

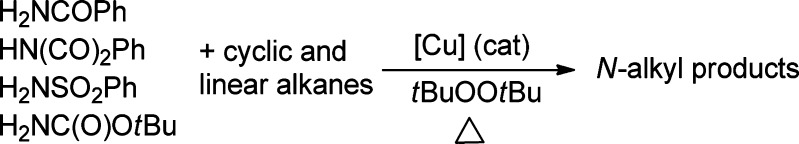
Abstract

We report a set of rare copper-catalyzed reactions of alkanes with simple amides, sulfonamides, and imides (i.e., benzamides, tosylamides, carbamates, and phthalimide) to form the corresponding N-alkyl products. The reactions lead to functionalization at secondary C–H bonds over tertiary C–H bonds and even occur at primary C–H bonds. [(phen)Cu(phth)] (1-phth) and [(phen)Cu(phth)2] (1-phth2), which are potential intermediates in the reaction, have been isolated and fully characterized. The stoichiometric reactions of 1-phth and 1-phth2 with alkanes, alkyl radicals, and radical probes were investigated to elucidate the mechanism of the amidation. The catalytic and stoichiometric reactions require both copper and tBuOOtBu for the generation of N-alkyl product. Neither 1-phth nor 1-phth2 reacted with excess cyclohexane at 100 °C without tBuOOtBu. However, the reactions of 1-phth and 1-phth2 with tBuOOtBu afforded N-cyclohexylphthalimide (Cy-phth), N-methylphthalimide, and tert-butoxycyclohexane (Cy-OtBu) in approximate ratios of 70:20:30, respectively. Reactions with radical traps support the intermediacy of a tert-butoxy radical, which forms an alkyl radical intermediate. The intermediacy of an alkyl radical was evidenced by the catalytic reaction of cyclohexane with benzamide in the presence of CBr4, which formed exclusively bromocyclohexane. Furthermore, stoichiometric reactions of [(phen)Cu(phth)2] with tBuOOtBu and (Ph(Me)2CO)2 at 100 °C without cyclohexane afforded N-methylphthalimide (Me-phth) from β-Me scission of the alkoxy radicals to form a methyl radical. Separate reactions of cyclohexane and d12-cyclohexane with benzamide showed that the turnover-limiting step in the catalytic reaction is the C–H cleavage of cyclohexane by a tert-butoxy radical. These mechanistic data imply that the tert-butoxy radical reacts with the C–H bonds of alkanes, and the subsequent alkyl radical combines with 1-phth2 to form the corresponding N-alkyl imide product.
Introduction
Amination reactions are crucial to the synthesis of medicinally important compounds. The amination of C–H bonds provides a route to N-alkyl and N-aryl amine derivatives that avoids typical functional group interconversions and reactants containing a functional group already present at the position where a C–N bond is desired. Intermolecular reactions that convert unactivated C–H bonds to C–N bonds are particularly challenging to achieve, but such reactions could directly modify complex molecules, create chemical feedstocks, or create functionalized polymers.1,2 Methods for the oxidation of alkanes by the combination of peroxides and iron complexes have been developed,3−12 but intermolecular functionalization of purely unactivated C–H bonds with reagents that form products containing common nitrogen-based functionality are rare.13 Most intermolecular amidations of C–H bonds have been catalyzed by copper complexes and occur at benzylic or allylic positions.
A few intermolecular aminations of C–H bonds occur with nitrene precursors to form sulfonamides.14−26 Reactions catalyzed by dirhodium complexes are the most developed for the synthesis of complex molecules.15,27 Although remarkable developments and applications have been reported for intramolecular reactions, intermolecular reactions are more limited. The selectivity of these reactions depends on the electron density at the C–H bond of the substrate, such that the preferred sites of reactions are tertiary, benzylic, and secondary C–H bonds.14,28 When a nitrene is the reactive intermediate, the nitrogen-based reagent is limited to those containing just one substituent.
We sought an alternative route to the intermolecular functionalization of alkyl C–H bonds that could form N-alkyl amides, carbamates, and imides, in addition to the formation of sulfonamides. Amides, carbamates, and imides are more common synthetic intermediates or final products than those generated by prior copper-catalyzed reactions with alkyl C–H bonds.29 We also sought to conduct these transformations with readily accessible complexes of first-row metals. Although copper-catalyzed reactions at allylic and benzylic C–H bonds with carboxylic acids and sulfonamides in the presence of peroxides is well-known (Karasch-Sosnovsky reaction),30−35 and the mechanism of these reactions has been studied,34,36−40 the amidation of C–H bonds with such reagents has been limited to reactions at benzylic C–H bonds.32,33
We report the reactions of common amides, carbamates, and imides with alkanes to form N-alkyl derivatives with simple copper catalysts and a peroxide (Scheme 1). The amidation of alkanes under our catalytic conditions preferentially forms the products from amidation at secondary sites over tertiary sites and even leads to the functionalization of primary C–H bonds in some cases. Mechanistic data from the stoichiometric reactions of isolated copper amidate or imidate complexes indicate that the transformation of alkanes to N-alkyl products likely occurs by the reactions of a alkyl radicals with copper(II) amidate and imidate complexes.
Scheme 1. Conversion of Alkanes to N-Alkyl Products.
Results and Discussion
1. Reaction Development of Intermolecular Amidation
To identify effective precatalysts and conditions for intermolecular amidation of unactivated C–H bonds, we evaluated the reactivity of benzamide (0.5 mmol) and cyclohexane (10 equiv) with 2.5–5.0 mol % of various copper precatalysts and oxidants. Results for the copper-catalyzed amidation of cyclohexane with benzamide are presented in Table 1. The combination of copper and tBuOOtBu was crucial for catalysis; other peroxide-based oxidants and redox-active 3d transition metals did not yield the amidation product.41
Table 1. Development of the Catalytic Amidation of Cyclohexanea.

| entry | catalyst | ligand | oxidant | yield (%)b |
|---|---|---|---|---|
| 1 | Cu(OAc)2 | tBuOOtBu | 10 | |
| 2 | CuCl2 | tBuOOtBu | 22 | |
| 3 | CuCl | tBuOOtBu | 24 | |
| 4 | CuI | tBuOOtBu | 27 | |
| 5 | CuI | bipy | tBuOOtBu | 36 |
| 6 | CuI | phen | tBuOOtBu | 95 |
| 7 | CuI | (MeO)2Phen | tBuOOtBu | 99 (76) |
| 8 | (L1)CuCl | tBuOOtBu | 83c | |
| 9 | [(phen)CuCl]2(μ2-Cl)2 | tBuOOtBu | 83c | |
| 10 | CuI | (MeO)2Phen | tBuOOAc | <5 |
| 11 | CuI | (MeO)2Phen | tBuOOH | <5 |
| 12 | CuI | (MeO)2Phen | H2O2 | <5 |
| 13 | (MeO)2Phen | tBuOOtBu | <5 |
Conditions: 0.5 mmol of benzamide, 5.0 mmol of cyclohexane, 0.0125 mmol of catalyst, 0.0125 mmol of ligand, 1.0 mmol of oxidant, 1 mL of PhH at 100 °C for 24 h.
GC yield with n-dodecane as the internal standard. Isolated yield in parentheses.
5 mol %. L1 = Me2NCH2CH2N=CH(2-HO–C6H4).
The reactions catalyzed by Cu(I) and Cu(II) salts without ligand gave the N-cyclohexylbenzamide in low yields (entry 1–4, 10–27%). However, the reactions catalyzed by Cu(I) and bipyridine (bipy), gave N-cyclohexylbenzamide in a measurably higher 36% yield. Reactions catalyzed by the well-defined [(L1)CuCl] precatalyst (entry 8) (L1 = (((2-(dimethylamino)ethyl)imino) methyl)phenoxide) provided 83% of N-cyclohexylbenzamide. Finally, we found that the reaction of benzamide with cyclohexane catalyzed by the simple combination of phenanthroline (phen) (entry 6) or 4,7-dimethoxy-phenantronline ((MeO)2phen) (entry 7) and CuI formed N-cyclohexylbenzamide in nearly quantitative yield.
2. Scope and Selectivity of Intermolecular Amidation
The scope of benzamides that undergo this C–H bond amination reaction is summarized in Table 2. Benzamides containing electron-withdrawing groups, such as Cl, Br, F, and CF3 (a–d), underwent the reaction to give good yields of the corresponding amidation product (73–82%). Benzamides containing electron-donating groups, such as Me, tBu, and OMe (e–k), also underwent the amidation process, but the isolated yields of the amidation products were slightly lower (55–78%) than those of the benzamides containing electron-withdrawing groups. Heteroaromatic amides, such as 2-thienylamide (m) and 2-pyridylamide (l), also underwent the oxidative coupling with cyclohexane to give the corresponding N-alkyl products. The reaction of the 2-pyridylamide is noteworthy because the 2-pyridylamide product could form a strong chelating ligand that would prevent catalysis. The position of the substituent on the benzamide (ortho, meta, or para) did not have a pronounced effect on the reaction yields.
Table 2. Amidation of Cyclohexane with Various Amides and Imidesa.
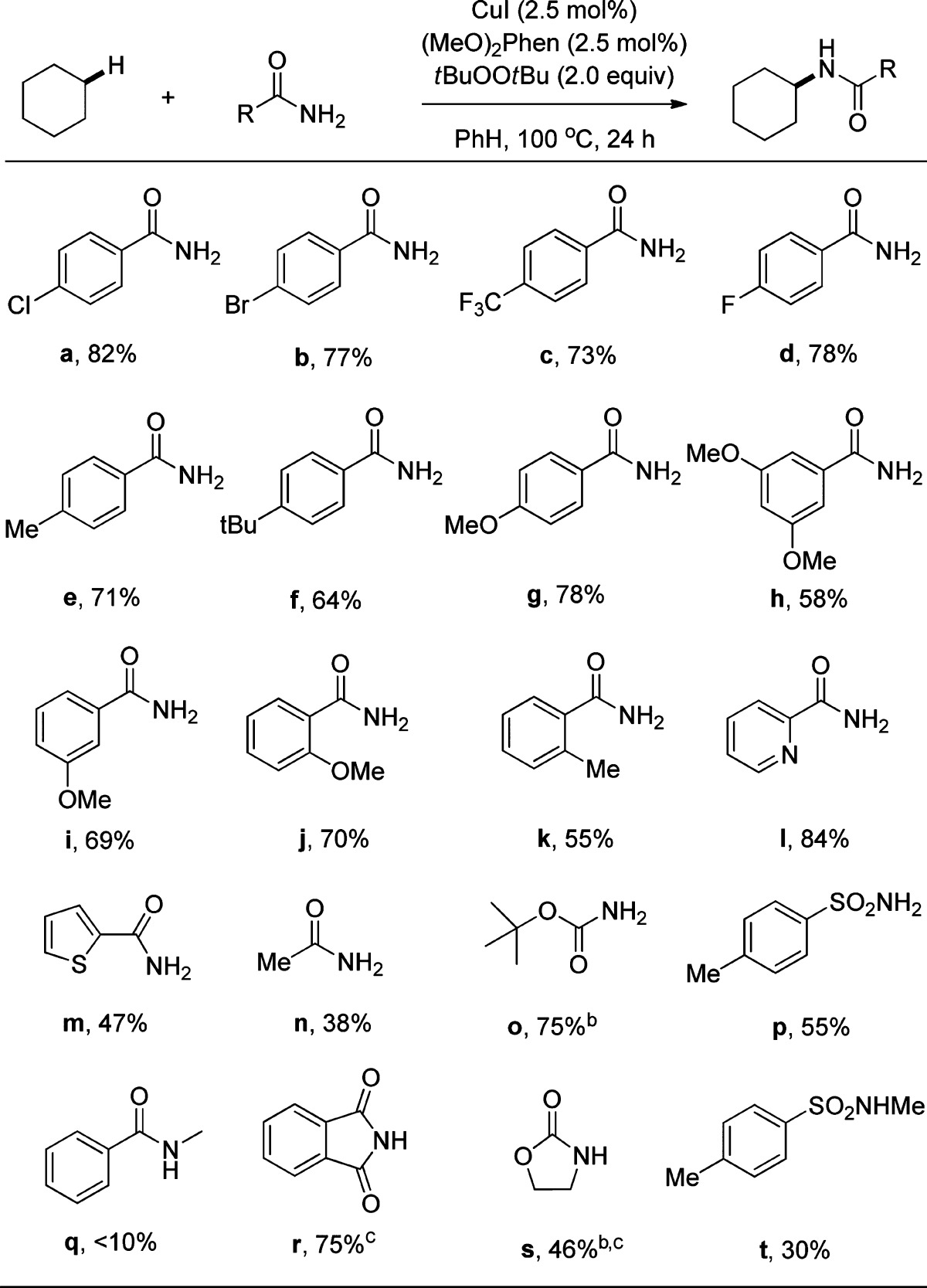
Conditions: 0.5 mmol of amide, 5.0 mmol of cyclohexane, 0.0125 mmol of CuI, 0.0125 mmol of (MeO)2Phen, 1.0 mmol of tBuOOtBu, 1 mL of PhH at 100 °C for 24 h.
Four equivalents of oxidant.
Reaction performed in o-C6H4Cl2 as solvent.
The reactions of cyclohexane with primary and secondary alkyl amides, carbamates and imides also occurred (Table 2). The yields of the reactions of cyclohexane with acetamide (n), tert-butylcarbamate (o), and toluenesulfonamide (p) to form N-cyclohexylacetamide, tert-butyl-N-cyclohexylcarbamate, and N-cyclohexyltoluenesulfonamide (38%, 42%, and 55%, respectively) were lower than those that formed N-alkyl benzamides. However, the reaction of tert-butylcarbamate, with excess tBuOOtBu (4 equiv) gave tert-butyl-N-cyclohexylcarbamate in 75% yield.
The reactions of cyclohexane with secondary nitrogen sources (N-methylbenzamide, phthalimide, pyrrolidinone, and N-Me-p-toluenesulfonamide) also occurred to form the corresponding N-alkyl products. The yields of these reactions were variable (10–75%) (q–t), but they do demonstrate that the mechanism cannot involve a nitrene intermediate. The reaction of phthalimide was improved from 20% to 75% by changing the solvent from benzene to 1,2-dichlorobenzene to increase the solubility of the reagent.
The selectivities for amidation of various alkanes with benzamide are presented in Table 3. The amidations of alkanes containing secondary and tertiary C–H bonds formed products from functionalization at the secondary C–H bonds (entry 1–5). For example, the major products of the reactions of benzamide with trans- and cis-1,4-dimethylcyclohexane (entry 1,2) resulted from amidation of secondary C–H bonds, while the minor products resulted from the amidation of primary C–H bonds. No product was formed from amidation of the tertiary C–H bond. Likewise, the reaction of 3-ethylpentane (entry 5) occurred preferentially at secondary and primary C–H bonds over tertiary C–H bonds. The reaction of 2,4-dimethylpentane (entry 7), in which the secondary C–H bond is hindered, occurred exclusively at the primary C–H bond. This product distribution contrasts that of most C–H oxidations by a radical mechanism.42,43 For reactions of cycloalkanes ranging from cyclopentane to cyclododecane, the ring size did not affect the yield of the corresponding N-alkyl products (69–80%) (entry 8).
Table 3. Selectivity for Amidation of Alkanesa.
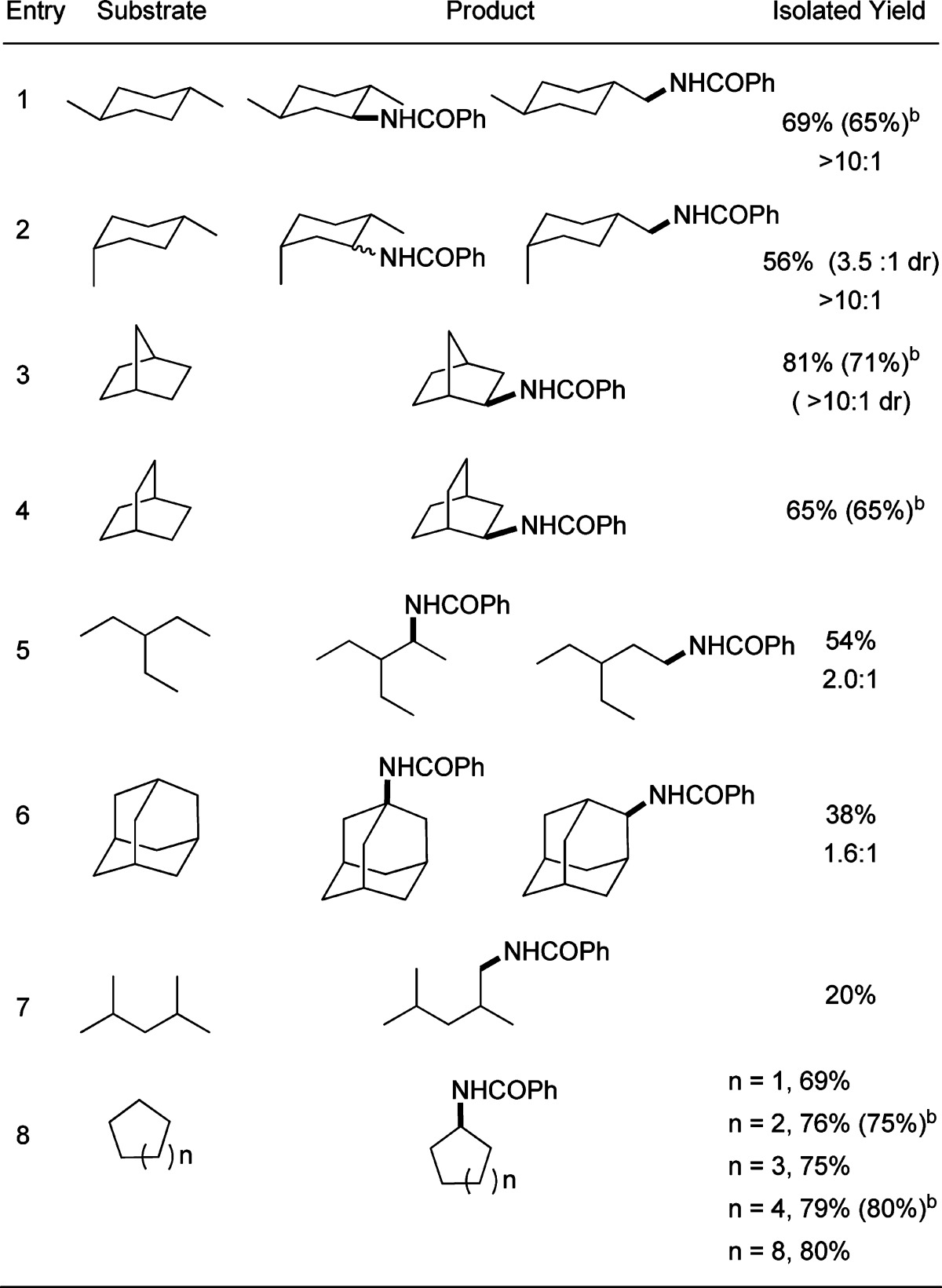
Conditions: 0.5 mmol of benzamide, 1 mL of alkane (10–17 equiv), 0.0125 mmol CuI, 0.0125 mmol of (MeO)2Phen, 2.0 mmol of tBuOOtBu, 1 mL of o-C6H4Cl2 at 100 °C for 36 h. Regioselectivity determined by crude 1H NMR.
Yield for the reaction with t-butylcarbamate.
3. Isolation and Characterization of Copper(I) and Copper(II) Imidate and Amidate Intermediates
To assess whether copper-amidate and copper-imidate complexes with Cu in the oxidation state of +1 and +2 are intermediates in the catalytic amidation reactions, we synthesized and characterized a variety of these amidate and imidate complexes supported by phen and 2-(dimethylamino)ethyl)imino)methyl) phenoxide (L1) (Scheme 2). Salt-metathesis reactions of [(phen)CuCl]2(μ-Cl)2 with potassium phthalimide (Kphth) or a combination of NaOtBu and H2NSO2Ph or H2NCOPh in THF at room temperature yielded air- and moisture-stable [(phen)Cu(phth)2] (1-phth2), [(phen)Cu(NHSO2Ph)2] (2), and [(phen)Cu(NHCOPh)2] (3), respectively.
Scheme 2. Syntheses of Copper(I) and Copper(II) Amidate and Imidate Complexes.

Phen-ligated Cu(I) complexes containing benzamidate and benzenesulfonamidate ligands were isolated from the reaction of [Cu(Mes)]n with phen, followed by the addition of benzenesulfonamide and benzamide to produce the corresponding products [(phen)2Cu][Cu(NHSO2Ph)2] (4) and [(phen)2Cu][Cu(NHCOPh)2] (5). The complex [(phen)Cu(phth)] (1-phth) was prepared by methods described previously.44
Salt-metathesis reactions of [(L1)CuCl]41 with phthalimide (Hphth), benzenesulfonamide, and benzamide produced the corresponding Cu(II) complexes of [(L1)Cu(phth)] (6), [(L1)Cu(NHSO2Ph)] (7), and [(L1)CuNHCOPh] (8) (Scheme 2).
Previous structural characterizations of phen-ligated copper(I) amidate and imidate complexes revealed that these complexes can exist as mononuclear or ion-pair species. Therefore, we assessed the atomic connectivity of 1-phth2 and 4 by single crystal X-ray diffraction (XRD) analysis.44 The solid-state structures of 1-phth2 and 4 are shown in Figure 1. All pertinent bond distances and angles for these complexes are listed in the caption of Figure 1. Phen-ligated Cu(II) phthalimidate 1-phth2 is mononuclear in the solid state with a square planar geometry around the Cu(II) ion (Figure 1).
Figure 1.
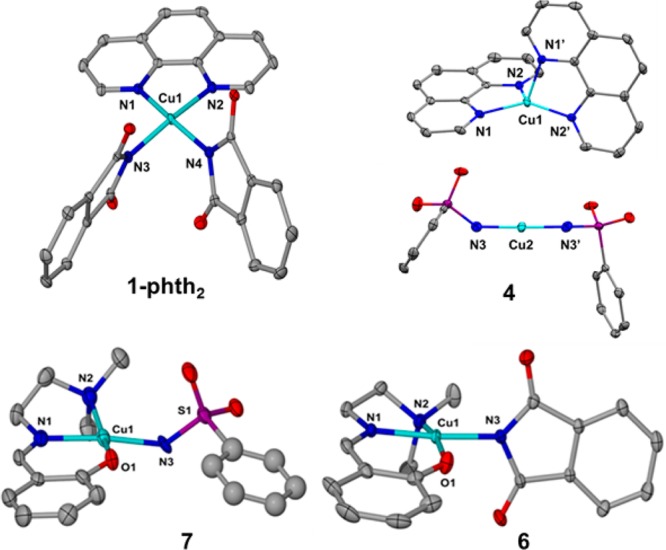
Molecular structures of [(phen)Cu(phth)2] (1-phth2), [(phen)2Cu] [Cu(NHSO2Ph)2] (4), [(L1)Cu(phth)] (6), and [(L1)Cu(NHSO2Ph)] (7) shown with 50% thermal ellipsoid. Selected bond lengths (Å) and angles (°) of 1-phth2: Cu1–N1 = 2.0480(14); Cu1–N2 = 2.0363(14); Cu1–N3 = 1.9667(14); N1–Cu1–N3 = 170.29(5); N1–Cu1–N2 = 93.45(5); N1–Cu1–O1 = 92.5(3). Selected bond lengths (Å) and angles (o) of 4: Cu1–N1 = 1.9920(15); Cu1–N2 = 2.1079(16); Cu1–N3 = 1.8553(17); N1–Cu1–N2 = 81.75(6); N3–Cu1–N3′ = 178.92(10). Selected bond lengths (Å) and angles (o) of 6: Cu1–N1 = 1.938(7); Cu1–N2 = 2.045(7); Cu1–N3 = 1.958(7); Cu1–O1 = 1.919(6); N1–Cu1–N3 = 175.2(3); N1–Cu1–N2 = 83.4(3); N1–Cu1–O1 = 92.5(3). Selected bond lengths (Å) and angles (o) of 7: Cu1–N1 = 1.956(6); Cu1–N2 = 2.104(6); Cu1–N3 = 1.972(6); Cu1–O1 = 1.993(5); N1–Cu1–N3 = 170.0(2); N1–Cu1–N2 = 83.7(2); N1–Cu1–O1 = 91.2(2).
In contrast, the copper(I) compounds were ion pairs in the solid state. The structure of phen-ligated benzenesulfonamidate 4 is an ion pair consisting of the cationic [(phen)2Cu]+, in which Cu(I) is ligated by two phen ligands in a distorted tetrahedral environment, and the linear anion [Cu(NHSO2Ph)2]− (Figure 1).
In contrast to the diverse molecular structures of phen-ligated copper species, complexes 6 and 7 containing the tridentate amino imine phenoxide ligands are mononuclear Cu(II) species with square-planar geometries in the solid state (Figure 1). These structures are also mononuclear in solution, as evidenced by X-Band EPR spectroscopy recorded at 20 K in CH2Cl2 indicating an axial environment.41
To determine whether the structures of 1-phth2 and 4 in solution differ from those in the solid state44 we measured the conductivities of 1-phth2, and 4 in DMSO. These measurements distinguish between ionic and neutral forms. The conductivities of 1-phth2, and 4 (1.0 mM in DMSO, 25 °C) were compared to those of a 1.0 mM (DMSO) solution of ferrocene (0.2 Ω–1 cm2 mol–1) and N(Bu)4Cl (11.1 Ω–1 cm2 mol–1). The conductivity of the solutions of Cu(I) complexes 4 and 5 in DMSO (1.0 mM) were 33.0 and 23.1 Ω–1 cm2 mol–1, respectively. These values are similar to the reported values for related (phen)Cu(I) amidate and imidate complexes.44 In contrast, the conductivity of the solution of 1-phth2 was 0.0 Ω–1 cm2 mol–1, showing that 1-phth2 retains its neutral, mononuclear structure in DMSO. Moreover, the results for conductivity of 6 (0.0 Ω–1 cm2 mol–1) and 7 (0.1 Ω–1 cm2 mol–1) also revealed that these complexes are neutral molecules in solution. Unfortunately, the insolubility of 1-phth2 and 4 in benzene prevented the measurement of their conductivities in a less polar solvent.
4. Catalytic and Stoichiometric Reactivity of Copper-Amidate and -Imidate Complexes
To gain information on the mechanism of the catalytic amidation of alkanes, we conducted catalytic and stoichiometric reactions with isolated [(phen)Cu(phth)n] (n = 1, 2) complexes. These well-defined complexes contain a reactive phthalimidate group bound to copper in the oxidation states of +1 and +2.
First, we investigated the effect of the oxidation state of the metal in the copper complexes lying on the catalytic reaction. The reactions of cyclohexane with Hphth catalyzed by 2.5 mol % of 1-phth2 and 2.5 mol % of 1-phth produced N-cyclohexylphthalimide (Cy-phth) in 54% and 80% yield, respectively (Scheme 3). N-Methylphthalimide (Me-phth) was observed in 13% and 20% yields in the reactions catalyzed by 1-phth2 and 1-phth, respectively. The observation of this side-product implies the intermediacy of a tert-butoxy radical because this radical is known to undergo β-Me scission to generate a methyl radical.42 This methyl radical can then combine with 1-phth2 to form Me-phth (Scheme 3).45 The observation of this side product suggests that the N-cyclohexylphthalimide forms from a reaction between 1-phth2 and a cyclohexyl radical generated from hydrogen atom abstraction of cyclohexane by a tert-butoxy radical.
Scheme 3. Catalytic C–H Amidation of Cyclohexane with 1-phth2 and 1-phth.

In the reaction catalyzed by 1-phth, the orange solution of 1-phth rapidly transformed to a light-blue suspension. A similar light-blue suspension was also observed in the reaction catalyzed by 1-phth2. Moreover, 1-phth2 was isolated from a reaction of 1-phth, tBuOOtBu, and Hphth in benzene at 100 °C for 0.5 h (Scheme 4). The identity of 1-phth2 was confirmed by comparison of its IR spectrum to that of the authentic sample of 1-phth2, prepared by the method in Scheme 2, and by elemental analysis. The isolation of 1-phth2 from the reaction catalyzed by 1-phth clearly demonstrates that a Cu(I)-imidate or amidate is a short-lived species in the presence of excess oxidant and imide or amide under catalytic conditions. Therefore, under the catalytic condition, the predominant species in the system is a Cu(II)-amidate or imidate complex.
Scheme 4. Isolation of 1-phth2 from 1-phth.

Second, we investigated the role of oxidant in the catalytic reaction by conducting a series of stoichiometric reactions with 1-phth and 1-phth2 in the presence and absence of tBuOOtBu. C–H bond amidations of cyclohexane by copper complexes have been proposed to occur by insertion of a copper-nitrene intermediate into a C–H bond or by hydrogen atom abstraction. In one study, a copper-nitrene species stabilized by Sc3+ and an isolated bridging copper-nitrene complex were shown to undergo stoichiometric reactions with cyclohexane to form N-cyclohexyl-4-methylbenzenesulfonamide and N-cyclohexyladamantane, respectively.46,47 In another study, an isolated, three-coordinate Cu(II)-NHAd (Ad = adamantane) complex was shown to react with cyclohexane to form N-cyclohexyladamantane; this reaction was proposed to occur by abstraction of a hydrogen atom from cyclohexane and capture of the cyclohexyl radical by Cu(II).29 A mechanism involving a nitrene intermediate in our system is ruled out by the reaction of secondary nitrogen reagents (i.e., phthalimide) with cyclohexane to form the corresponding N-alkyl product, but reaction of the amidate and imidate complexes with the alkane could occur.
Thus, we tested the potential reactions of the copper amidate and imidate complexes with cyclohexane. To do so, we performed stoichiometric reactions of 1-phth and 1-phth2 with cyclohexane. These reactions would assess whether these complexes are capable of abstracting a hydrogen atom from cyclohexane. The reaction of 1-phth or 1-phth2 with cyclohexane (80 equiv) in MeCN or benzene without tBuOOtBu at 100 °C for 24–72 h produced no Cy-phth detectable by GC (Scheme 5, A). The absence of any Cy-phth product from these reactions rules out a mechanism involving hydrogen atom abstraction by the Cu(I) or Cu(II) phthalimidates and combination of the resulting alkyl radical with the copper complexes.29
Scheme 5. Stoichiometric Reactions of 1-phth2 and 1-phth with Cyclohexane.

However, the stoichiometric reaction of 1-phth2 with cyclohexane and tBuOOtBu (16 equiv) for 24 h produced Cy-phth (70% yield based on copper), Me-phth, and Cy-OtBu in a 70:25:35 ratio (Scheme 5, B). The analogous reaction of 1-phth with cyclohexane and tBuOOtBu (16 equiv) also produced Cy-phth (73% yield based on copper), Me-phth, and Cy-OtBu in a 73:21:32 ratio (Scheme 5, C). Formation of the ether product has been observed from Cu(I)-catalyzed decomposition of tBuOOtBu and recombination of a cyclohexyl radical with a Cu–OtBu functionality.48 Moreover, the observation of Cy-OtBu suggests that [(phen)Cu(phth)(OtBu)] is a likely intermediate in these reactions lacking free phthalimide; reaction of the cyclohexyl radical with this complex would form Cy-phth and Cy-OtBu.
The formation of N-alkyl amides in the presence of tBuOOtBu, but not in the absence of tBuOOtBu, was observed for copper complexes 1-phth2 or 1-phth. Complexes 2–6 also reacted with cyclohexane to produce their corresponding N-alkyl products in the presence of tBuOOtBu, but not in the absence of tBuOOtBu.41 These studies provide further evidence that tBuOOtBu leads to formation of tert-butoxy radical, which cleaves the alkane C–H bond.
To investigate if the Cu(II) complex 1-phth2 and the Cu(I) complex 1-phth react by a common pathway, we measured the kinetic isotope effect of the amination reactions catalyzed by these two complexes. Reactions of the combination of cyclohexane and cyclohexane-d12 in a single vessel containing phth, tBuOOtBu, and 2.5 mol % 1-phth2 occurred with a primary KIE of 2.7 ± 0.1. The reaction of 1-phth under the same experimental conditions occurred with a primary KIE of 3.0 ± 0.3. The similarity of these values is consistent with reaction of Cu(I) and Cu(II) complexes through the same C–H bond cleavage step. Furthermore, the catalytic reaction of cyclohexane and cyclohexane-d12 in the same vessel with benzamide and 2.5 mol % of the phen-ligated Cu(II) benzamidate complex 5 occurred with a primary KIE value of 2.8 ± 0.4. These results imply that the same C–H bond cleavage step occurs for reactions of the different copper-amidate complexes.
5. Trapping of Proposed Radical Intermediates
Results from the stoichiometric and catalytic reactions of 1-phth2 and 1-phth strongly suggest that a tert-butoxy radical is responsible for the C–H abstraction of cyclohexane. To assess this hypothesis further, we performed experiments that would trap the tert-butoxy and alkyl radicals.
First, we conducted the catalytic reaction in the presence of diphenylmethanol (2.5 equiv) (Scheme 6, A), a common probe for tert-butoxyl radical.45,49 This alcohol can react with an alkoxy radical by abstraction of the C–H hydrogen atom from Cu(II)-diphenylmethoxide to form a Cu(II)-ketyl radical that is then oxidized to form benzophenone to regenerate Cu(I).50 Alternatively, an alkoxy radical abstracts the C–H bond of diphenylmethanol to form a ketyl radical. This ketyl radical undergoes another hydrogen atom abstraction at the OH group by another alkoxy radical to form benzophenone.51 Consistent with the generation of an alkoxy radical, the catalytic reaction conducted in the presence of diphenylmethanol formed the N-cyclohexyl benzamide product in only 47% yield, based on benzamide. Instead of the N-cyclohexyl amide, a large amount of benzophenone (2.4:1.0 ratio of benzophenone to N-cyclohexyl benzamide) formed. This reaction to form benzophenone decreases the amount of available tert-butoxy radical for hydrogen atom abstraction of cyclohexane and thus decreased the yield of the N-cyclohexyl benzamide product.
Scheme 6. Trapping of tert-Butoxy Radical.

Second, we probed for the formation of tert-butoxy radical by conducting the reaction in the presence of the hydrogen atom donor 9,10-dihydroanthracene. The tert-butoxy radical should abstract the weak C–H bond of 9,10-dihydroanthracene over the stronger C–H bonds in cyclohexane. Indeed, the catalytic reaction of benzamide with cyclohexane conducted under the standard conditions, except for the addition of 3 equiv of 9,10-dihydroanthracene, yielded anthracene as the only organic product besides tert-butanol (Scheme 6, B).
Finally, to probe for a transient cyclohexyl radical in the reaction, we performed the catalytic reaction in the presence of CBr4 (Scheme 7, A), which would trap a cyclohexyl radical to form bromocyclohexane.52−54 The reaction of benzamide with cyclohexane and tBuOOtBu at 100 °C for 24 h did not form any product from the amidation of cyclohexane. Instead, bromocyclohexane formed in 90% yield, based on benzamide. This result is consistent with the intermediacy of a cyclohexyl radical in the catalytic reaction.53,54
Scheme 7. Trapping of the Putative Alkyl Radical.

The analogous reaction of trans-1,4-dimethylcyclohexane was conducted. The reaction of this alkane in the presence of CBr4 and 2.5 mol % CuI/(MeO)2Phen in benzene at 100 °C for 24 h (Scheme 7, B) formed the products from bromination at the tertiary and the secondary sites: cis- and trans-1-bromo-1,4-dimethylcyclohexane and cis- and trans-2-bromo-1,4-dimethylcyclohexane. The two sets of constitutional isomers formed in a ratio of 1.7:1 and a combined 86% yield based on CBr4. The 1-bromo-1,4-dimethylcyclohexane diastereomers were obtained as a 1.2:1 mixture, indicating that the transient tertiary radical formed under this condition undergoes facile racemization.55 To investigate whether an alkyl radical would react with the copper imidates to form an N-alkyl product, we conducted reactions of the phthalimidate complex 1-phth2 with precursors to alkyl radicals. We chose to use peroxides as the precursors to alkyl radicals, and we performed these reactions with 1-phth2 instead of 1-phth because our data indicate that 1-phth2 is the predominant complex under catalytic conditions. The reaction of 1-phth2 with tBuOOtBu and (Ph(Me)2CO)2 at 100 °C for 18 h generated Me-phth in 64% and 81% yield based on copper, respectively (Scheme 8). The rate of β-Me scission of the cumyloxide radical is faster than that of the tert-butoxy radical. Thus, the higher yield of N-alkyl product from the reaction of dicumyl peroxide than from the reaction of tBuOOtBu is consistent with the rate of generation of the methyl radical.52,56
Scheme 8. Trapping of the Putative Alkyl Radical with 1-phth2.

These results with radical probes and alkyl radicals provide strong evidence that an alkoxy radical generates an alkyl radical by hydrogen atom abstraction and that the alkyl radical combines with a copper(II)-amidate and -imidate species to produce the amidation and imidation products. The formation of products from halogenation at the tertiary C–H bond of the dimethylcyclohexane indicates that the absence of the products from amination at the tertiary C–H bonds results from the ability of these radicals to form the C–N bond (vide infra).
Reactions in acetonitrile provide evidence against a carbocationic intermediate that could form by oxidation of the radical and combination of the cation with the copper amide or imidates.57 Acetonitrile is well known to trap carbocations by a Ritter-type reaction.4,57,58 The amidation of cyclohexane with Hphth in wet acetonitrile (Scheme 3) produced Me-phth (80%), and this result argues against a transient carbocation in our system.
6. Determination of the Turnover-Limiting Step and Order of Reagents
Additional measurements of KIE values were conducted to determine the turnover-limiting step (TLS) of the catalytic reaction. The KIE determined from independent measurements of the rates of amidation of cyclohexane and cyclohexane-d12 catalyzed by 10 mol % of 3 was 2.9 ± 0.1.41 This primary KIE indicates that hydrogen atom abstraction of cyclohexane by a tert-butoxy radical is the turnover-limiting step in the catalytic process.
To identify the TLS and to explain the observed primary KIE, we determined the order of the reaction in each component. We obtained the kinetic data by measuring the concentration of cyclohexylbenzamide and N-methylbenzamide versus time by the method of initial rates with varied concentrations of oxidant, cyclohexane, and benzamide.41 At copper concentrations of 23 mM, the reaction was found to be first-order in oxidant, first-order in cyclohexane, and zero-order in benzamide. The reaction order of oxidant and cyclohexane is consistent with turnover-limiting C–H abstraction of cyclohexane by a tert-butoxy radical. The dependence of the reaction rate on the concentration of oxidant and cyclohexane was lower for reactions conducted with higher concentrations of these two reagents. When the concentration of oxidant and cyclohexane exceeded 1.0 M, there was little dependence of the rate of cyclohexane amidation on the concentration of these two species.41
The order of Cu is complex; our data indicates that the reaction rate is positively dependent on the concentration of Cu. However, the order of the reaction in the concentration of copper varied between first-order and essentially zeroth-order.41 A similar dependence of reaction rate on the concentration of copper was reported for copper-catalyzed allylic oxidation of cyclohexene by tert-butyl perbenzoate in benzene.59 At low concentration of copper, the rate of allylic oxidation increased with increasing concentration of copper; however, the rate of this reaction was less dependent on the concentration at higher concentrations of copper.40,59
In addition to the difference in dependence of the rate on the concentration of copper, we observed a difference in yield of the amination products as a function of the concentration of copper. Reactions conducted with a higher mol % of the Cu catalyst formed the N-alkyl amide product in lower yields than the reactions conducted with a lower loading of the Cu catalyst. Specifically, reactions of cyclohexane conducted with 2.84–17.0 mM 5 yielded 94–99% of the N-cyclohexy benzamide product, while those conducted with 34.0–45.5 mM 5 gave the N-cyclohexyl benzamide product in 76% yield, along with MeNHCOPh (10%) and unreacted benzamide. Perhaps, at higher loading of Cu(I), faster decomposition of peroxide leads to a higher concentration of tert-butoxy radical, relative to the concentration of copper complex, and the tert-butoxy radical undergoes processes that do not lead to the N-alkyl amide product.
The tert-butoxy radical is known to undergo multiple nonproductive pathways: (1) β-Me scission for the methylation of the bound Cu(II)-amidate; (2) coupling of methyl radicals;60 and (3) reaction of tert-butoxy radical with Cu(I) to form Cu(II)-OtBu.42,48 The quenching of the tert-butoxy radical at high concentration of copper offers one possible explanation for the lower yield of product at high loading of Cu(I) compared to low loading of Cu(I) and the lower order in copper.
Regardless of the origin of the effect of the concentration of copper on yield, a copper complex does not directly participate in the C–H bond cleavage of cyclohexane. Several pieces of data provide evidence that copper is not required for C–H cleavage of cyclohexane. For example, in the absence of copper, the reaction of adamantane with tBuOOtBu and CBr4 at 100 °C for 24 h produced a mixture of 1-bromoadamantane and 2-bromoadamantane in a ratio of 2.6:1, respectively.41 The same reaction conducted in the presence of copper, formed a mixture of products containing 1-bromoadamantane and 2-bromoadamantane in a 2:1 ratio. The similarity of the ratio of products in the presence and absence of copper suggest that copper is not directly involved in the C–H cleavage step.
Thus, copper appears to play two roles in our system. First, copper catalyzes the decomposition of tBuOOtBu to generate tert-butoxy radical, which is the species that cleaves the C–H bond of the alkanes. Specifically, reaction of 1-phth with tBuOOtBu and Hphth in the absence of a hydrogen atom donor (i.e., cyclohexane) leads to rapid oxidation of 1-phth to 1-phth2 and exclusive formation of Me-phth, in which the methyl radical is derived from tert-butoxy radical by β-Me scission.42 Additional evidence for copper-catalyzed formation of tert-butoxy radical is the observation of benzophenone from diphenylmethanol under catalytic conditions.45 Second, copper participates in the events after the turnover-limiting step of C–H activation by tert-butoxy radical to form the N-alkyl product. This proposal is corroborated by the reaction of methyl radicals, derived from the tertiary alkoxy radical precursors RO-OR (R = tBu, Ph(Me)2C), with 1-phth2 to generate the Me-phth product.
7. Proposed Mechanism for Amidation of Cyclohexane
Our proposed mechanism for C–H amidation of cyclohexane by 1-phth is summarized in Scheme 9. An analogous cycle is assumed to operate for the amidation with benzamide. Our proposed mechanism begins with the decomposition of tBuOOtBu initiated by 1-phth to produce a tert-butoxy radical and a potential intermediate containing an alkoxide and a phthalimidate ligand [(phen)Cu(phth)(OtBu)].29,61 This complex would react rapidly with Hphth to generate 1-phth2. The tert-butoxy radical can regenerate tBuOOtBu by secondary geminate recombination or can abstract a hydrogen atom from an alkane to form an alkyl radical.62 In this case, the turnover-limiting step is abstraction of a hydrogen atom from cyclohexane by a tert-butoxy radical to produce a cyclohexyl radical, which recombines with 1-phth2 to expel Cy-phth and regenerate 1-phth to close the cycle.
Scheme 9. Proposed Mechanism of Copper-Catalyzed Intermolecular Amidation of Cyclohexane.

The identity of the species that forms the C–N bond is not defined by our studies. This species could be a transient Cu(III) intermediate ligated by a cyclohexyl and amidate ligand. Such a species could undergo rapid reductive elimination to release the N-alkyl product and to regenerate Cu(I).63,64 Prior mechanistic and DFT investigations of the Kharasch-Sosnovsky reaction have led to the conclusion that these oxidative reactions occur by formation of a Cu(III) species, which forms the C–O bond by reductive elimination.34,36,38,39,65 Conversely, DFT calculations on the combination of a cyclohexyl radical with the three-coordinate [(nacnac)Cu(II)-OtBu] (nacnac– = [ArNC(Me)]2CH; Ar = 2,6-Cl2C6H3) to form Cy-OtBu suggest that capture of the alkyl radical at the oxygen atom of the OtBu ligand is thermodynamically favorable (ΔG = −13.4 kcal/mol), whereas combination of the alkyl radical at Cu(II) to form the square planar complex [(nacnac)Cu(III)(Cy)(OtBu)] is thermodynamically disfavored (ΔG = +14.9 kcal/mol).48 Thus, it is possible that the alkyl radical reacts directly with the imidate or amidate ligands to form the N-alkyl imide and amide products.
The selectivity of secondary (2°) and primary sites (1°) over tertiary (3°) sites for the amidation of alkanes by this copper system is different from the selectivity of 3° > 2° > 1° for hydrogen abstraction by a tert-butoxy radical.43 This selectivity for secondary over primary and tertiary sites has been observed in the copper-catalyzed amidation of benzylic C–H bonds, and the rationale for such selectivity was based on steric effects.33 We propose that the selectivity for the amidation of secondary over tertiary C–H bonds results from the recombination of alkyl radicals with phen-ligated Cu(II) amidate and imidate complexes; we propose that the tertiary radical is too hindered to recombine with phen-ligated Cu(II) amidate and imidate complexes and the alkyl radical undergoes decomposition.
To address this proposal, we determined the mass balance of the reaction. This information would provide insight into the fate of all the alkyl radicals that could form. The amidation of adamantane afforded a ratio of 1.6 N-(adamantan-1-yl)benzamide to 1.0 N-(adamantan-2-yl)benzamide and 1-phenyladamantane. The 1-phenyladamantane product likely results from the direct reaction of 1-adamantyl radical with benzene.66 The observation of 1-phenyladamantane reveals another pathway by which tertiary alkyl radicals decompose in the reaction. Moreover, the amidation of trans-1,4-dimethylcyclohexane that produced trans-2,5-dimethylcyclohexyl benzamide also produced 1,4-dimethylcyclohex-1-ene.41 The alkene likely forms from the decomposition of the tertiary radical, and this decomposition likely occurs because the combination of the tertiary radical with the phen copper amidate or imidate is sterically disfavored.
Summary and Conclusions
In summary, we have described a copper-catalyzed amidation of unactivated alkanes by benzamide, sulfonamide, carbamate, phthalimide, and their derivatives. The amidation of alkanes under our catalytic conditions preferentially forms the products from amidation at secondary sites over tertiary sites. The amidation of primary C–H bonds to form N-alkyl products is observed when the secondary C–H bonds are hindered. Potential Cu(II)-amidate and -imidate intermediates were isolated, characterized, and demonstrated to be intermediates for C–H amidation of cyclohexane. Our mechanistic data imply that the tert-butoxy radical, not a copper-nitrene or copper-aminyl species, leads to the activation of aliphatic C–H bonds.
Acknowledgments
We gratefully acknowledge financial support by the Einstein Foundation Berlin (J.F.H.), the NIH-NIGMS (R29-55382 to J.F.H.), and the Cluster of Excellence UniCat (financed by the DFG and administered by the TU Berlin to M.D.) for research support. We thank Paula Nixdorf and Dr. Antonio DiPasquale for assistance with XRD. We thank Dr. Ruchira Chatterjee (LBNL) for access and assistance with EPR spectroscopy.
Supporting Information Available
Experimental procedures, characterization of new compounds, and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ B.L.T. and B.L. contributed equally to the work presented.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sàchez C.; Mèndez C.; Salas J. A. Nat. Prod. Rep. 2006, 23, 1007. [DOI] [PubMed] [Google Scholar]
- Yamaguchi J.; Yamaguchi A. D.; Itami K. Angew. Chem., Int. Ed. 2012, 51, 8960. [DOI] [PubMed] [Google Scholar]
- Li Z.; Capretto D. A.; Rahaman R.; He C. Angew. Chem., Int. Ed. 2007, 119, 5276. [DOI] [PubMed] [Google Scholar]
- Michaudel Q.; Thevenet D.; Baran P. S. J. Am. Chem. Soc. 2012, 2012, 2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C.; Collet F.; Robert-Peillard F.; Müller P.; Dodd R. H.; Dauban P. J. Am. Chem. Soc. 2008, 130, 343. [DOI] [PubMed] [Google Scholar]
- Sun C.-L.; Li B.-J.; Shi Z.-J. Chem. Rev. 2011, 111, 1293. [DOI] [PubMed] [Google Scholar]
- Chen M. S.; White M. C. Science 2007, 318, 783. [DOI] [PubMed] [Google Scholar]
- Gormisky P. E.; White M. C. J. Am. Chem. Soc. 2013, 135, 14052. [DOI] [PubMed] [Google Scholar]
- Li C.-J. Acc. Chem. Res. 2009, 42, 335. [DOI] [PubMed] [Google Scholar]
- Gómez L.; Canta M.; Font D.; Prat I.; Ribas X.; Costas M. J. Org. Chem. 2013, 78, 1421. [DOI] [PubMed] [Google Scholar]
- Barton D. H. R.; Doller D. Acc. Chem. Res. 1992, 25, 504. [Google Scholar]
- Barton D. H. R.; Bévière S. D.; Hill D. R. Tetrahedron 1994, 50, 2665. [Google Scholar]
- Díaz-Requejo M. M.; Pérez P. J. Chem. Rev. 2008, 108, 3379. [DOI] [PubMed] [Google Scholar]
- Roizen J. L.; Harvey M. E.; Bois J. D. Acc. Chem. Res. 2012, 45, 911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dequirez G.; Pons V.; Dauban P. Angew. Chem., Int. Ed. 2012, 51, 7384. [DOI] [PubMed] [Google Scholar]
- Gephart R.; Warren T. H. Organometallics 2012, 31, 7728. [Google Scholar]
- Barman D. N.; Nicholas K. M. Eur. J. Org. Chem. 2011, 908. [Google Scholar]
- Barman D. N.; Liu P.; Houk K. N.; Nicholas K. M. Organometallics 2010, 29, 3404. [Google Scholar]
- Srivastava R. S.; Tarver N. R.; Nicholas K. M. J. Am. Chem. Soc. 2007, 129, 15250. [DOI] [PubMed] [Google Scholar]
- Yu X.-Q.; Huang J.-S.; Zhou X.-G.; Che C.-M. Org. Lett. 2000, 2, 2233. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Guan X.; Wong E. L.-M.; Liu P.; Huang J.-S.; Che C.-M. J. Am. Chem. Soc. 2013, 135, 7194. [DOI] [PubMed] [Google Scholar]
- Lebel H.; Trudel C.; Spitz C. Chem. Commun. 2012, 48, 7799. [DOI] [PubMed] [Google Scholar]
- Lebel H.; Huard K.; Lectard S. J. Am. Chem. Soc. 2005, 127, 14198. [DOI] [PubMed] [Google Scholar]
- Lu H.; Hu Y.; Jiang H.; Wojtas L.; Zhang X. P. Org. Lett. 2012, 14, 5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyaskovskyy V.; Suarez A. I. O.; Lu H.; Jiang H.; Zhang X. P.; Bruin B. d. J. Am. Chem. Soc. 2011, 133, 12264. [DOI] [PubMed] [Google Scholar]
- Paradine S. M.; White M. C. J. Am. Chem. Soc. 2012, 134, 2036. [DOI] [PubMed] [Google Scholar]
- Zalatan D. N.; Bois J. D. Top. Curr. Chem. 2009, 292, 347. [DOI] [PubMed] [Google Scholar]
- Fiori K. W.; Espino C. G.; Brodsky B. H.; Bois J. D. Tetrahedron 2009, 65, 3042. [Google Scholar]
- Wiese S.; Badiei Y. M.; Gephart R. T.; Mossin S.; Varonka M. S.; Melzer M. M.; Meyer K.; Cundari T. R.; Warren T. H. Angew. Chem., Int. Ed. 2010, 49, 8850. [DOI] [PubMed] [Google Scholar]
- Kharasch M. S.; Sosnovsky G. J. Am. Chem. Soc. 1958, 80, 756. [Google Scholar]
- Kohmura Y.; Kawasaki K.-i.; Katsuki T. Synlett 1997, 1456. [Google Scholar]
- Pelletier G.; Powell D. A. Org. Lett. 2006, 8, 6031. [DOI] [PubMed] [Google Scholar]
- Powell D. A.; Fan H. J. Org. Chem. 2010, 75, 2726. [DOI] [PubMed] [Google Scholar]
- Andrus M. B.; Lashley J. C. Tetrahedron 2002, 58, 845. [Google Scholar]
- Eames J.; Watkison M. Angew. Chem., Int. Ed. 2001, 40, 3567. [DOI] [PubMed] [Google Scholar]
- Beckwith A. L. J.; Zavitsas A. A. J. Am. Chem. Soc. 1986, 108, 8230. [Google Scholar]
- Kochi J. K.; Mains H. E. J. Org. Chem. 1965, 30, 1862. [Google Scholar]
- Smith K.; Hupp C. D.; Allen K. L.; Slough G. A. Organometallics 2005, 24, 1747. [Google Scholar]
- Mayoral J. A.; Rodríguez-Rodríguez S.; Salvatella L. Chem.—Eur. J. 2008, 14, 9274. [DOI] [PubMed] [Google Scholar]
- Walling C.; Zavitsas A. A. J. Am. Chem. Soc. 1963, 85, 2084. [Google Scholar]
- See the Supporting Information.
- Walling C.Free Radicals in Solution; Wiley: New York, 1957. [Google Scholar]
- Walling C.; Jacknow B. B. J. Am. Chem. Soc. 1960, 82, 6108. [Google Scholar]
- Tye J. W.; Weng Z.; Johns A. M.; Incarvito C. D.; Hartwig J. F. J. Am. Chem. Soc. 2008, 130, 9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul H.; R. D. Small J.; Scaiano J. C. J. Am. Chem. Soc. 1978, 100, 4520. [Google Scholar]
- Kundu S.; Miceli E.; Farquhar E.; Pfaff F. F.; Kuhlmann U.; Hildebrandt P.; Braun B.; Greco C.; Ray K. J. Am. Chem. Soc. 2012, 134, 14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiei Y. M.; Dinescu A.; Dai X.; Palomino R. M.; Heinemann F. W.; Cundari T. R.; Warren T. H. Angew. Chem., Int. Ed. 2008, 47, 9961. [DOI] [PubMed] [Google Scholar]
- Gephart R. T.; McMullin C. L.; Sapiezynski N. G.; Jang E. S.; Aguila M. J. B.; Cundari T. R.; Warren T. H. J. Am. Chem. Soc. 2012, 134, 17350. [DOI] [PubMed] [Google Scholar]
- Finn M.; Friedline R.; Suleman N. K.; Wohl C. J.; Tanko J. M. J. Am. Chem. Soc. 2004, 126, 7578. [DOI] [PubMed] [Google Scholar]
- Steves J. E.; Stahl S. S. J. Am. Chem. Soc. 2013, 135, 15742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konaka R.; Terabe S.; Kuruma K. J. Org. Chem. 1969, 34, 1334. [Google Scholar]
- Avila D. V.; Brown C. E.; Ingold K. U.; Lusztyk J. J. Am. Chem. Soc. 1993, 115, 466. [Google Scholar]
- MacFaul P. A.; Ingold K. U.; Wayner D. D. M.; Que L. J. Am. Chem. Soc. 1997, 119, 10594. [Google Scholar]
- Zavitsas A. A.; Ehrenson S. J. Am. Chem. Soc. 1965, 87, 2841. [Google Scholar]
- Carey F. A.; Sundberg R. J.. Advanced Organic Chemistry, 5th ed.; Springer: New York, 2007. [Google Scholar]
- Kochi J. K. Tetrahedron 1962, 18, 483. [Google Scholar]
- Kochi J. K. J. Am. Chem. Soc. 1962, 84, 3271. [Google Scholar]
- Ritter J. J.; Minieri P. P. J. Am. Chem. Soc. 1948, 70, 4045. [DOI] [PubMed] [Google Scholar]
- Barnard P. W. C.; Yang N. C. Chem. Ind. 1961, 1573. [Google Scholar]
- Xia Q.; Liu X.; Zhang Y.; Chen C.; Chen W. Org. Lett. 2013, 15, 3326. [DOI] [PubMed] [Google Scholar]
- Kochi J. K. J. Am. Chem. Soc. 1961, 83, 3162. [Google Scholar]
- Niki E.; Kamiya Y. J. Am. Chem. Soc. 1974, 96, 2129. [Google Scholar]
- Casitas A.; King A. E.; Parella T.; Costas M.; Stahl S. S.; Ribas X. Chem. Sci 2010, 1, 326. [Google Scholar]
- Huffman L. M.; Stahl S. S. J. Am. Chem. Soc. 2008, 130, 9196. [DOI] [PubMed] [Google Scholar]
- Andrus M. B.; Argade A. B.; Chen X.; Pamment M. G. Tetrahedron Lett. 1995, 36, 2945. [Google Scholar]
- Stavropoulos P.; Çelenligil-Çetin R.; Tapper A. E. Acc. Chem. Res. 2001, 34, 745. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.