depending on the residues affected. On one hand, H2O2-mediated oxidation of C245 and C487 (according to the numbering of the chicken sequence) may lead to the formation of an intramolecular disulfide bond and c-Src activation.15 By contrast, H2O2-mediated oxidation of C277 may result in the formation of an intermolecular disulfide bridge with C277 of a second c-Src molecule, producing an inactive homodimer.16 It has also been suggested that the formation of a disulfide bond between c-Src and vascular endothelial growth factor receptor may activate c-Src.5 The mechanisms underlying the seemingly contradictory consequences of H2O2-mediated disulfide formation for c-Src activity depending on the affected cysteine residues have yet to be elucidated. One partial explanation may involve unique binding partners of c-Src that alter its conformation and subcellular localization, thus influencing the cysteines in c-Src that are exposed to H2O2. However, specific c-Src partners that might mediate such effects remain unidentified.5,17 A second point that may bear on the outcome of redox signaling through c-Src is that c-Src is also known to be directly and indirectly influenced by the PTPs and their oxidation status,5,18 as described below. Most studies of c-Src oxidation have revealed H2O2 to be the relevant oxidizing agent. However, in some tumor cells, O2– instead appears to play the major role,19 even though it (but not H2O2) can act as a reducing agent.20 The chemical mechanism underlying this difference has not been determined.
CSK: Possible Layers of Redox Control
Notably, the terminology for c-Src in the literature can lead to some confusion. Because c-Src is a tyrosine kinase, it has frequently been called both “tyrosine kinase Src” and “Src tyrosine kinase”.18,21 However, c-Src is not to be confused with a different protein, “tyrosine protein kinase CSK”, which is more commonly termed simply CSK (for “c-Src kinase”). The primary known function of CSK is to phosphorylate c-Src at a specific tyrosine residue in the C-terminal tail and thereby inactivate c-Src. Interestingly, like c-Src, CSK is regulated by PTPs.18 Thus, c-Src may be redox-regulated at numerous levels.
Adding to the confusion over the terminology is the fact that a great deal of the research on c-Src has been based on the proteins from two organisms: mouse and chicken. These proteins, of course, have slightly different amino acid sequences, and the numbering of the relevant cysteines and tyrosines varies somewhat among species. Thus, although very important findings have been made using the two protein sequences, it is important to be aware of the different terminologies when following these advances.22
II. Redox Signaling Enzymes That Produce ROS
Mammalian cells express a great number of enzymes that produce either O2– or H2O2. Among these are the protein complexes of the mitochondrial transport chain, xanthine oxidase, cytochrome P450 proteins, cyclooxygenases, NADPH oxidases (NOXes), lipoxygenases, nitric oxide synthases, and others. Many of these enzymes are implicated in subcellular redox signaling at specific membrane-bound organelles. However, we will focus primarily on NOXes because they are the only ROS-producing enzymes for which an association with redoxosomes and the nuclear envelope has been demonstrated.
NADPH Oxidases
The NOX family of ROS-producing enzymes includes transmembrane proteins NOX1, NOX2, NOX3, NOX4, NOX5, Duox1, and Duox2. NOX2 was the first of these to be described and was first discovered in a nonsignaling role. That is, this enzyme (also known as gp91phox) produces O2– during the microbicidal respiratory (oxidative) burst of neutrophils. NOX2 utilizes reduced nicotinamide adenine dinucleotide phosphate (NADPH) as its substrate. It transfers an electron from NADPH on the cytosolic side of a phagosomal membrane, via two hemes, to O2 on the lumenal side of the membrane and thus releases O2– directly into the phagosome. Downstream derivatives of the O2– then kill microbes. In unmodified form, NOX2 has a molecular mass of ∼65 kDa, but once it is glycosylated, it weighs 91 kDa (hence, the name gp91phox, where “phox” stands for “phagocyte oxidase”).23 The microbicidal activity of NOX2 in phagosomes depends on several binding partners, namely, p22phox, p47phox, p40phox, p67phox, and Rac2. In nonphagocytic cells, NOX2 produces O2– during nonmicrobicidal signaling activity by interacting with almost all of these binding partners, the exceptions being that Rac2 is replaced by Rac1, which is expressed in most cell types (Table 1), and that p40phox is primarily found in phagocytes. Except for the transmembrane protein p22phox, these binding partners are cytosolic and interact with the cytoskeleton, a very dynamic structure that controls their subcellular localization.24 Schematic diagrams of p22phox have frequently been drawn with only two transmembrane helices, but this view may be incorrect. Recently, in silico modeling suggested that the most stable form of p22phox has three transmembrane helices.25 According to this new model, when p22phox is at the plasma membrane, its N-terminus is extracellular and its C-terminus is intracellular.25 As for NOX2 itself, it has six transmembrane domains with both N- and C-termini in the cytoplasm.26
NOX1 was identified after NOX2, and shortly thereafter NOX3, -4, and -5 as well as Duox1 and 2 were discovered (see ref (27) and references cited therein). All seven of these proteins have several shared features: six transmembrane domains (minimally), two hemes, one FAD-binding domain, and one NADPH-binding domain. They all transfer electrons from NADPH to O2 across a membrane, producing O2– on the opposite side of the membrane (Table 1). With regard to the formation of oligomers, NOX1–4 bind p22phox, but NOX5 and Duox1 and -2 do not seem to do so.28,29 NOX1 has several additional binding partners not known to interact with NOX2. For example, NOX1 is capable of binding not only p47phox but also the p47phox homologues NOXO1, Tks4, and Tks5.30 Furthermore, NOX1 can bind either p67phox or its homologue, NOXA1.30 Rac1 and Rac2 can bind and activate either NOX1 or NOX2.31,32 Importantly, the interactions of the various binding partners with NOX1 are not static but dynamic and are controlled by many cellular processes. For example, phosphorylation of NOXA1 at either Ser-282 or Ser-172 has been observed to cause a decrease in the level of binding of NOXA1 to NOX1 and Rac1, decreasing NOX1 activity.33 NOX3 also seems to bind several of the cytosolic proteins mentioned above, although this is not yet well characterized. NOX5 and the Duox proteins have calcium-binding domains; thus, their activity is strongly regulated by the intracellular calcium concentration. Instead of binding p22phox, the Duoxes bind DuoxA1 and DuoxA2, each of which probably has five transmembrane helices rather than three like p22phox.34 To date, studies of redoxosomes have identified only NOX1 and NOX2 activity in these organelles; however, the other NOX family members might be involved in as yet unidentified redoxosomal signaling pathways. By contrast, as described below, NOX4 at the nuclear envelope appears to control superoxide production.35
Redox-Signaling Organelles
Although this review focuses primarily on redox signaling by endosomes, these are by no means the only intracellular organelles with significant redox signaling functions. The best-studied ROS-producing organelle is the mitochondrion. The production of ROS by this organelle was once thought to be a dangerous and unwanted side effect of respiration.36 However, more recent reports have conclusively demonstrated that mitochondrial ROS have many signaling roles.7,37,38
The plasma membrane is also a site of local ROS signaling, even in the absence of endocytosis.39 In this case, specific domains of and structures within the plasma membrane (such as rafts, caveolae, focal adhesions, cell–cell junctions, or leading-edge lamellipodia in the case of migrating cells) appear to be the locations of ROS release.39
The endoplasmic reticulum (ER) is heavily involved in redox signaling. As a major site of protein folding, which in many cases requires the formation of disulfide bonds, the ER lumen is a much more oxidizing environment than the cytosol. Proteins that promote this oxidizing environment include the well-known Ero1 and peroxiredoxin IV, (40) and also NADPH oxidases, in particular NOX4.41 ROS in the ER may directly oxidize thiols in glutathione peroxidases and peroxiredoxins, allowing these proteins to promote oxidative folding.42
Another organelle that is probably important in redox cell signaling is the perinuclear space (PNS), located between the nuclear envelope membranes. Although the PNS is continuous with the ER lumen, the inner nuclear membrane incorporates transmembrane proteins that are not found in the ER and that directly bind heterochromatin, nuclear pores, and other nuclear structures. Thus, redox functions of this membrane-enclosed compartment may differ from those of the non-nuclear ER. Recent evidence suggests that ROS may influence chromatin function through the epigenetic modifications made by histone- and DNA-modifying enzymes (i.e., DNA methyltransferases, histone demethylases, histone acetyltransferases, and histone deacetylases).43−48 For example, H2O2 recruits DNA methyltransferases to chromatin,45 and this process has been implicated in redox-dependent transcriptional alterations in atherosclerosis.44 Thus, it is plausible that ROS production in the nuclear envelope could directly regulate nuclear transcription and play an important role in health and disease. Furthermore, production of ROS in the nuclear envelope may directly regulate the activity of redox-sensitive transcription factors such as HIF-1α, NF-κB, and AP-1.47,49−51 It is interesting to note that the distance across the PNS, ranging from <50 nm to several hundred nanometers,52,53 is comparable to the 100–300 nm diameter of a typical redoxosome.54 Thus, local signaling events regulated by diffusion or transport of ROS across nuclear membranes may function in some ways like redoxosomes.
Both endosomal membranes and nuclear membranes are major sites of redox activity and, in particular, NOX activity. NOX4-dependent ROS production at the nucleus is confined to the PNS.35 However, unlike NOX1 and NOX2 that can be regulated by recruitment of cytosolic subunits to redoxosomes, NOX4 is not always regulated in this fashion and has been shown in certain instances to have constitutive activity.55 Interestingly, NOX4 activity in the nuclear envelope appears to be, at least in part, controlled by the local production of its substrate (NADPH) through glucose-6-phosphate dehydrogenase (G6PD).35 It has been demonstrated that NADPH is 2–3 orders of magnitude more dilute in the nucleus than in the cytoplasm.56 This observation suggests that NADPH is not able to diffuse freely in and out of the nucleus. G6PD, a primarily cytoplasmic enzyme, exists at low concentrations in nuclei.35 In isolated nuclei, G6PD produces NADPH from NADP+ and G6P, leading to NOX4-dependent O2– production.35 The ability of isolated hepatic nuclei to produce NOX4-dependent O2– in the presence of NADP+ and G6P is also decreased in G6PD mutant mice and following shRNA-mediated knockdown of G6PD. Immunofluorescence staining of nuclei demonstrated colocalization of punctate areas of G6PD and NOX4 expression, in agreement with the hypothesis that the direct supply of NADPH to NOX4 is from G6PD.35
Other organelles such as the Golgi57 and lysosomes58 are also involved in redox signaling, but their precise roles have not been extensively characterized. Redox signaling also occurs in trafficking pathways that involve endocytosis but in which ROS generation and redox reactions occur at a cellular site other than the endosome. One example of this is signaling by granulocyte colony stimulating factor (G-CSF), a cytokine that activates hematopoietic cells. When G-CSF is endocytosed, the signaling endosome that forms does not have any known redox activity. However, when that endosome is brought into the proximity of the ER, where NOX4 is active, G-CSF endosomal signaling is moderated.59 Similarly, in an endothelin 1-mediated pathway that was recently described in endothelial cells, the endolysosome contributes to activation of downstream ER pathways, eventually stimulating endothelial nitric oxide synthase to produce NO•, causing vascular smooth muscle to relax.60
III. The Biology of Redoxosomal Signaling
Endosomal Signaling
Before continuing on the subject of redox signaling and delving specifically into the details of redoxosomal signaling, we will take a step back at this point to describe general non-redox endosome-mediated signaling to demonstrate how redoxosomes fit into the broader picture of cell signaling. In a discussion of the nascent field of redoxosomal signaling, it is important to recognize that the concept of endosome-mediated signaling, regardless of whether it is redox-mediated, is still fairly new. In fact, endocytosis was long considered a cellular mechanism for turning signals off rather than on.61 Most endosomal signaling begins with receptors on the plasma membrane. Of course, endosomal signaling is only one of several mechanisms by which such cell-surface receptors transduce signals. As described in a previous review,22 receptor-mediated signaling can involve various types of pathways, some of which are more complex than others. For example, in the case of type I signals, ligand binding to a receptor results in direct signal transduction to the cell interior. Type II signaling, in contrast, involves a second messenger that is first recruited to the receptor and initiates downstream signaling. Type III signaling involves endosomes and can be even more complex: the endosome, with all of its associated proteins, may recruit cytosolic proteins to transduce a signal (type IIIA); alternatively, the endosome may fuse with another intracellular vesicle before transducing a signal (type IIIB).
The existence of signaling endosomes first came to light in 1994, with studies of liver parenchyma demonstrating that epidermal growth factor receptor (EGFR) is internalized in response to the administration of epidermal growth factor, and that the resultant signaling is specific to the EGFR pathway.62 Soon afterward, researchers found that transforming growth factor β (TGF-β) signaling, nerve growth factor (NGF) signaling, and G-protein-coupled receptor (GPCR) signaling involve additional distinct endosomal pathways.63 Another early study, demonstrating that tumor necrosis factor α [TNFα (see Table 1)] signaling proceeds by endocytosis,64 was of special relevance to the field of cytokine signaling. Because TNFα can sometimes cause apoptosis, the study focused on the pathway responsible for this phenomenon. It demonstrated that the following sequence of events occurs following ligand binding (compare also Table 1). First, RIP-1 and TRAF-2 are recruited to TNF receptor 1 (TNFR1). Next, TNFR1 is internalized (this step is dependent on the presence of an internalization signal between amino acids 205 and 214 of TNFR1), and then TRADD, FADD, and caspase-8 are recruited to the complex to form a “receptosome”. Finally, the receptosome fuses with the Golgi to further transduce the apoptotic death signal.65
The receptosome versus redoxosome terminology is useful in distinguishing between specific categories of endosomes. A receptosome is a signaling endosome that has endocytosed a receptor after ligand binding. In many cases, redoxosomes represent a subcategory of receptosome in which ROS production within the endosome is required for signal transduction. However, as discussed below in relation to hypoxia and reoxygenation, some redoxosomes may not require ligand binding for endocytosis to proceed and thus would not fall into the category of receptosomes.
Receptor-based endosomal signaling is now well accepted as a means of controlling the interconnectivity of numerous cellular signaling pathways.61 Endosomes can either isolate pathways from one another or mediate crosstalk between them, and at least 49 distinct endosomal “scaffolds” (i.e., endosome-mediated organizations of distinct pathways) have been identified.61 Not all of these have been thoroughly characterized, and as the field advances, additional complexities may be uncovered. Describing the crosstalk between endosomal signaling pathways as “scaffold-mediated” should not be misconstrued to indicate that these organelles are static in space or time. On the contrary, endosomal pathways are dynamic and involve rapidly changing membrane structures.
Endosome Dynamics
Historically, endocytosis has been classified on the basis of whether the endocytic process was clathrin-dependent or -independent. The clathrin-dependent endocytic pathways require the formation of a polygonal lattice of clathrin proteins, which are recruited from the cytosol to the site of vesicle formation.66 This results in the formation of a clathrin-coated pit that encloses an area of the membrane with a high concentration of phosphatidylinositol 4,5-bisphosphate. Dynamin, in conjunction with other proteins, then pinches the neck of the vesicle to form a new subcellular compartment (Table 1), after which the nascent vesicle loses its clathrin coating. By contrast, clathrin-independent pathways of endocytosis do not necessarily require dynamin for the pinching off of a new vesicle, although many other proteins such as ADP-ribosylation factors and actin are sometimes involved.67 In some of these clathrin-independent endocytic events, a caveolin protein (Table 1) coats the pit (often localized at a lipid raft region of the plasma membrane), and the pit is pinched off in a dynamin-dependent manner, much as in clathrin-dependent endocytosis. Indeed, clathrin-independent endocytosis in many cases, though not all, generates the endosome from an area of the plasma membrane where a high concentration of cholesterol or lipid raft is found (Table 1).67,68 A lipid raft is a specialized region of a membrane that will be discussed in more detail below. Many other combinations of proteins and lipids can also lead to endocytic events.68
In the endosome signaling literature, a nascent vesicle (formed by a clathrin-dependent or -independent mechanism) is sometimes identified as an “early endosome”. In other cases, the nascent vesicle is described as maturing into the organelle called the early endosome, or fusing with a preexisting, membrane-bound organelle and only then becoming known as the early endosome.66 This confusing terminology arises because the events immediately following vesicle formation in many signaling pathways are not yet well understood. In any event, a newly formed endosome or “early endosome” has several marker proteins, including EEA1 and Rab5 (Table 1),67,69 and its membrane is marked by a high concentration of the phospholipid phosphatidylinositol 3-phosphate.66 After the early endosome forms, complex molecular interactions determine the fates of “cargo”, endocytosed membrane and nonmembrane molecules (i.e., proteins, lipids, and any other molecules that were endocytosed). Some cargoes (including some receptors) are recycled to the cell surface by either rapid or slow recycling systems, often via “tubular recycling endosomes”.70 Other cargoes are sorted and moved to compartments such as late endosomes, the trans-Golgi network, multivesicular bodies, and lysosomes. Although Rab5 and EEA1 are present in early endosomes, they are absent in late and recycling endosomes, where Rab7 and Rab11 are present, respectively. These Rab proteins serve as “zip codes” for endosomal movement within cells. The membranes of individual endosomes contain specific domains in which these marker proteins cluster.71 Depending on the exact combination of protein, lipid, and other molecules present in a particular early endosome, very different signaling or metabolic pathways are activated. Thus, as mentioned above, signaling endosomes may interact with other endosomes or organelles in dynamic scaffolds that may either prevent or facilitate crosstalk between pathways.61
Lipid Rafts
Given that mammalian cells contain many thousands of types of lipids,72 lipid rafts have proven to be difficult to investigate. The current general consensus is that lipid rafts exist as dynamic, transient domains within lipid bilayers of the cell, primarily in the plasma membrane but possibly also in the membranes of various organelles. These raft domains can be described as “liquid-ordered” because they have a higher percentage of saturated lipids than other membrane regions and are thus more tightly packed.73 The lipids within these domains include cholesterol, sphingolipids, and saturated phospholipids (Table 1); because of the size of these molecules, a raft is generally slightly thicker than other areas of the plasma membrane.73,74 A raft includes lipids of both leaflets of the lipid bilayer, but the reasons for this are not completely clear. One proposed explanation is that the extra length of some sphingolipids may allow them to extend from the exoplasmic leaflet into the opposite (cytosolic) leaflet of a bilayer membrane, thus holding both leaflets together.75 The major chemical and physical impact of lipid rafts is that they bring specific proteins and glycoproteins together within a dynamic and ordered, yet easily movable, area. In neutrophils, the transmembrane protein NOX2 resides in lipid rafts constitutively, whereas the cytosolic subunits p40phox, p47phox, and p67phox are recruited to the raft (and to NOX2) in response to stimuli of phagocytosis.76 The cholesterol of lipid rafts is required for NOX2 activation in neutrophils, as demonstrated by the observation that cholesterol sequestration inhibits NOX2 activation.77 NOXes also are found in lipid rafts of other cell types, and their presence in these rafts is necessary for NOX signaling.78 Rafts are also collection points for palmitoylated and myristoylated proteins and cholesterol-binding proteins.79 For example, acylation of tyrosine kinases of the Src family triggers the localization of these proteins to lipid rafts.73
Redox-Active Endosomes (Redoxosomes)
Because of the rapidly growing bodies of knowledge on the subjects of both endosomal signaling in nonredox pathways and redox signaling by such transmembrane proteins as the NOXes, it was natural to investigate the possibility that endosomal membranes could serve as sites of NOX or redox signaling. Redox-active signaling endosomes were first discovered by Li et al. in 200632 and were named “redoxosomes” by Oakley et al. in 2009.22,80 Prior to the discovery of redoxosomes, many extracellular ligands, in particular cytokines, had been shown to induce cellular ROS production. Indeed, indirect evidence of this was available more than 20 years ago, as treatment of Jurkat T cells with either the cytokine TNFα or H2O2 was sufficient to trigger the same response, an increase in NF-κB activity.81 Because TNFα is endocytosed,64 this finding was important to the eventual discovery of redox-signaling endosomes.
The redoxosomes that have been studied to date have all been found to utilize either NOX1 or NOX2. The cell types that have been demonstrated to use redoxosomal signaling pathways include MCF-7 (an immortalized human mammary carcinoma cell line),32,54,80,82 mouse and human aortic smooth muscle cells (SMCs),83−86 human neutrophils,87 primary mouse embryonic fibroblasts,54,88 primary mouse dermal fibroblasts,54,88 HeLa cells (immortalized human cervical cancer cells),89 brain cells, spinal cord cells, immortalized glial and neuronal cells,88,90 Kupffer cells, hepatocytes, and additional liver cell types that have not yet been clearly identified.91,92 Another cell type in which redoxosome signaling might occur is the renal mesangial cell, where a high level of glucose stimulates NOX activity in a caveolin-dependent manner.93 This was observed to lead to the activation of both RhoA and protein kinase C and thus to upregulation of the cytokine TGF-β, a known cause of renal matrix overproduction and diabetic nephropathy.93 This study did not address the question of whether the caveolae involved in the observed ROS-mediated damage were pinched off by dynamin to form endosomes; although caveolae were certainly implicated, it seems possible that redoxosomal pathways may also have been activated in this high-glucose model. This wide range of cell types indicates that redoxosomal signaling is probably fairly universal.
Dynamin
Studies of cytokine signaling in MCF-7 cells have shown that endosomal fractions from cells treated with TNFα or IL-1β (Table 1) contain greatly elevated levels of Rac, p47phox, and p67phox, as compared to such fractions from untreated cells, and that the recruitment of these proteins is largely dependent on dynamin-mediated endocytosis.32,54 Specifically, the expression of dominant-negative dynamin greatly inhibited the recruitment of these proteins to endosomes.32,54 Dynamin was also found to be essential for ROS formation in these redoxosomes. Similar, though not identical, results have been reported for SMCs. TNFα treatment of these cells led to dynamin- and NOX1-dependent ROS production in endosomes and, in turn, to activation of the phosphatidylinositol 3-kinase (PI3K)–Akt-activating transcription factor-1 (ATF-1) pathway.85
A dynamin-independent, nonendocytic mechanism whereby ROS-producing membrane-bound organelles could conceivably enter the cell from the plasma membrane is pinocytosis. Additionally, pinocytosis of membrane-associated regulatory proteins could constitute part of a system whereby NOX activity, and thus redoxosomal signaling, is terminated.94
ROS Diffusion
Throughout the past two decades, the study of how specificity is conferred to ROS signaling has been plagued by the problem of H2O2 diffusion and membrane permeability. The concern was that O2– dismutates extremely rapidly to form H2O2, which then might freely diffuse through lipid bilayers and indiscriminately oxidize many molecules; thus, O2– and H2O2 would be too nonspecific for most signaling functions. It was suggested that ROS have signaling properties at low concentrations, but only host-defense functions at high concentrations (as in phagosomes).95 Indeed, it is now clear that ROS concentrations have an important influence on outcomes. However, concentration alone is not as simple a consideration as once thought, because the H2O2 concentrations at different sites within a single cell can differ significantly. In fact, this is the case even within the cytoplasm. Notably, the H2O2 concentration increases slightly at protein surfaces, with the mathematical probability of H2O2 diffusion decreasing as it nears the surface of a protein.96 Because the cytoplasm is not a well-mixed homogeneous aqueous solution, but rather a dynamic and complex arrangement of the protein machinery, the concentration and rate of diffusion of H2O2 vary across the cytoplasm.96 Furthermore, membranes (both the plasma membrane and the organelle membranes) significantly inhibit H2O2 diffusion. Recent research shows that, although H2O2 is uncharged, it is not as freely membrane permeable as was once thought.97,98 The membrane permeability of H2O2 depends on the biophysical properties of the membrane in question and, in many cases, requires the presence of an appropriate aquaporin channel.99,100 The concentration of H2O2 therefore can vary significantly between membrane-enclosed subcellular compartments. As an anion, O2– is not membrane permeable and cannot freely diffuse across membranes. However, specific anion channels may be able to transport O2– across membranes.82 The fledgling field of redoxosomal signaling has greatly broadened the research area of the transfer of O2– across membranes, suggesting that in specific instances O2– may be transported out of redoxosomes in a controlled manner (see below).
ROS Concentration via Compartmentalization in Redoxosomes
As we have previously demonstrated, the rate of ROS production in the redoxosome is in the micromolar per second range,54 whereas that in the phagosome is in the millimolar per second range.101 In both of these organelles, the ROS concentration is much higher inside than outside, because of the topology of the NOX transmembrane domains. One interesting consideration regarding NOX activity at the membrane of any organelle is the placement of negatively charged O2– within the lumen, while the H+ produced remains at the other side of the membrane (i.e., in the cytoplasm). This not only introduces the possibility of ROS accumulation and thus redox chemistry but also affects the electrogenic potential of the membrane across which O2– is formed. If positive charge builds up in the cytoplasm, while negative charge builds up in the endosome, there might be signaling effects even in the absence of ROS-triggered redox chemistry. Studies investigating this possibility have focused primarily on the chloride channel ClC-3,82,102 which is essential for the respiratory bursts of neutrophils because it facilitates the necessary charge neutralization of the phagosome.103 The data that are currently available indicate that this channel might also direct O2– out of some redoxosomal lumens. In these cases, O2– might compete with Cl– for flux across the endosomal membrane.82 In addition to ClC-3, several other membrane ion transporters, including IClswell (Table 1), may possibly allow charge neutralization of redoxosomes, by either transporting cations into the lumen or transporting O2– (or other anions) out.86,102 The activities of these ion transporters could, at least theoretically, affect the lumenal pH and thereby the rate of spontaneous dismutation of O2– and ultimately H2O2 levels. If O2– dismutation were significantly slowed by an increase in the redoxosomal lumenal pH, the increasing concentration of O2– within the redoxosome would act as a chemical driving force for movement of O2– out of the redoxosome via ClC-3 or another anion channel, and a corresponding decrease in the rate of diffusion of the competing ion, Cl–. By contrast, a decrease in the redoxosomal lumenal pH, as might be observed following fusion with the late endosomal compartment, would increase the rate of spontaneous dismutation of O2– and might enhance the diffusion of H2O2 across the endosomal membrane. Thus, the downstream effects of redoxosome signaling may depend on the number and types of anion channels in their membranes. The membrane composition of anion channels may also be influenced by the fusion of redoxosomes with other vesicular compartments. Interestingly, when anion channels (including ClC-3) in MCF-7 cells were blocked by applying 4′-diisothiocyano-2,2′-disulfonic acid stilbene (DIDS) or niflumic acid (NFA), O2– was prevented from exiting redoxosomes.82 These anion channel inhibitors did not significantly change the level of ROS production in these endosomes. In contrast, in the redoxosomes of SMCs, ClC-3 knockout led to a block of ROS production. Furthermore, the application of DIDS and NFA to these cells blocked downstream signaling by NF-κB.84 Why blocking anion channels inhibited ROS production in the redoxosomes of SMCs, but not those of MCF-7 cells, is not clear. One possible explanation relates to the fact that redoxosomes in MCF-7 cells use NOX232 whereas those in SMCs use NOX1.84 Thus, the two signaling pathways probably utilize different downstream proteins; also, the anion channels involved in charge neutralization of endosomal membranes may differ among cell types.102
Cytokines and Redoxosomes
Cytokines are secreted peptides or proteins with signaling functions and may act on either the cells that secrete them (autocrine) or others (paracrine or endocrine). They do so by binding to receptors on the cell surface, which in turn leads to the stimulation of intracellular signaling pathways. Hundreds of cytokines exist, and it is possible that many of them stimulate specific redoxosomal signaling pathways. However, only a few have been studied in this regard. Those best studied for their roles in redoxosome signaling are IL-1β and TNFα. The application of these cytokines to cultured cells was first shown to elicit ROS production more than 20 years ago.104,105 Nevertheless, the steps involved in redoxosomal signaling by these cytokines were delineated only recently. These are outlined below (see also Table 1).
IL-1β Redoxosome Signaling
In the case of IL-1β, its binding to the IL1 receptor (IL-1R) on the plasma membrane triggers the following complex chain of signaling events (see also Figure 1 and Table 1). Specific proteins (described below) are recruited to the plasma membrane. The receptor–ligand complex is endocytosed. Additional specific proteins are recruited, and the signal is transduced to the nucleus. IL-1R is a heterodimer composed of IL-1R1 and IL-1RAcP. The binding of IL-1β to this IL-1R complex is followed by recruitment of both MyD88 [a cytosolic protein (see Table 1)] and the GTPase Rac1. Prior to its recruitment, Rac1 in its inactive, GDP-bound form is held in the cytosol, through binding by a Rho guanine nucleotide dissociation inhibitor [RhoGDI (see Table 1)]. When IL-1β binds to IL-1R, the RhoGDI becomes phosphorylated by mechanisms that are not entirely clear and releases Rac1-GDP. Rac1 in this form is subject to two events: its translocation to the plasma membrane and the exchange of GTP in place of its bound GDP. Which of these occurs first is not known. The replacement of GDP with GTP is essential for Rac to become “activated” (see below and Table 1). This exchange requires a guanine exchange factor (GEF) protein. Some studies have indicated that Vav2, phosphorylated by c-Src, acts in this capacity,106 whereas other studies have implicated Vav1 or another GEF in this role (Table 1).107 In the case of MCF-7 redoxosomes, Vav1 has been characterized as a GEF capable of activating Rac1 after IL-1β stimulation,80 but whether Vav1 is phosphorylated by c-Src in this pathway remains unknown. Differences in the GEF protein involved and the other factors that act in these pathways are probably dependent on cell type.
Figure 1.
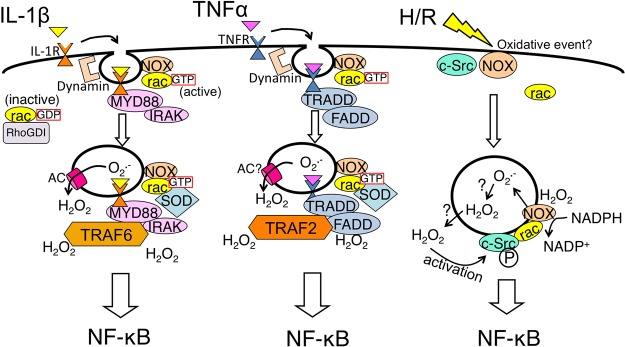
Schematic illustration of redoxosomal pathways: IL-1β pathway (left), TNFα pathway (middle), and hypoxia/reoxygenation (H/R) pathway (right). Major proteins known or strongly supported by data are shown. In all three pathways, a triggering event (receptor binding by cytokine IL-1β or TNFα, or an H/R event) initiates endocytosis (often dynamin-dependent). AC denotes anion channel. For more details, see Table 1 and the text.
Rac bound to GTP is in its “active” conformation and theoretically able to promote NOX activity. However, as mentioned above, Rac must be recruited to the plasma membrane, specifically to a site near the IL-1R complex, to conduct its signaling function. Rac associates with membranes by means of its prenylated tail, which is inaccessible as long as Rac is bound to RhoGDI. Lipid raft studies in MCF-7 cells have demonstrated that Vav1 is recruited to these domains simultaneously with Rac1; it is thus possible that the GDP to GTP exchange occurs after Rac has been recruited to the raft.80
Once Rac1 is recruited to IL-1R within the lipid raft, it facilitates recruitment of NOX2 to the receptor complex prior to endocytosis; this ultimately results in assembly of the entire NOX2 complex (including p67phox, p47phox, and other associated proteins), though whether the recruitment of these cytosolic subunits is completed at the plasma membrane or at the endocytosed vesicle remains unclear. It is clear, however, that endocytosis of IL-1R requires MyD88, but not Rac1, and that Rac1 is required for recruitment of NOX2 into the endosome with the IL-1R–MyD88–Rac-1 complex (see Table 1).32 In the absence of Rac1, the IL-1β-stimulated IL-1R–MyD88 complex will undergo endocytosis without NOX2. In the absence of MyD88, IL-1β-stimulated IL-1R remains at the plasma membrane. Endocytosis of the IL-1β–IL-1R–IL-1RAcP–MyD88–Rac-1–NOX2 receptor complex is often caveolin- and dynamin-dependent and leads to the formation of Rab5 positive endosomes.32,80
Following endocytosis, additional protein recruitment takes place. Specifically, superoxide dismutase 1 (SOD1) is recruited to the redoxosome surface and binds to Rac-1,32,88 and IRAK/TRAF6 is recruited and binds to IL-1R (Table 1). Key redox-dependent processes are required for this last step; NOX-dependent ROS produced in the interior of the endosomal compartment is required for recruitment of TRAF6 to the IL-1R complex.32 Neutralization of ROS within newly formed redoxosomes, using an excess of extracellular ROS scavenger enzymes (SOD and catalase), prevents TRAF6 recruitment and downstream signal activation (Table 1). H2O2 appears to be the key ROS involved in the redox regulation of TRAF6 and its recruitment to IL-1R;32 however, it remains unclear if the redox changes occur on TRAF6 and/or IL-1R. Either way, O2– produced by IL-1R-activated redoxosomes will be dismutated to H2O2 (either spontaneously or through SOD1 on its surface), providing a local redox driving force for the recruitment of TRAF6 to the IL-1R complex. This mechanism is thought to spatially restrict the recruitment of TRAF6 to the surface of ligand-activated redoxosomes. Subsequently, the IKKk protein is recruited, IKK is phosphorylated and activated, IκB is phosphorylated and degraded, and NF-κB is liberated to enter the nucleus and initiate transcriptional responses.
The recruitment of SOD1 to the surface of redoxosomes,32 where it binds Rac-1 (see Table 1), is of substantial importance to redoxosome function and has at least two functions in addition to the possible dismutation of O2– molecules (Figure 2). First, SOD1 inhibits the conversion of Rac-GTP to Rac-GDP;88 this decrease in the intrinsic GTPase activity of Rac promotes NOX activation. Second, the binding of SOD1 to Rac-1 is redox-regulated; specifically, when Rac is oxidized by H2O2, SOD1 dissociates from Rac-1.88 This dissociation of SOD1 enhances the intrinsic GTPase activity of Rac, leading to the inactivation of Rac-1 and thus also NOX. Thus, the SOD1–Rac-1 complex on redoxosomes serves as a self-limiting redox sensor for NOX-derived superoxide production. Whether SOD1 dismutase activity is required for the function of this sensor remains to be determined.
Figure 2.
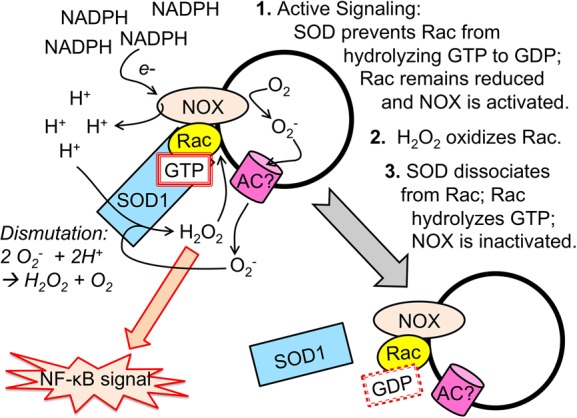
Schematic illustration of SOD1 and Rac interactions on the surface of redoxosomes and how this interaction controls NOX-mediated ROS production. In step 1, following redoxosome formation, SOD1 is recruited to the surface of the endosome and binds Rac-GTP. Binding of SOD1 to Rac stabilizes the GTP-bound conformation by inhibiting the intrinsic GTPase activity of Rac. NOX is active while bound to Rac-GTP, and NADPH supplies electrons to NOX, which transfers electrons onto oxygen inside the endosome. Protons may remain in the cytoplasm and thus perhaps build up a membrane potential with the lumenal side becoming more negative as O2– accumulates. This membrane potential may help to facilitate movement of O2– out of the redoxosome via an anion channel (AC). Upon exiting, the O2– may be dismutated by SOD1 at the redoxosomal surface. O2– may also spontaneously dismutate within the redoxosomal lumen and passively diffuse as H2O2 across the redoxosomal membrane (not shown). The accumulation of H2O2 at the redoxosomal surfaces transduces oxidative signals that allow for activation of the receptor complex (not shown) and the induction of pathways such as NF-κB. In step 2, as the concentration of H2O2 at the redoxosomal surface increases, Rac becomes oxidized. In step 3, the oxidation of Rac leads to the dissociation of SOD1. In the absence of binding of SOD1 to Rac, the intrinsic GTPase activity of Rac leads to rapid hydrolysis of GTP to GDP and the inactivation of the NOX complex. In this state, the activated receptor complex can likely still transduce its signal (not shown) but ROS production is terminated. In this manner, redoxosomes self-regulate the local production of ROS required to transduce its redox signal.
At least two additional mechanisms whereby redoxosomal signals may be terminated have been proposed. According to one, the early endosome fuses with a macropinosome and the proteins of the redoxosomal complex are recycled.94 In the other, negative charges build up inside the redoxosome whereas the cytoplasm is positively charged, until NOX can no longer pump electrons against the charge gradient.102 These three mechanisms for turning off a redoxosomal signal (dissociation of SOD1 from Rac, recycling, and the buildup of charge gradients) are not necessarily mutually exclusive, and additional mechanisms may also exist. Furthermore, the function of the SOD1/Rac-1 sensor on redoxosomes will be influenced by the rate of spontaneous dismutation within the redoxosome lumen, and thus as this endosomal compartment becomes more acidic (which occurs following fusion with late endosomes), the extent of uncoupling of SOD1 from Rac-1 would likely increase, leading to inactivation of NOX. Future studies will be necessary to identify specific events that dictate which mechanisms are utilized at the end of any given redoxosomal signaling event.
TNFα Redoxosome Signaling
When TNFα binds to the TNF receptor (TNFR1), redoxosome signaling is activated in a manner similar to that described above for binding of IL-1β to IL-1R. However, the two pathways differ in a few important respects. In particular, a distinct set of proteins is recruited, with TRADD, TRAF2, and RIP replacing MyD88, IRAK, and TRAF6 (Table 1). Moreover, TRAF2 recruitment is redox-dependent, as demonstrated by the fact that the loading of endosomes with SOD1 and catalase greatly decreased this recruitment;54 notably, levels of TRADD recruitment and TNFR1 endocytosis did not decrease with this treatment. Furthermore, TRAF2 recruitment is mediated by H2O2 formed from NOX2-generated O2–. This leads to initiation of TNFα-specific downstream signals.
Consistent with the differences in IL-1β- and TNFα-triggered endocytosis described above, Rac1-containing endosomes isolated after IL-1β stimulation were positive for IL-1R1 and TRAF6, but not for TNFR1 or TRAF2 (see Table 1). Conversely, Rac1-containing endosomes isolated after TNFα stimulation were positive for TNFR1 and TRAF2, but not for IL-1R1 or TRAF6.54 These results demonstrated that redoxosomal signaling specificity is dependent on the triggering ligand and reinforces the idea that ROS generated by redoxosomes acts locally to facilitate signal transduction.
IV. Redoxosome Signaling in the Context of Disease
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a motor neuron disease characterized by neurodegeneration and loss of voluntary muscle action. A small minority of ALS cases harbor specific mutations of SOD1. This fact, in combination with recent findings about the importance of SOD1 in the control of redoxosomal ROS in neuronal tissues,88 suggests that redoxosomal pathways are of importance in ALS. Notably, another protein that has been associated with this disease, alsin (see Table 1), is an early endosome-associated RhoGEF that is involved in redoxosomal signaling during ALS, particularly in the context of TNFα signaling (see Table 1). Overexpression of either alsin or the ALS-associated SOD1 mutant SOD1G93A in glial cells was sufficient to cause pro-inflammatory TNFα secretion.90 Overexpression of either alsin or SOD1G93A in glial cells also caused increases in endosomal ROS levels and Rac1 activation, and a decrease in the number of surviving of neuronal cells cocultured with these glial cells. Knockdown of NOX2 in glial cells in the context of glial SOD1G93A overexpression was protective to neuronal cocultured cells. Interestingly, when alsin and SOD1G93A were co-overexpressed in such glial cells, the pro-inflammatory effects were attenuated and neuronal toxicity decreased.90 The RhoGEF activity of alsin is probably targeted to Rac1 and/or Rab5 and likely mediates at least some of these effects. Furthermore, the recruitment of alsin specifically to the endosomal compartment appeared to regulate endosomal ROS induced by SOD1G93A. These findings indicate that alsin and SOD1 interact in multiple and complex pro-inflammatory and anti-inflammatory redoxosomal pathways during redoxosomal signaling, in both glia and neurons. Thus, targeting endocytic events that activate NOX2 in glia may be an effective means of decreasing the symptoms and severity of ALS.
Liver Ischemia/Reperfusion Injury
Ischemia followed by reperfusion of liver tissue, as occurs during liver transplantation, leads to the activation of inflammatory pathways, liver tissue damage, and increased TNFα levels in the plasma. In models of this condition using hypoxia/reoxygenation (H/R) treatment of cells in culture (see Figure 1), c-Src is recruited to the early endosomes and activated at the redoxosomal level in the absence of known ligands.89 This ligand-independent endocytic event is dynamin-dependent and leads to NF-κB activation as a consequence of NOX activation in redoxosomes.89 Both PTP and c-Src can be activated in the context of H/R,89,108 and the activation of c-Src in this context may therefore involve either direct oxidative activation or indirect activation as a consequence of PTP oxidation; this has not been tested yet. Recently, we demonstrated that the c-Src-dependent H/R-mediated redoxosomal pathways that are activated in cell culture are relevant in vivo in the context of liver ischemia/reperfusion (I/R).92 Wild-type mice subjected to surgically induced liver I/R produced higher levels of redoxosomal ROS than c-Src knockout counterparts. These higher levels of ROS correlated with an increased level of NF-κB activation in the liver, as well as with increased TNFα levels in plasma. Mouse strains in which NOX1 and NOX2, p47phox, or Rac-1 and/or Rac-2 are knocked out were also studied. In agreement with previous studies on redoxosome signaling, we found that these proteins were all important in the overall inflammatory response to I/R; however, several of these genes appear to play specific roles in hepatocytes and Kupffer cells of the liver.92 We found that hepatocytes themselves could produce low levels of TNFα after H/R and that this facilitated much higher levels of TNFα production by Kupffer cells. Thus, it appears that “ligand-independent” (I/R-dependent) redoxosomal signaling in hepatocytes leads to the production of TNFα, and that the endocytosis of this TNFα by nearby Kupffer cells leads to “ligand-dependent”, redoxosome-mediated NF-κB signaling in Kupffer cells. These cells in turn produce much higher levels of plasma TNFα. The molecular mechanism whereby endocytosis is initiated in hepatocytes after an I/R or H/R event is not known.
We refer to I/R- or H/R-triggered endocytosis as ligand-independent because no initiating ligand has been described, although we cannot exclude the possibility that an unidentified ligand may be involved.22 Cell types as diverse as HeLa cells, primary mouse dermal fibroblasts, and primary mouse hepatocytes can initiate endocytosis in response to reoxygenation.54,92 Is this endocytosis a direct response to extracellular O2 molecules that flood the medium during reoxygenation? Perhaps, as it is conceivable that endocytosis is triggered by oxidative modifications of lipids or proteins that are brought to the cell surface during hypoxia. Alternatively, unidentified autocrine or paracrine factors secreted during hypoxia may function as oxygen-dependent ligands, activating receptor-mediated endocytosis upon reoxygenation. It seems likely that such modifications or factors would also be produced during liver I/R in vivo. Our data indicate that hepatocytes utilize redoxosomal pathways to initiate a rapid cascade of TNFα production in response to reperfusion.92 Similarly, a study using redox-sensitive green fluorescent protein, roGFP, in a mouse I/R model has revealed that the overall level of oxidation in the mouse liver increases in vivo within minutes of reperfusion.109 Therefore, the time course of reperfusion probably allows almost instant induction of some ROS during initial formation of the first postreoxygenation redoxosomes. Specific reperfusion-activated autocrine or paracrine ligands or cell-surface modifications remain to be identified. Even TNFα itself could conceivably be one such reperfusion-activated ligand. For example, although the de novo synthesis of TNFα is NF-κB-dependent, in some cells, a small amount of precursor protein might already be present and available for release in soluble form. Given that the release or cleavage of the soluble ligand by TNFα-converting enzyme is indirectly redox-dependent,110 this cleavage may involve some of the as yet uncharacterized initiating redox events that occur during the first moments after reperfusion. Determining whether TNFα or other ligands function as postreoxygenation initiating factors will be important therapeutically. Such initiating factors could prove to be useful targets in preventing liver I/R inflammatory injury in the surgical setting. Endocytosis itself, and the signaling pathways that are activated during this process, may also prove to be useful targets in this context.
Cardiac Hypertrophy and Hypertension
Dysregulated angiotensin II signaling is a major contributor to hypertension and subsequent cardiac hypertrophy, which involves enlargement of cardiomyocytes. Redoxosomal signaling is very likely an important event in both healthy and diseased cardiomyocytes. In these cells, Rac1-regulated NOX2 produces O2– in response to stimulation with angiotensin II (Table 1), and this leads to activation of the Akt signaling pathway and cardiomyocyte hypertrophy.111 It is possible that this ROS production occurs in early endosomes, because a recent report indicates that, at least in vascular SMCs, Akt signaling occurs at EEA1-positive early endosomes and regulates p38 MAP kinases.112 If a similar pathway is active in cardiomyocytes, the redoxosome may be an important component of their Akt activation pathway, as well. However, the pathways may be somewhat different in these cell types, as suggested by the fact that NOX2 is important for ROS production in cardiomyocytes, leading to cardiac hypertrophy,111 whereas it is NOX1 activation that leads to Akt signaling in the endosomes of SMCs, leading to neointimal hyperplasia that can be associated with hypertension.83,85
Studies of cardiomyocyte responses to angiotensin II have suggested that redox signaling may occur not only in the canonical early endosome type of redoxosomes but also in some late endosomes. This concept is based on studies of the activation of CamKII in these cells, an event that requires the oxidation of specific methionines in CamKII. Recent data showed that when angiotensin II activates the p38K pathway, CamKII methionines are oxidized in biphasic fashion.113 The delayed phase of this signaling was found to depend on endocytosis; although the upstream trigger has not been identified, it could be NOX-generated ROS within early, or perhaps even late, endosomes. This delayed phase of signaling led to the expression of specific mRNAs several hours after the initial endocytic event.113 Thus, although early endosomes are the best characterized of the redoxosomes, the maturation and fusion of NOX-containing early endosomes with the later endosomal compartment may perhaps also play a part in some oxidative redox signaling pathways. Further studies to investigate these possibilities are needed. It should be noted that angiotensin II signaling involving redox chemistry is not limited to endosomal pathways; in neuronal cells, angiotensin II-dependent mitochondrial NOX4 signaling has been demonstrated, as well.114 For the development of specific therapies for hypertension and cardiac hypertrophy, a much improved understanding of the redox pathways in SMC and cardiomyocyte endosomes is needed.
Infection and Immunity
Recently, redoxosome activity was described as part of the neutrophil-mediated immune response to invading pathogens. Neutrophils not only form their canonical NOX2-containing phagosomes but also can form NOX2-containing redoxosomes in response to endotoxin.87 The redoxosomal signaling pathway thus triggered leads to neutrophil priming activity.87
Redoxosomes might also act within the adaptive branch of the immune system, in antigen-presenting cells. This possibility is implied by the discovery that the redox state of late endosomes in antigen-presenting cells is regulated by γ-interferon-inducible lysosomal thiol reductase (GILT),115 which reduces disulfide bonds in proteins in the late endosomes of monocytes.115 The need for reduction of these cysteine thiols could imply that these residues had been oxidized upstream of this late endosome event, perhaps during an early redoxosomal NOX-mediated oxidizing event. Thus, it may be that not only IL-1β and TNFα but also IFNγ can activate redoxosomal signaling pathways (Table 1).
Chronic hepatitis B currently infects ∼1 million Americans, and several thousand die of this disease every year in this country alone. During hepatitis B infection of human HepaRG cell lines, the transport of viral particles subjects the virus to reducing conditions in late endosomes.116 Thus, as in the case of GILT, reduction of proteins is observed specifically in late endosomes.
A Pattern of Redox Function in Early and Late Endosomes
In pathological states as diverse as liver I/R, bacterial infection, ALS, and cardiac hypertrophy, production of ROS and oxidation of signaling proteins take place specifically in early endosomes, as described above. In contrast, in the studies of antigen-presenting cells and hepatitis B mentioned above, reduction of previously oxidized thiols occurs in the late endosomes. Fluorescence microscopy studies have also shown reduction of disulfide bonds in late endosomes.117 Thus, comparing the findings regarding early versus late endosomes suggests that the former most frequently facilitate oxidative redox reactions that contribute to cell signaling pathways, whereas the late endosomes typically support reducing reactions that may or may not qualify as “signaling” events. Possibly, exceptions to this pattern may exist (see the description of CamKII above). In any event, therapies targeted at blocking either oxidative signaling specifically at the early endosome or reduction of thiols specifically at the late endosome, in specific cell types, may prove to be beneficial in future drug development.
V. Future Directions
The activation of cytokine-dependent signaling pathways by redoxosomes is enormously complex, and the differences between cell types have only begun to be evident. The therapeutic potential of uncovering these details is tremendous. For example, diseases as diverse as ALS and liver I/R injury, both of which involve TNFα-mediated redoxosomes, could potentially be prevented or at least ameliorated if specific targets in or related to their specific redoxosomes can be identified. IL-1β-mediated disease states are also numerous and diverse.118 Moreover, hundreds of other cytokines might be able to initiate redoxosome-mediated signaling, and the potential links to cardiac, vascular, and infectious diseases are further indications of the huge diversity of processes that may potentially be treated by therapies targeting specific mediators of redoxosomal pathways.
Clearly, effective targets will have to have very specialized roles and be highly localized within the cell, as systemic administration of “antioxidants”, which indiscriminately and loosely target all ROS, has failed repeatedly in clinical trials.119,120 One major challenge will be to find cellular redoxosome-associated targets that are specific enough to avoid disrupting major signaling pathways that are crucial to good health. In this regard, the current state of redoxosome research strongly points to NF-κB activation and possibly a few other signaling pathways. Determining exactly how these diverse redoxosome signals contribute to the transduction of specific NF-κB and/or other signals will be extremely important. As shown in Figure 1, all known redoxosome signaling pathways activate NF-κB signaling. Conversely, not all TNFα-mediated NF-κB signaling leads to disease states such as ALS or reperfusion-type liver damage. Thus, a precise understanding of the downstream effects of redoxosomal signals in specific cell types will be vital to identifying appropriate targets of therapy. A second challenge will be to engineer drugs or other therapies that affect these precise targets in very specific ways. As shown in Table 1, the majority of proteins involved in redoxosomal signaling are not known to be redox-modified. However, research on these proteins is generally incomplete, and if some are in fact redox-modified, their redox-active cysteines, methionines, or other residues might represent useful therapeutic targets. As such, the possibilities for future research and discovery on cytokine-mediated redoxosomal signaling appear to be numerous.
Acknowledgments
We express gratitude to Dr. Christine Blaumueller for her expert editorial assistance in the preparation of the manuscript.
Glossary
Abbreviations
- ALS
amyotrophic lateral sclerosis
- ATF-1
Akt-activating transcription factor-1
- CSK
c-Src kinase
- DIDS
4′-diisothiocyano-2,2′-disulfonic acid stilbene
- EGFR
epidermal growth factor receptor
- ER
endoplasmic reticulum
- G-CSF
granulocyte colony stimulating factor
- GEF
guanine exchange factor
- GILT
γ-interferon-inducible lysosomal thiol reductase
- GPCR
G-protein-coupled receptor
- H/R
hypoxia/reoxygenation
- IFNγ
interferon γ
- IL-1β
interleukin-1β
- IL-1R
IL1 receptor
- I/R
ischemia/reperfusion
- NFA
niflumic acid
- NGF
nerve growth factor
- NOX
NADPH oxidase
- PI3K
phosphatidylinositol 3-kinase
- PNS
perinuclear space
- PTP
protein tyrosine phosphatase
- RhoGDI
rho guanine nucleotide dissociation inhibitor
- ROS
reactive oxygen species
- SMC
smooth muscle cell
- SOD
superoxide dismutase
- TGF-β
transforming growth factor β
- TNF
tumor necrosis factor
- TNFR
TNF receptor.
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01 DK051315 and K01 DK083429, the Animal Models Core funded through the Center for Gene Therapy (P30 DK54759), and the Roy J. Carver Chair in Molecular Medicine.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- van’t Erve T. J.; Wagner B. A.; Ryckman K. K.; Raife T. J.; Buettner G. R. (2013) The Concentration of Glutathione in Human Erythrocytes is a Heritable Trait. Free Radical Biol. Med. 65, 742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh J. L.; Wagner B. A.; van’t Erve T. J.; Zehr P. S.; Berg D. J.; Halfdanarson T. R.; Yee N. S.; Bodeker K. L.; Du J.; Roberts L. J. II; Drisko J.; Levine M.; Buettner G. R.; Cullen J. J. (2013) Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): Results from a phase I clinical trial. Cancer Chemother. Pharmacol. 71, 765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B., and Gutteridge J. C. M. (1989) Free Radicals in Biology and Medicine, 2nd ed., Clarendon Press, Oxford, U.K. [Google Scholar]
- Burgoyne J. R.; Oka S.; Ale-Agha N.; Eaton P. (2013) Hydrogen peroxide sensing and signaling by protein kinases in the cardiovascular system. Antioxid. Redox Signaling 18, 1042–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran A.; Cotter T. G. (2013) Redox regulation of protein kinases. FEBS J. 280, 1944–1965. [DOI] [PubMed] [Google Scholar]
- Rhee S. G.; Kang S. W.; Jeong W.; Chang T. S.; Yang K. S.; Woo H. A. (2005) Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr. Opin. Cell Biol. 17, 183–189. [DOI] [PubMed] [Google Scholar]
- Ray P. D.; Huang B. W.; Tsuji Y. (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signalling 24, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner J. J.; Parsons Z. D.; Cummings A. H.; Zhou H.; Gates K. S. (2011) Redox regulation of protein tyrosine phosphatases: Structural and chemical aspects. Antioxid. Redox Signaling 15, 77–97. [DOI] [PubMed] [Google Scholar]
- Seth D.; Rudolph J. (2006) Redox regulation of MAP kinase phosphatase 3. Biochemistry 45, 8476–8487. [DOI] [PubMed] [Google Scholar]
- Buhrman G.; Parker B.; Sohn J.; Rudolph J.; Mattos C. (2005) Structural mechanism of oxidative regulation of the phosphatase Cdc25B via an intramolecular disulfide bond. Biochemistry 44, 5307–5316. [DOI] [PubMed] [Google Scholar]
- Salmeen A.; Barford D. (2008) Methods for preparing crystals of reversibly oxidized proteins: Crystallization of protein tyrosine phosphatase 1B as an example. Methods Mol. Biol. 476, 101–116. [DOI] [PubMed] [Google Scholar]
- Goldstein B. J.; Mahadev K.; Wu X. (2005) Redox paradox: Insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes 54, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez J. C.; Manuia M.; Burnett M. E.; Betancourt O.; Boivin B.; Shaw D. E.; Tonks N. K.; Mazar A. P.; Donate F. (2008) Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. U.S.A. 105, 7147–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunati A. M.; Pagano M. A.; Bindoli A.; Rigobello M. P. (2010) Thiol redox systems and protein kinases in hepatic stellate cell regulatory processes. Free Radical Res. 44, 363–378. [DOI] [PubMed] [Google Scholar]
- Giannoni E.; Buricchi F.; Raugei G.; Ramponi G.; Chiarugi P. (2005) Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 25, 6391–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemble D. J.; Sun G. (2009) Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc. Natl. Acad. Sci. U.S.A. 106, 5070–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H.; Hao Q.; Rutherford S. A.; Low B.; Zhao Z. J. (2005) Inactivation of SRC family tyrosine kinases by reactive oxygen species in vivo. J. Biol. Chem. 280, 23918–23925. [DOI] [PubMed] [Google Scholar]
- Giannoni E.; Taddei M. L.; Chiarugi P. (2010) Src redox regulation: Again in the front line. Free Radical Biol. Med. 49, 516–527. [DOI] [PubMed] [Google Scholar]
- Zhu P.; Tan M. J.; Huang R. L.; Tan C. K.; Chong H. C.; Pal M.; Lam C. R.; Boukamp P.; Pan J. Y.; Tan S. H.; Kersten S.; Li H. Y.; Ding J. L.; Tan N. S. (2011) Angiopoietin-like 4 protein elevates the prosurvival intracellular O2–:H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell 19, 401–415. [DOI] [PubMed] [Google Scholar]
- Buettner G. R. (1993) The pecking order of free radicals and antioxidants: Lipid peroxidation, α-tocopherol, and ascorbate. Arch. Biochem. Biophys. 300, 535–543. [DOI] [PubMed] [Google Scholar]
- Yu X.-M.; Askalan R.; Keil G. J.; Salter M. W. (1997) NMDA Channel Regulation by Channel-Associated Protein Tyrosine Kinase Src. Science 275, 674–678. [DOI] [PubMed] [Google Scholar]
- Oakley F. D.; Abbott D.; Li Q.; Engelhardt J. F. (2009) Signaling components of redox active endosomes: The redoxosomes. Antioxid. Redox Signaling 11, 1313–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn M. T.; Gauss K. A. (2004) Structure and regulation of the neutrophil respiratory burst oxidase: Comparison with nonphagocyte oxidases. J. Leukocyte Biol. 76, 760–781. [DOI] [PubMed] [Google Scholar]
- Shao D.; Segal A. W.; Dekker L. V. (2010) Subcellular localisation of the p40phox component of NADPH oxidase involves direct interactions between the Phox homology domain and F-actin. Int. J. Biochem. Cell Biol. 42, 1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijles D. N.; Howlin B. J.; Li J. M. (2012) Consensus in silico computational modelling of the p22phox subunit of the NADPH oxidase. Comput. Biol. Chem. 39, 6–13. [DOI] [PubMed] [Google Scholar]
- Picciocchi A.; Debeurme F.; Beaumel S.; Dagher M. C.; Grunwald D.; Jesaitis A. J.; Stasia M. J. (2011) Role of putative second transmembrane region of Nox2 protein in the structural stability and electron transfer of the phagocytic NADPH oxidase. J. Biol. Chem. 286, 28357–28369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G.; Cao Z.; Xu X.; van Meir E. G.; Lambeth J. D. (2001) Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 269, 131–140. [DOI] [PubMed] [Google Scholar]
- Ameziane-El-Hassani R.; Morand S.; Boucher J. L.; Frapart Y. M.; Apostolou D.; Agnandji D.; Gnidehou S.; Ohayon R.; Noel-Hudson M. S.; Francon J.; Lalaoui K.; Virion A.; Dupuy C. (2005) Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. J. Biol. Chem. 280, 30046–30054. [DOI] [PubMed] [Google Scholar]
- Kawahara T.; Ritsick D.; Cheng G.; Lambeth J. D. (2005) Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J. Biol. Chem. 280, 31859–31869. [DOI] [PubMed] [Google Scholar]
- Montezano A. C.; Touyz R. M. (2012) Oxidative stress, Noxs, and hypertension: Experimental evidence and clinical controversies. Ann. Med. 44(Suppl. 1), S2–S16. [DOI] [PubMed] [Google Scholar]
- Cheng G.; Diebold B. A.; Hughes Y.; Lambeth J. D. (2006) Nox1-dependent reactive oxygen generation is regulated by Rac1. J. Biol. Chem. 281, 17718–17726. [DOI] [PubMed] [Google Scholar]
- Li Q.; Harraz M. M.; Zhou W.; Zhang L. N.; Ding W.; Zhang Y.; Eggleston T.; Yeaman C.; Banfi B.; Engelhardt J. F. (2006) Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol. Cell. Biol. 26, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroviarski Y.; Debbabi M.; Bachoual R.; Perianin A.; Gougerot-Pocidalo M. A.; El-Benna J.; Dang P. M. (2010) Phosphorylation of NADPH oxidase activator 1 (NOXA1) on serine 282 by MAP kinases and on serine 172 by protein kinase C and protein kinase A prevents NOX1 hyperactivation. FASEB J. 24, 2077–2092. [DOI] [PubMed] [Google Scholar]
- Morand S.; Ueyama T.; Tsujibe S.; Saito N.; Korzeniowska A.; Leto T. L. (2009) Duox maturation factors form cell surface complexes with Duox affecting the specificity of reactive oxygen species generation. FASEB J. 23, 1205–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer N. Y.; Yan Z. Y.; Boudreau R. L.; Zhang Y. L.; Luo M. H.; Li Q. A.; Tian X.; Shah A. M.; Davisson R. L.; Davidson B.; Banfi B.; Engelhardt J. F. (2011) Control of Hepatic Nuclear Superoxide Production by Glucose 6-Phosphate Dehydrogenase and NADPH Oxidase-4. J. Biol. Chem. 286, 8977–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes P. S. (1998) Mitochondrial proton leak and superoxide generation: An hypothesis. Biochem. Soc. Trans. 26, S331. [DOI] [PubMed] [Google Scholar]
- Collins Y.; Chouchani E. T.; James A. M.; Menger K. E.; Cocheme H. M.; Murphy M. P. (2012) Mitochondrial redox signalling at a glance. J. Cell Sci. 125, 801–806. [DOI] [PubMed] [Google Scholar]
- Wang S. B.; Murray C. I.; Chung H. S.; Van Eyk J. E. (2013) Redox regulation of mitochondrial ATP synthase. Trends Cardiovasc. Med. 23, 14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushio-Fukai M. (2009) Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signaling 11, 1289–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito E.; Melo E. P.; Yang Y.; Wahlander A.; Neubert T. A.; Ron D. (2010) Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell 40, 787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R. F.; Ma Z.; Liu Z.; Terada L. S. (2010) Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol. Cell. Biol. 30, 3553–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakihana T.; Nagata K.; Sitia R. (2012) Peroxides and peroxidases in the endoplasmic reticulum: Integrating redox homeostasis and oxidative folding. Antioxid. Redox Signaling 16, 763–771. [DOI] [PubMed] [Google Scholar]
- Cyr A. R.; Domann F. E. (2011) The redox basis of epigenetic modifications: From mechanisms to functional consequences. Antioxid. Redox Signaling 15, 551–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G. H.; Ryan J. J.; Archer S. L. (2013) The role of redox signaling in epigenetics and cardiovascular disease. Antioxid. Redox Signaling 18, 1920–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hagan H. M.; Wang W.; Sen S.; Destefano Shields C.; Lee S. S.; Zhang Y. W.; Clements E. G.; Cai Y.; Van Neste L.; Easwaran H.; Casero R. A.; Sears C. L.; Baylin S. B. (2011) Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG islands. Cancer Cell 20, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. C.; Wang K. Y.; Shen C. K. (2013) DNA 5-methylcytosine demethylation activities of the mammalian DNA methyltransferases. J. Biol. Chem. 288, 9084–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adcock I. M.; Cosio B.; Tsaprouni L.; Barnes P. J.; Ito K. (2005) Redox regulation of histone deacetylases and glucocorticoid-mediated inhibition of the inflammatory response. Antioxid. Redox Signaling 7, 144–152. [DOI] [PubMed] [Google Scholar]
- Rahman I.; Marwick J.; Kirkham P. (2004) Redox modulation of chromatin remodeling: Impact on histone acetylation and deacetylation, NF-κB and pro-inflammatory gene expression. Biochem. Pharmacol. 68, 1255–1267. [DOI] [PubMed] [Google Scholar]
- Haddad J. J. (2002) Oxygen-sensing mechanisms and the regulation of redox-responsive transcription factors in development and pathophysiology. Respir. Res. 3, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Colavitti R.; Rovira I. I.; Finkel T. (2005) Redox-dependent transcriptional regulation. Circ. Res. 97, 967–974. [DOI] [PubMed] [Google Scholar]
- Yang J.; Marden J. J.; Fan C.; Sanlioglu S.; Weiss R. M.; Ritchie T. C.; Davisson R. L.; Engelhardt J. F. (2003) Genetic redox preconditioning differentially modulates AP-1 and NF κB responses following cardiac ischemia/reperfusion injury and protects against necrosis and apoptosis. Mol. Ther. 7, 341–353. [DOI] [PubMed] [Google Scholar]
- Mazzanti M.; Bustamante J. O.; Oberleithner H. (2001) Electrical Dimension of the Nuclear Envelope. Physiol. Rev. 81, 1–19. [DOI] [PubMed] [Google Scholar]
- Buser C.; Walther P.; Mertens T.; Michel D. (2007) Cytomegalovirus Primary Envelopment Occurs at Large Infoldings of the Inner Nuclear Membrane. J. Virol. 81, 3042–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Spencer N. Y.; Oakley F. D.; Buettner G. R.; Engelhardt J. F. (2009) Endosomal Nox2 Facilitates Redox-Dependent Induction of NF-κB by TNFα. Antioxid. Redox Signaling 11, 1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hordijk P. L. (2006) Regulation of NADPH oxidases: The role of Rac proteins. Circ. Res. 98, 453–462. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Piston D. W.; Goodman R. H. (2002) Regulation of Corepressor Function by Nuclear NADH. Science 295, 1895–1897. [DOI] [PubMed] [Google Scholar]
- Mesecke N.; Spang A.; Deponte M.; Herrmann J. M. (2008) A novel group of glutaredoxins in the cis-Golgi critical for oxidative stress resistance. Mol. Biol. Cell 19, 2673–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M.; Zhang C.; Boini K. M.; Thacker A. M.; Li P. L. (2011) Membrane raft-lysosome redox signalling platforms in coronary endothelial dysfunction induced by adipokine visfatin. Cardiovasc. Res. 89, 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palande K.; Roovers O.; Gits J.; Verwijmeren C.; Iuchi Y.; Fujii J.; Neel B. G.; Karisch R.; Tavernier J.; Touw I. P. (2011) Peroxiredoxin-controlled G-CSF signalling at the endoplasmic reticulum-early endosome interface. J. Cell Sci. 124, 3695–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deliu E.; Brailoiu G. C.; Mallilankaraman K.; Wang H.; Madesh M.; Undieh A. S.; Koch W. J.; Brailoiu E. (2012) Intracellular endothelin type B receptor-driven Ca2+ signal elicits nitric oxide production in endothelial cells. J. Biol. Chem. 287, 41023–41031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfy M.; Remenyi A.; Korcsmaros T. (2012) Endosomal crosstalk: Meeting points for signaling pathways. Trends Cell Biol. 22, 447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Guglielmo G. M.; Baass P. C.; Ou W. J.; Posner B. I.; Bergeron J. J. (1994) Compartmentalization of SHC, GRB2 and mSOS, and hyperphosphorylation of Raf-1 by EGF but not insulin in liver parenchyma. EMBO J. 13, 4269–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miaczynska M.; Pelkmans L.; Zerial M. (2004) Not just a sink: Endosomes in control of signal transduction. Curr. Opin. Cell Biol. 16, 400–406. [DOI] [PubMed] [Google Scholar]
- Schneider-Brachert W.; Tchikov V.; Neumeyer J.; Jakob M.; Winoto-Morbach S.; Held-Feindt J.; Heinrich M.; Merkel O.; Ehrenschwender M.; Adam D.; Mentlein R.; Kabelitz D.; Schutze S. (2004) Compartmentalization of TNF receptor 1 signaling: Internalized TNF receptosomes as death signaling vesicles. Immunity 21, 415–428. [DOI] [PubMed] [Google Scholar]
- Tchikov V.; Bertsch U.; Fritsch J.; Edelmann B.; Schutze S. (2011) Subcellular compartmentalization of TNF receptor-1 and CD95 signaling pathways. Eur. J. Cell Biol. 90, 467–475. [DOI] [PubMed] [Google Scholar]
- Le Roy C.; Wrana J. L. (2005) Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat. Rev. Mol. Cell Biol. 6, 112–126. [DOI] [PubMed] [Google Scholar]
- Grant B. D.; Donaldson J. G. (2009) Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 10, 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandvig K.; Torgersen M. L.; Raa H. A.; van Deurs B. (2008) Clathrin-independent endocytosis: From nonexisting to an extreme degree of complexity. Histochem. Cell Biol. 129, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigerer A.; Gilleron J.; Bogorad R. L.; Marsico G.; Nonaka H.; Seifert S.; Epstein-Barash H.; Kuchimanchi S.; Peng C. G.; Ruda V. M.; Del Conte-Zerial P.; Hengstler J. G.; Kalaidzidis Y.; Koteliansky V.; Zerial M. (2012) Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature 485, 465–470. [DOI] [PubMed] [Google Scholar]
- Sharma M.; Giridharan S. S.; Rahajeng J.; Naslavsky N.; Caplan S. (2009) MICAL-L1 links EHD1 to tubular recycling endosomes and regulates receptor recycling. Mol. Biol. Cell 20, 5181–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial M.; McBride H. (2001) Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117. [DOI] [PubMed] [Google Scholar]
- van Meer G.; de Kroon A. I. (2011) Lipid map of the mammalian cell. J. Cell Sci. 124, 5–8. [DOI] [PubMed] [Google Scholar]
- Rajendran L.; Simons K. (2005) Lipid rafts and membrane dynamics. J. Cell Sci. 118, 1099–1102. [DOI] [PubMed] [Google Scholar]
- Pike L. J. (2009) The challenge of lipid rafts. J. Lipid Res. 50(Suppl.), S323–S328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K.; Sampaio J. L. (2011) Membrane organization and lipid rafts. Cold Spring Harbor Perspect. Biol. 3, a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao D.; Segal A. W.; Dekker L. V. (2003) Lipid rafts determine efficiency of NADPH oxidase activation in neutrophils. FEBS Lett. 550, 101–106. [DOI] [PubMed] [Google Scholar]
- Vilhardt F.; van Deurs B. (2004) The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. EMBO J. 23, 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A. Y.; Yi F.; Zhang G.; Gulbins E.; Li P. L. (2006) Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 47, 74–80. [DOI] [PubMed] [Google Scholar]
- Drisdel R. C.; Alexander J. K.; Sayeed A.; Green W. N. (2006) Assays of protein palmitoylation. Methods 40, 127–134. [DOI] [PubMed] [Google Scholar]
- Oakley F. D.; Smith R. L.; Engelhardt J. F. (2009) Lipid rafts and caveolin-1 coordinate interleukin-1β (IL-1β)-dependent activation of NFκB by controlling endocytosis of Nox2 and IL-1β receptor 1 from the plasma membrane. J. Biol. Chem. 284, 33255–33264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreck R.; Rieber P.; Baeuerle P. A. (1991) Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-κB transcription factor and HIV-1. EMBO J. 10, 2247–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbengegwi D. R.; Li Q.; Li C.; Bear C. E.; Engelhardt J. F. (2008) Evidence for a Superoxide Permeability Pathway in Endosomal Membranes. Mol. Cell. Biol. 28, 3700–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu X.; Filali M.; Stanic B.; Takapoo M.; Sheehan A.; Bhalla R.; Lamb F. S.; Miller F. J. Jr. (2011) A critical role for chloride channel-3 (CIC-3) in smooth muscle cell activation and neointima formation. Arterioscler., Thromb., Vasc. Biol. 31, 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller F. J. Jr.; Filali M.; Huss G. J.; Stanic B.; Chamseddine A.; Barna T. J.; Lamb F. S. (2007) Cytokine activation of nuclear factor κB in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ. Res. 101, 663–671. [DOI] [PubMed] [Google Scholar]
- Miller F. J. Jr.; Chu X.; Stanic B.; Tian X.; Sharma R. V.; Davisson R. L.; Lamb F. S. (2010) A differential role for endocytosis in receptor-mediated activation of Nox1. Antioxid. Redox Signaling 12, 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda J. J.; Filali M. S.; Moreland J. G.; Miller F. J.; Lamb F. S. (2010) Activation of swelling-activated chloride current by tumor necrosis factor-α requires ClC-3-dependent endosomal reactive oxygen production. J. Biol. Chem. 285, 22864–22873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb F. S.; Hook J. S.; Hilkin B. M.; Huber J. N.; Volk A. P.; Moreland J. G. (2012) Endotoxin priming of neutrophils requires endocytosis and NADPH oxidase-dependent endosomal reactive oxygen species. J. Biol. Chem. 287, 12395–12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harraz M. M.; Marden J. J.; Zhou W.; Zhang Y.; Williams A.; Sharov V. S.; Nelson K.; Luo M.; Paulson H.; Schoneich C.; Engelhardt J. F. (2008) SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Invest. 118, 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Zhang Y.; Marden J. J.; Banfi B.; Engelhardt J. F. (2008) Endosomal NADPH oxidase regulates c-Src activation following hypoxia/reoxygenation injury. Biochem. J. 411, 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Spencer N. Y.; Pantazis N. J.; Engelhardt J. F. (2011) Alsin and SOD1(G93A) proteins regulate endosomal reactive oxygen species production by glial cells and proinflammatory pathways responsible for neurotoxicity. J. Biol. Chem. 286, 40151–40162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marden J. J.; Zhang Y.; Oakley F. D.; Zhou W.; Luo M.; Jia H. P.; McCray P. B. Jr.; Yaniv M.; Weitzman J. B.; Engelhardt J. F. (2008) JunD protects the liver from ischemia/reperfusion injury by dampening AP-1 transcriptional activation. J. Biol. Chem. 283, 6687–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer N. Y.; Zhou W.; Li Q.; Zhang Y.; Luo M.; Yan Z.; Lynch T. J.; Abbott D.; Banfi B.; Engelhardt J. F. (2013) Hepatocytes produce TNFα following hypoxia-reoxygenation and liver ischemia-reperfusion in a NADPH oxidase- and c-Src-dependent manner. Am. J. Physiol. 305, G84–G94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Peng F.; Gao B.; Ingram A. J.; Krepinsky J. C. (2012) High glucose-induced RhoA activation requires caveolae and PKCβ1-mediated ROS generation. Am. J. Physiol. 302, F159–F172. [DOI] [PubMed] [Google Scholar]
- Carter B. J.; Anklesaria P.; Choi S.; Engelhardt J. F. (2009) Redox modifier genes and pathways in amyotrophic lateral sclerosis. Antioxid. Redox Signaling 11, 1569–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal V. J.; Fanburg B. L. (2000) Reactive oxygen species in cell signaling. Am. J. Physiol. 279, L1005–L1028. [DOI] [PubMed] [Google Scholar]
- Chung Y. H.; Xia J.; Margulis C. J. (2007) Diffusion and residence time of hydrogen peroxide and water in crowded protein environments. J. Phys. Chem. B 111, 13336–13344. [DOI] [PubMed] [Google Scholar]
- Miller E. W.; Dickinson B. C.; Chang C. J. (2010) Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 15681–15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco M. R.; Marinho H. S.; Cyrne L.; Antunes F. (2004) Decrease of H2O2 plasma membrane permeability during adaptation to H2O2 in Saccharomyces cerevisiae. J. Biol. Chem. 279, 6501–6506. [DOI] [PubMed] [Google Scholar]
- Folmer V.; Pedroso N.; Matias A. C.; Lopes S. C.; Antunes F.; Cyrne L.; Marinho H. S. (2008) H2O2 induces rapid biophysical and permeability changes in the plasma membrane of Saccharomyces cerevisiae. Biochim. Biophys. Acta 1778, 1141–1147. [DOI] [PubMed] [Google Scholar]
- Bienert G. P.; Moller A. L.; Kristiansen K. A.; Schulz A.; Moller I. M.; Schjoerring J. K.; Jahn T. P. (2007) Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 282, 1183–1192. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C.; Hampton M. B.; Livesey J. H.; Kettle A. J. (2006) Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: Implications for microbial killing. J. Biol. Chem. 281, 39860–39869. [DOI] [PubMed] [Google Scholar]
- Lamb F. S.; Moreland J. G.; Miller F. J. Jr. (2009) Electrophysiology of reactive oxygen production in signaling endosomes. Antioxid. Redox Signaling 11, 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreland J. G.; Davis A. P.; Bailey G.; Nauseef W. M.; Lamb F. S. (2006) Anion channels, including ClC-3, are required for normal neutrophil oxidative function, phagocytosis, and transendothelial migration. J. Biol. Chem. 281, 12277–12288. [DOI] [PubMed] [Google Scholar]
- Rhee S. G.; Bae Y. S.; Lee S. R.; Kwon J. (2000) Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Sci. STKE 2000, pe1. [DOI] [PubMed] [Google Scholar]
- Sulciner D. J.; Irani K.; Yu Z. X.; Ferrans V. J.; Goldschmidt-Clermont P.; Finkel T. (1996) rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-κB activation. Mol. Cell. Biol. 16, 7115–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni D.; Bohl B.; Courtneidge S. A.; Bokoch G. M. (2008) The involvement of the tyrosine kinase c-Src in the regulation of reactive oxygen species generation mediated by NADPH oxidase-1. Mol. Biol. Cell 19, 2984–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming W.; Li S.; Billadeau D. D.; Quilliam L. A.; Dinauer M. C. (2007) The Rac effector p67phox regulates phagocyte NADPH oxidase by stimulating Vav1 guanine nucleotide exchange activity. Mol. Cell. Biol. 27, 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.; Zhang Z.; Wang L. (2008) Small interference RNA against PTP-1B reduces hypoxia/reoxygenation induced apoptosis of rat cardiomyocytes. Apoptosis 13, 383–393. [DOI] [PubMed] [Google Scholar]
- Haga S.; Remington S. J.; Morita N.; Terui K.; Ozaki M. (2009) Hepatic ischemia induced immediate oxidative stress after reperfusion and determined the severity of the reperfusion-induced damage. Antioxid. Redox Signaling 11, 2563–2572. [DOI] [PubMed] [Google Scholar]
- Scott A. J.; O’Dea K. P.; O’Callaghan D.; Williams L.; Dokpesi J. O.; Tatton L.; Handy J. M.; Hogg P. J.; Takata M. (2011) Reactive oxygen species and p38 mitogen-activated protein kinase mediate tumor necrosis factor α-converting enzyme (TACE/ADAM-17) activation in primary human monocytes. J. Biol. Chem. 286, 35466–35476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingtgen S. D.; Tian X.; Yang J.; Dunlay S. M.; Peek A. S.; Wu Y.; Sharma R. V.; Engelhardt J. F.; Davisson R. L. (2006) Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol. Genomics 26, 180–191. [DOI] [PubMed] [Google Scholar]
- Nazarewicz R. R.; Salazar G.; Patrushev N.; San Martin A.; Hilenski L.; Xiong S.; Alexander R. W. (2011) Early endosomal antigen 1 (EEA1) is an obligate scaffold for angiotensin II-induced, PKC-α-dependent Akt activation in endosomes. J. Biol. Chem. 286, 2886–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.; Cavolo S. L.; Levitan E. S. (2012) Delayed endosome-dependent CamKII and p38 kinase signaling in cardiomyocytes destabilizes Kv4.3 mRNA. J. Mol. Cell. Cardiol. 52, 971–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case A. J.; Li S. M.; Basu U.; Tian J.; Zimmerman M. C. (2013) Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons. Am. J. Physiol. 305, H19–H28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings K. T.; Cresswell P. (2011) Disulfide reduction in the endocytic pathway: Immunological functions of γ-interferon-inducible lysosomal thiol reductase. Antioxid. Redox Signaling 15, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macovei A.; Petrareanu C.; Lazar C.; Florian P.; Branza-Nichita N. (2013) Regulation of hepatitis B virus infection by rab5, rab7, and the endolysosomal compartment. J. Virol. 87, 6415–6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Chen H.; Vlahov I. R.; Cheng J. X.; Low P. S. (2006) Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proc. Natl. Acad. Sci. U.S.A. 103, 13872–13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C. A. (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhubl S. R. (2008) Why have antioxidants failed in clinical trials?. Am. J. Cardiol. 101, 14D–19D. [DOI] [PubMed] [Google Scholar]
- Galasko D. R.; Peskind E.; Clark C. M.; Quinn J. F.; Ringman J. M.; Jicha G. A.; Cotman C.; Cottrell B.; Montine T. J.; Thomas R. G.; Aisen P. (2012) Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 69, 836–841. [DOI] [PMC free article] [PubMed] [Google Scholar]