

Abstract
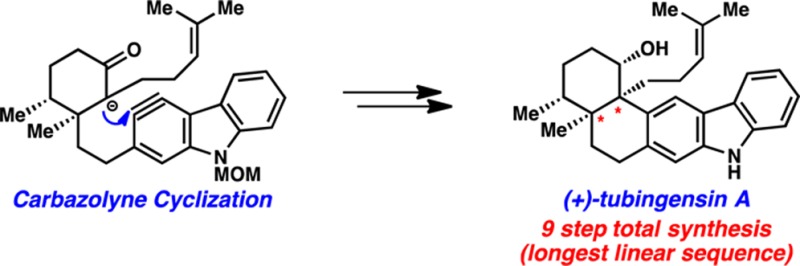
We report the enantiospecific total synthesis of (+)-tubingensin A. Our synthesis features an aryne cyclization to efficiently introduce the vicinal quaternary stereocenters of the natural product and proceeds in only nine steps (longest linear sequence) from known compounds.
Carbazole-containing natural products have long provided a fruitful area for scientific discovery.1 The complex alkaloid tubingensin A (1)2 and its structural isomer tubingensin B (2)3 are members of this class that were isolated from the fungus Aspergillus tubingensis by Gloer and co-workers in 1989 (Figure 1). Compounds 1 and 2 are both postulated to arise biosynthetically from the indole diterpenoid anominine (3)4 and have been shown to display antiviral, anticancer, and insecticidal activity.2,3 Structurally, tubingensin A (1) possesses a disubstituted carbazole that is fused to a densely functionalized cis-decalin framework.5 Moreover, the decalin core contains four contiguous stereocenters, two of which are vicinal quaternary centers. With regard to synthetic efforts toward tubingensin A (1), Bonjoch and co-workers reported an approach to assemble the natural product’s scaffold,6 and Li and Nicolaou recently completed an elegant bio-inspired total synthesis of 1 that proceeds in 23 linear steps from a cyclohexyl precursor.4a
Figure 1.
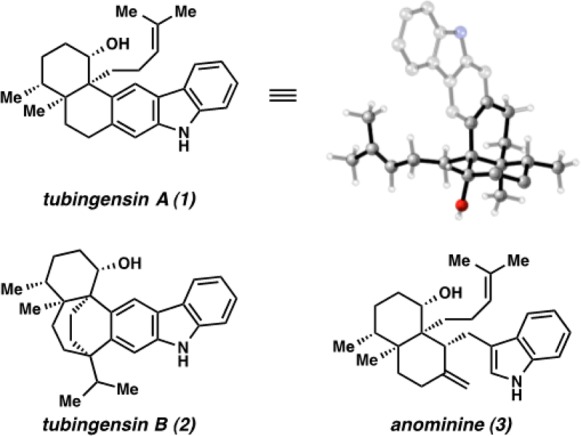
Tubingensin A (1) and related family members 2 and 3.
Given our interest in assembling congested scaffolds using heterocyclic aryne intermediates,7,8 we questioned if the vicinal quaternary stereocenters of tubingensin A (1) could be introduced using aryne cyclization methodology. Although arynes have been used in an array of interesting trapping experiments,9 their use in the assembly of sterically congested linkages is limited.10 Furthermore, to our knowledge, arynes have not previously been employed for the introduction of vicinal quaternary stereocenters. In this Communication, we report the use of a late-stage carbazolyne cyclization to efficiently construct the tubingensin A framework. Our approach not only demonstrates that arynes can be used for the assembly of challenging molecular scaffolds but also provides (+)-1 in just nine steps (longest linear sequence) from known compounds.
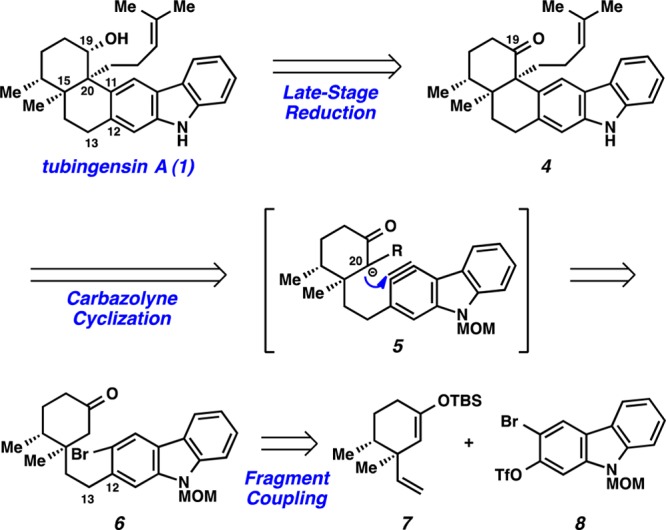
Our retrosynthetic analysis for the synthesis of tubingensin A (1) is shown in Scheme 1. It was envisioned that the natural product would arise by late-stage reduction of ketone 4. This intermediate could be constructed through a key carbazolyne11 cyclization (see transition structure 5). If successful, this transformation would fashion the challenging vicinal quaternary stereocenters and provide the cis-decalin framework of the natural product. Ketone 6, the precursor to potential carbazolyne cyclization substrates, would ultimately arise from the coupling of two simple fragments, cyclohexyl derivative 7 and carbazole triflate 8.
Scheme 1.
The key coupling partners were prepared in a straightforward manner (Figure 2). Commercially available hydroxycarbazole 9 was brominated using a known procedure.12 Subsequent O-triflation and N-protection afforded 8 in 61% yield over three steps. The synthesis of cyclohexyl fragment 7 began from commercially available (+)-dihydrocarvone (10), which was elaborated to enone 11 using a known three-step procedure.13 Vinyl cuprate addition and trapping of the resulting enolate furnished silyl enol ether 7 as a single diastereomer.14
Figure 2.
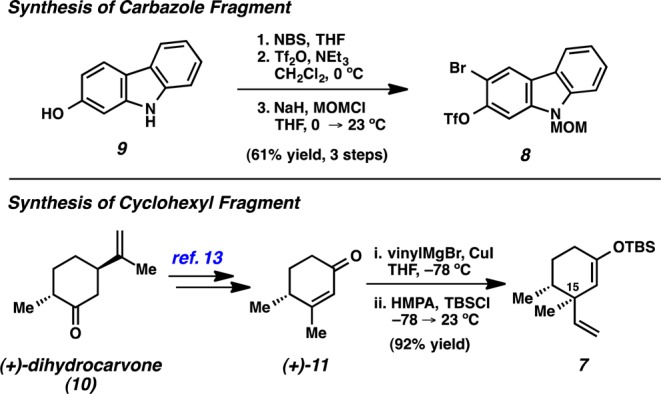
Synthesis of carbazole and cyclohexyl fragments.
With cyclohexyl fragment 7 and carbazole triflate 8 in hand, we pursued their coupling and the ensuing carbazolyne cyclization (Scheme 2). Treatment of 7 with 9-BBN15 led to chemoselective hydroboration of the terminal olefin. Subsequent Pd-catalyzed cross-coupling16 with 8 proceeded smoothly to generate coupled product 12. Addition of TBAF to the reaction mixture then delivered ketone 6. The highly optimized conversion of 7 and 8 to 6 in one pot17 provides a rare example of a silyl enol ether being employed as a ketone protecting group in a complex setting.14,18 To control regioselectivity of enolate formation in the forthcoming carbazolyne cyclization,19 ketone 6 was oxidized to enone 13 using IBX and NMO.20 To our delight, exposure of enone 13 to NaNH2 and t-BuOH in THF8c,21 facilitated the desired cyclization and afforded C20-epimeric products 14 and 15 in 44% combined yield (1:2.3 ratio). We hypothesize that 14 forms kinetically as the major product but is prone to epimerization under the reaction conditions.22
Scheme 2.

Having demonstrated the feasibility of the carbazolyne cyclization, we next prepared a C20-substituted substrate to test if the method could be used to forge the critical vicinal quaternary stereocenters (Scheme 3). Sequential treatment of enone 13 with LiHMDS and Eschenmoser’s salt led to C20 alkylation.23 Exposure of the crude adduct to mCPBA furnished enone 16.24 Addition of prenylcuprate23 provided 17 as an inconsequential mixture of diastereomers. Unfortunately, attempts to effect the carbazolyne cyclization of substrate 17 were unsuccessful. Analysis of the major product suggested that a skeletal rearrangement had occurred, rather than the desired formation of 18.
Scheme 3.
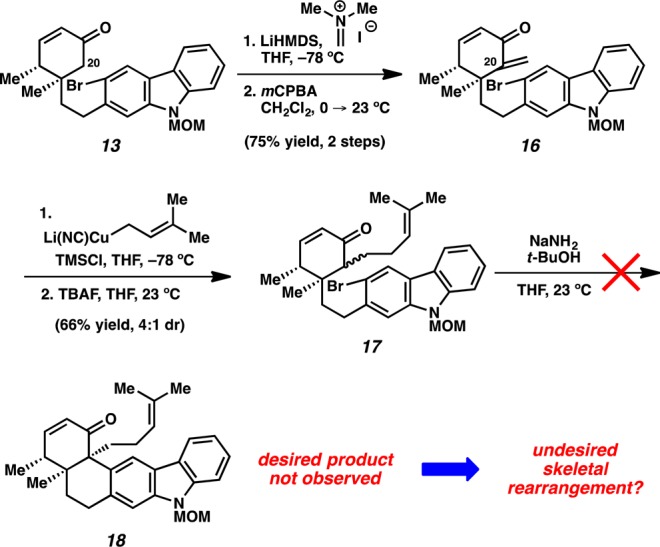
To identify the skeletal rearrangement product, we prepared the simpler C20 methylated substrate 19 and exposed it to aryne cyclization conditions (Scheme 4). The major product obtained was analogous to that formed in the attempted cyclization of 17. Interestingly, although the desired C11–C20 linkage was intact, a bond was unexpectedly formed between the carbazole and C19. In addition, the C15–C16 and C19–C20 linkages had been severed. 2D NMR analysis eventually revealed the rearranged structure to be compound 20. We propose that the desired enolate–carbazolyne intermediate indeed undergoes cyclization, but the resulting anion cyclizes onto C19, analogous to C–C aryne insertion reactions of 1,3-dicarbonyls (see transition structure 21).10e,25 A subsequent retro-Diels–Alder reaction, as shown in transition structure 22, and protonation yield 20.
Scheme 4.
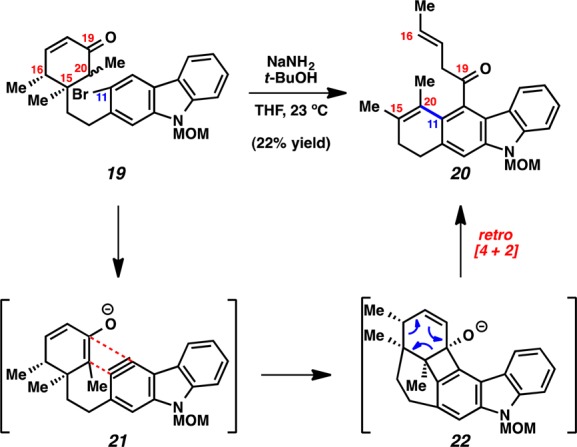
As the attempted cyclization indeed led to the desired C11–C20 bond formation, we postulated that a modified substrate might be more likely to undergo the desired transformation without rearrangement. Specifically, we envisioned that a substrate lacking the alkene in the six-membered ring could not suffer from the retro [4+2] cycloaddition and would thus not be prone to cyclohexyl ring fragmentation.26 Therefore, we elected to prepare enol ether 26 (Scheme 5). In contrast to enone substrates 13 and 17, the selectivity for C20 enolate formation for the critical carbazolyne cyclization would be dictated by the controlled formation of 26 as a single enol ether isomer.
Scheme 5.

The design and synthesis of substrate 26 ultimately proved fruitful and enabled an efficient enantiospecific total synthesis of tubingensin A (1), which is summarized in Scheme 5.27 Vinyl cuprate addition to enone 11, followed by trapping with Me2(i-Pr)SiCl (DMIPSCl), gave enol ether 23. Next, a one-pot hydroboration/Suzuki coupling (analogous to that described previously) afforded the desired coupled product 24 in 64% yield. Using the labile DMIPS enol ether, enone 25 was generated via a two-step sequence.28 Subsequent cuprate addition and trapping with TESCl supplied aryne cyclization precursor 26. Fortuitously, in the key step, aryne cyclization delivered the desired product 28 in 84% yield and established the vicinal quaternary centers with complete diastereoselectivity. The structure of 28 was verified by X-ray crystallography. Only two steps remained to complete the total synthesis: N-deprotection and ketone reduction. The removal of the MOM group of 28 provided ketone 4 smoothly. However, reduction proved more challenging. An array of reducing agents were examined, all of which gave the undesired C19 epimer as the major product.29 The best conditions using Na/i-PrOH delivered a 1:2 ratio of tubingensin A (1) and its C19 epimer. It should be noted that oxidation of epi-1 gives 4, so the undesired epimer can be readily recycled.30 Nonetheless, synthetic (+)-1 was found to be identical to an authentic sample in all respects.
In summary, we have achieved a concise enantiospecific total synthesis of tubingensin A (1). Our synthesis features an aryne cyclization to efficiently introduce the vicinal quaternary stereocenters of the natural product and proceeds in only nine steps (longest linear sequence) from known compounds. Further efforts to probe the use of arynes for the assembly of challenging molecular scaffolds are currently underway in our laboratory.
Acknowledgments
The authors are grateful to the NIH-NIGMS (R01 GM090007), Boehringer Ingelheim, DuPont, Eli Lilly, Amgen, AstraZeneca, Roche, Bristol-Myers Squibb, the A. P. Sloan Foundation, the Dreyfus Foundation, the S.T. Li Foundation, UCLA, the NSF (M.A.C., DGE-1144087), the ACS Division of Organic Chemistry (A.E.G.), UCLA DYF program (A.E.G.), and the Foote Family (A.E.G. and A.L.S.) for financial support. We thank Steven Loskot for experimental assistance, the Garcia-Garibay laboratory (UCLA) for access to instrumentation, Dr. Saeed Khan (UCLA) for X-ray analysis, and Professor Gloer (University of Iowa) for an authentic sample of tubingensin A. These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the National Center for Research Resources (S10RR025631).
Supporting Information Available
Detailed experimental procedures, compound characterization, and crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† A.E.G. and A.L.S. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Schmidt A. W.; Reddy K. R.; Knölker H.-J. Chem. Rev. 2012, 112, 3193–3328. [DOI] [PubMed] [Google Scholar]; b Knölker H.-J.; Reddy K. R. Chem. Rev. 2002, 102, 4303–4428. [DOI] [PubMed] [Google Scholar]; c Bergman J.; Pelcman B. Pure Appl. Chem. 1990, 62, 1967–1976. [Google Scholar]
- TePaske M. R.; Gloer J. B.; Wicklow D. T.; Dowd P. F. J. Org. Chem. 1989, 54, 4743–4746. [Google Scholar]
- TePaske M. R.; Gloer J. B.; Wicklow D. T.; Dowd P. F. Tetrahedron Lett. 1989, 30, 5965–5968. [Google Scholar]
- For biosynthetic proposals, see:; a Bian M.; Wang Z.; Xiong X.; Sun Y.; Matera C.; Nicolaou K. C.; Li A. J. Am. Chem. Soc. 2012, 134, 8078–8081. [DOI] [PubMed] [Google Scholar]; b Marcos I. S.; Moro R. F.; Costales I.; Escola M. A.; Basabe P.; Díez D.; Urones J. G. Tetrahedron 2009, 65, 10235–10242. [Google Scholar]
- The 3D representation of 1 was obtained using B3LYP/6-31G* calculations (geometry optimization), using MacSpartan software.
- Bradshaw B.; Etxebarria-Jardí G.; Bonjoch J. Org. Biomol. Chem. 2008, 6, 772–778. [DOI] [PubMed] [Google Scholar]
- For reviews regarding hetarynes, see:; a Bronner S. M.; Goetz A. E.; Garg N. K. Synlett 2011, 18, 2599–2604. [Google Scholar]; b Reinecke M. G. Tetrahedron 1982, 38, 427–498. [Google Scholar]; c Kauffmann T.; Wirthwein R. Angew. Chem., Int. Ed. 1971, 10, 20–33. [Google Scholar]
- a Bronner S. M.; Bahnck K. B.; Garg N. K. Org. Lett. 2009, 11, 1007–1010. [DOI] [PubMed] [Google Scholar]; b Bronner S. M.; Goetz A. E.; Garg N. K. J. Am. Chem. Soc. 2011, 133, 3832–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Huters A. D.; Quasdorf K. W.; Styduhar E. D.; Garg N. K. J. Am. Chem. Soc. 2011, 133, 15797–15799. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Goetz A. E.; Bronner S. M.; Cisneros J. D.; Melamed J. M.; Paton R. S.; Houk K. N.; Garg N. K. Angew. Chem., Int. Ed. 2012, 51, 2758–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Goetz A. E.; Garg N. K. Nat. Chem. 2013, 5, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Styduhar E. D.; Huters A. D.; Weires N. A.; Garg N. K. Angew. Chem., Int. Ed. 2013, 52, 12422–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tadross P. M.; Stoltz B. M. Chem. Rev. 2012, 112, 3550–3577. [DOI] [PubMed] [Google Scholar]; b Gampe C. M.; Carreira E. M. Angew. Chem., Int. Ed. 2012, 51, 3766–3778. [DOI] [PubMed] [Google Scholar]
- For the use of arynes to construct sp2–sp3 linkages with a newly formed tertiary carbon center, see refs (8a,8c) and the following:; a Kametani T.; Kigasawa K.; Hiiragi M.; Kusama O. J. Heterocycl. Chem. 1973, 10, 31–33. [Google Scholar]; b Semmelhack M. F.; Chong B. P.; Jones L. D. J. Am. Chem. Soc. 1972, 94, 8629–8630. [DOI] [PubMed] [Google Scholar]; c Semmelhack M. F.; Chong B. P.; Stauffer R. D.; Rogerson T. D.; Chong A.; Jones L. D. J. Am. Chem. Soc. 1975, 97, 2507–2516. [DOI] [PubMed] [Google Scholar]; d Soorukram D.; Qu T.; Barrett A. G. M. Org. Lett. 2008, 10, 3833–3835. [DOI] [PubMed] [Google Scholar]; e Danheiser R. L.; Helgason A. L. J. Am. Chem. Soc. 1994, 116, 9471–9479. [Google Scholar]; f Tambar U. K.; Ebner D. C.; Stoltz B. M. J. Am. Chem. Soc. 2006, 128, 11752–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Best W. M.; Wege D. Tetrahedron Lett. 1981, 22, 4877–4880. [Google Scholar]; h Best W. M.; Wege D. Aust. J. Chem. 1986, 39, 647–666. [Google Scholar]; i Gribble G. W.; Saulnier M. G.; Sibi M. P.; Obaza-Nutaitis J. A. J. Org. Chem. 1984, 49, 4518–4523. [Google Scholar]; j Buszek K. R.; Bixby D. L. Tetrahedron Lett. 1995, 36, 9129–9132. [Google Scholar]; k Chen C.-L.; Sparks S. M.; Martin S. F. J. Am. Chem. Soc. 2006, 128, 13696–13697. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Sparks S. M.; Chen C.-L.; Martin S. F. Tetrahedron 2007, 63, 8619–8635. [DOI] [PMC free article] [PubMed] [Google Scholar]; m McManus H. A.; Fleming M. J.; Lautens M. Angew. Chem., Int. Ed. 2007, 46, 433–436. [DOI] [PubMed] [Google Scholar]; n Buszek K. R.; Brown N.; Luo D. Org. Lett. 2009, 11, 201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Candito D. A.; Dobrovolsky D.; Lautens M. J. Am. Chem. Soc. 2012, 134, 15572–15580. [DOI] [PubMed] [Google Scholar]
- Brown R. F. C.; Choi N.; Coulston K. J.; Eastwood F. W.; Ercole F.; Horvath J. M.; Mattinson M.; Mulder R. J.; Ooi H. C. Liebigs Ann. Recl. 1997, 1931–1940. [Google Scholar]
- Bonesi S. M.; Ponce M. A.; Erra-Balsells R. J. Heterocycl. Chem. 2004, 41, 161–171. [Google Scholar]
- White J. D.; Grether U. M.; Lee C.-S. Org. Synth. 2005, 82, 108–114. [Google Scholar]
- a Jung M. E.; Berliner J. A.; Angst D.; Yue D.; Koroniak L.; Watson A. D.; Li R. Org. Lett. 2005, 7, 3933–3935. [DOI] [PubMed] [Google Scholar]; b Jung M. E.; Berliner J. A.; Koroniak L.; Gugiu B. G.; Watson A. D. Org. Lett. 2008, 10, 4207–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaura N.; Ishiyama T.; Sasaki H.; Ishikawa M.; Satoh M.; Suzuki A. J. Am. Chem. Soc. 1989, 111, 314–321. [Google Scholar]
- For a pertinent review, see:Chemler S. R.; Trauner D.; Danishefsky S. J. Angew. Chem., Int. Ed. 2001, 40, 4544–4568. [DOI] [PubMed] [Google Scholar]
- Attempts to utilize alternative Pd sources provided <10% yield of 6. The use of alternative bases and/or solvents in the coupling step also led to minimal formation of the desired product.
- a Potter G. A.; Barrie S. E.; Jarman M.; Rowlands M. G. J. Med. Chem. 1995, 38, 2463–2471. [DOI] [PubMed] [Google Scholar]; b Grosse A. C.; Moorhoff C. M. Heteroat. Chem. 1997, 8, 361–369. [Google Scholar]; c Moorhoff C. M. J. Chem. Soc., Perkin Trans. 1 1997, 1987–1996. [Google Scholar]
- Attempts to cyclize ketone 6 were unsuccessful due to a lack of regioselectivity during enolate formation; see the SI for details.
- a Nicolaou K. C.; Montagnon T.; Baran P. S. Angew. Chem., Int. Ed. 2002, 41, 993–996. [DOI] [PubMed] [Google Scholar]; b Annette P.-J. C.; Müller C. C.; Cooper H. M.; Williams C. M. Tetrahedron 2010, 66, 6842–6850. [Google Scholar]
- Caubere P. Acc. Chem. Res. 1974, 7, 301–308. [Google Scholar]
- Re-exposure of 14 to the reaction conditions predominantly yielded 15. Attempts to further elaborate 14 or 15 were unsuccessful.
- Piers E.; Marais P. C. J. Chem. Soc., Chem. Commun. 1989, 1222–1223. [Google Scholar]
- Lipshutz B. H.; Ellsworth E. L.; Dimock S. H.; Smith R. A. J. J. Am. Chem. Soc. 1990, 112, 4404–4410. [Google Scholar]
- a Guyot M.; Molho D. Tetrahedron Lett. 1973, 14, 3433–3436. [Google Scholar]; b Tambar U. K.; Stoltz B. M. J. Am. Chem. Soc. 2005, 127, 5340–5341. [DOI] [PubMed] [Google Scholar]
- Although this strategy would not preclude the corresponding cyclobutyl ring formation, it was plausible that such an intermediate would fragment to give the desired carbazolyne cyclization product.
- Alternatively, 26 could be derived from 17; see the SI for details.
- Analogous attempts to employ TBS ether 12 were unsuccessful.
- Reduction of 4 with LiAlH4 provided a ∼1:7.7 mixture favoring epi-1. Attempts to reduce 28 with sodium naphthalenide gave a ∼1:5 ratio favoring the undesired epimer, while borohydride reducing agents (e.g., L-Selectride, NaBH4) were found to be unreactive. Meerwein–Ponndorf–Verley reduction was also unsuccessful.
- Dess–Martin oxidation of epi-1 gives 4 (60% yield, unoptimized). Exhaustive efforts to effect Mitsunobu inversion of epi-1 failed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.