

Abstract

Inorganic nanoscale X-ray contrast agents (XCAs) offer many potential advantages over currently used intravascular molecular contrast agents, including longer circulation and retention times, lower administration volumes, and greater potential for site directed imaging. Elemental bismuth nanoparticles (BiNPs) are particularly attractive candidate XCAs due to the low cost, the high atomic number and high density of bismuth, and the likelihood that BiNPs will oxidatively decompose to biocompatible bismuth(III) ions at controlled rates for renal excretion. Herein we describe the synthesis of ultrahigh payload BiNPs in 1,2-propanediol using a borane reducing agent and glucose as a biocompatible surface stabilizer. Both synthetic solvent (1,2-propanediol) and surfactant (glucose) are evident on the BiNP surfaces when analyzed by 1H NMR and FT-IR spectroscopies. These particles contain ∼6 million Bi atoms per NP and have large inorganic cores (74 nm by TEM) compared to their hydrodynamic size (86 nm by DLS). Thus, the dense BiNP core constitutes the majority (∼60%) of each particle’s volume, a necessary property to realize the full potential of nanoscale XCAs. Using quantitative computed tomography in phantom and in vitro imaging studies, we demonstrate that these BiNPs have greater X-ray opacity than clinical small molecule iodinated contrast agents at the same concentrations. We furthermore demonstrate a favorable biocompatibility profile for these BiNPs in vitro. Altogether, these studies indicate that these ultrahigh payload BiNPs, synthesized from known biocompatible components, have promising physical and cytotoxicological properties for use as XCAs.
Introduction
Nanoscale X-ray contrast agents are an attractive new class of potentially non-nephrotoxic contrast materials, which have been demonstrated to evade kidney filtration because of their large size1 and which can offer the prospect of blood pool and site directed X-ray imaging.2,3 A number of recent publications concerning nanoparticle XCAs focus on the use of inorganic nanocrystals, based on elements such as gold, platinum, tantalum, and bismuth.4−12 The higher atomic numbers of these elements confer higher X-ray attenuation compared to iodinated molecules, the element upon which many clinically approved agents are based.13 Furthermore, the density of inorganic nanocrystals is substantially greater than the density of iodinated molecules, producing higher X-ray attenuation per unit volume of contrast material, which is beneficial for site directed X-ray imaging.2
Bismuth nanoparticles (BiNPs) have several attractive features as XCAs. First, the salt precursors from which metal nanoparticles are typically prepared by chemical reduction are significantly less expensive for bismuth vs many other high Z elements, e.g., gold and platinum, owing to the relatively low price of the bulk metal (bulk bismuth is currently approximately $4/mol).14 Furthermore, examination of Pourbaix diagrams (reduction potential vs pH) indicates that BiNPs are expected to oxidatively decompose to soluble bismuth(III) species under physiological conditions, while other NPs based on heavy elements, such as gold and platinum, are oxidatively stable under physiological conditions, thus posing challenges for excretion. If the rate of oxidative decomposition of BiNPs in vivo is slow, BiNPs could offer both long circulation and retention times based on the large molecular weight of the NPs and renal clearance via excretion of soluble bismuth(III) ions. The evolution of soluble bismuth(III) ions from BiNPs is expected to be biologically safe, since bismuth(III) compounds have been used medicinally for centuries.15 Bismuth(III) plasma concentrations up to 50 μg/mL (0.2 mM) are generally considered safe,16 and thus the development of bismuth-based XCAs has recently started to receive attention. Reports of bismuth-based nanoparticle XCAs have included Bi2S3 nanoparticles stabilized by polyvinylpyrrolidone (PVP)4 or phospholipids,5 and bismuth ions loaded into polymer nanoclusters17 for blood pool imaging, as well as ∼20 nm PVP-coated elemental BiNPs, which were used in X-ray fluorescence detection of circulating tumor cells.18,19 Of these examples, the latter represents the most ideal nanoparticle XCA because its X-ray opaque portion is composed entirely of densely packed, high Z atoms; thus it can be considered to have a relatively high X-ray opaque payload.
Despite their advantages as XCAs, BiNPs have one major drawback - synthetic methods to produce BiNPs are in their infancy relative to other inorganic nanocrystals (e.g., gold, silver, cadmium selenide, iron oxide, etc.). The best size and morphology controlled BiNP preparations have been demonstrated using a chemical reduction of Bi(N(SiMe3)2)3 in apolar solvents,20−22 and the resultant products are not water-soluble. Thermal polyol reduction reactions of dissolved bismuth(III) ions in polyalcohol solvents have been accomplished, but these methods generally result in poor morphology and size control over the bismuth nanocrystals.23,24 Aqueous preparations of BiNPs are uncommon, since they have also suffered from poor morphology and size control, most likely due to the poor and pH-dependent solubility of bismuth(III) ions in water. Three successful examples of aqueous BiNP preparations are the aqueous reduction of bismuth(III) salts by either hydrazine hydrate in the presence of sodium oleate25 or sodium-hypophosphite in the presence of tartaric acid and sodium oleate,26 both of which ultimately yield 25–40 nm hydrophobic particles, and our pH-dependent synthesis, which yields small (BiNP core diameter = 20 nm), somewhat polydisperse BiNP cores that are embedded in a large dextran–polymer shell (hydrodynamic diameter = 60 nm).27 The products of our previous method had relatively small X-ray opaque BiNP cores as a fraction of their total volume (Figure 1), with bismuth atoms constituting ∼4% of the particle volume, resulting in a low payload of X-ray attenuating atoms (∼100 000 bismuth atoms/NP). Such a low payload is undesirable due to the low sensitivity (∼5 mM)13 of X-ray based imaging methods such as computed tomography (CT), which has hampered the extension of molecular imaging techniques to CT, despite the large and increasing clinical application of CT imaging.
Figure 1.
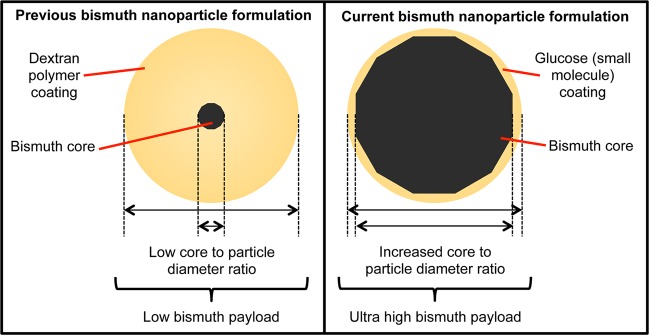
Schematic illustration of the BiNP XCA formulation presented herein compared to our previously reported BiNP XCA formulation. Due to the increased BiNP core to total diameter ratio, the payload of the current formulation is much higher.
In this study, we sought to maximize the X-ray opaque payload by increasing the BiNP core size and decreasing the organic shell size to produce ultrahigh payload BiNPs (Figure 1). In addition, we set out to achieve a scalable (in quantity) and controllable (in size and morphology) synthesis method for BiNPs that employs biologically benign reagents and results in noncytotoxic, aqueous BiNP colloids. Herein, we report the synthesis of large BiNPs (74 ± 14 nm BiNP cores) that uses 1,2-propanediol and glucose as biocompatible small molecule surfactants (1,2-propanediol also serves as solvent), which results in highly uniform, aqueous nanoparticles with a mean hydrodynamic diameter of 86 nm. These BiNPs are ∼60% bismuth by volume and carry ∼6 million bismuth atoms per nanoparticle. For BiNPs synthesized using this method at 80 °C, we demonstrate a high clinical X-ray opacity in body phantom and cellular CT imaging studies in addition to retained cell viability with short exposures (1 h) to either HeLa or macrophage cells but a decreased viability for macrophage cells at long (24 h) exposures.
Materials and Methods
Synthesis of BiNPs
In a 250 mL beaker, 54 g of α-d-glucose, (Acros Organics, >99+%, anhydrous) was partially dissolved in 84 mL of 1,2-propanediol (PPD, Acros Organics, 99%) by immersion of the beaker in a temperature-controlled oil bath with mechanical stirring. To this mixture, 4 mL of a 250 mM bismuth nitrate pentahydrate (Acros Organics, 98%) solution in PPD was added, and the temperature was increased to a synthetic reaction temperature varying between 60 and 100 °C. At the synthetic reaction temperature, borane morpholine (Acros Organics, 97%) dissolved in PPD (250 mM) was added (12.2 mL) to rapidly and homogenously initiate nanoparticle formation. The reaction was stirred for 60 s after addition of reducing agent and then quenched by pouring the beaker contents into 200 mL of electrophoretically pure ice water. To obtain larger quantities of particles for biological and imaging assays, batches of particles were pooled prior to work up and purification. To optimize synthetic conditions in terms of BiNP size and morphological uniformity, reagent concentrations, synthesis temperature, and particle growth times were varied. Size and uniformity as a function of these reaction conditions was assessed qualitatively using transmission electron microscopy (TEM). The effect of the small molecule, sugar surfactant additive on size and morphology control was also examined qualitatively by TEM. For syntheses using surfactants other than glucose, an equimolar amount of surfactant was used in place of glucose.
Purification of BiNPs
After reaction quenching, 20 mL aqueous solutions of the black BiNPs were diluted to 50 mL with electrophoretically pure water followed by centrifugation at 3.0 krcf for 30 min. After centrifugation, the supernatant was decanted off and the precipitated BiNPs were resuspended with sonication and subsequently dialyzed (SnakeSkin regenerated cellulose dialysis tubing, 10k MWCO) against electrophoretically pure water to remove excess reagents (e.g., PPD and glucose) and reaction byproducts (e.g., borate). Finally the BiNPs were passed through a 450 nm syringe filter (Chromafil Xtra PA-45/25 polyamide) and either stored at 4 °C or flash frozen on liquid nitrogen followed by lyophilization.
BiNP Characterization
Transmission Electron Microscopy (TEM)
TEM was performed on an FEI Tecnai F-20 TEM operating at 200 kV. Purified BiNPs were briefly sonicated and then drop-cast onto holey carbon Cu supported TEM grids (Ted Pella) and dried at 100 °C for 1 h prior to imaging. To determine the core size distribution we report for BiNPs synthesized with glucose surfactant at 80 °C, the longest diameter internal to the nanocrystal was measured for 1124 faceted (anisotropic) particles.
Scanning Electron Microscopy (SEM)
SEM was performed on a FEI Sirion XL30 FEG SEM. Aqueous BiNPs samples were drop-cast onto an aluminum support stub and sputter coated with gold (PELCO 91000 Sputter Coater) for 60 s prior to imaging.
Dynamic Light Scattering (DLS)
DLS measurements were performed on a Horiba LB-550 dynamic light scattering instrument and an instrumental algorithm was used to supply the hydrodynamic diameters as number distributions. For these measurements, freshly syringed filtered samples were dispersed in water and measured at five dilutions to ensure size distributions independent of concentration effects. To determine the core loading (i.e., the volume percent bismuth), the same sample used to determine BiNP core size by TEM (synthesized at 80 °C with glucose surfactant) was also measured by both SEM and DLS.
X-ray Diffraction (XRD)
Lyophilized BiNPs were pressed onto a glass support slide, and XRD data were collected in focused beam (Bragg–Brentano) geometry on a Rigaku Ultima IV X-ray diffraction system using graphite monochromatized Cu Kα radiation. Scans were performed over the angular range 10–70° 2θ at a scan rate of 0.25°/min at room temperature.
Fourier Transform Infrared Spectroscopy (FT-IR)
FT-IR spectra were collected on a Thermo Scientific Nicolet iS10 spectrophotometer equipped with a single-bounce diamond attenuated total reflectance (ATR) attachment. Aqueous or lyophilized BiNPs were drop-cast or pressed with a glass slide onto the ATR crystal to deposit sample films for analysis. Glucose solid and liquid PPD were measured in their neat forms for comparison by placing them in physical contact with the ATR crystal for measurement.
Proton Nuclear Magnetic Resonance Spectroscopy (1H NMR)
Lyophilized BiNPs, glucose, and PPD (250 μL) were separately dissolved in 750 μL of D2O (Cambridge Isotope Laboratories, 99.9%), and 1H NMR spectra were obtained on a Bruker 600 MHz AVANCE-III Nuclear Magnetic Resonance (NMR) spectrometer using a standard pulse sequence (Zg30). Spectra were processed using the Bruker TopSpin 2.1 software package.
Inductively Coupled Plasma–Optical Emission Spectroscopy (ICP-OES)
BiNP stocks (5, 10, and 25 μL) were each dissolved in 1 mL of a 1:1 mixture of concentrated nitric acid and water. The final volume of each sample was brought to 10 mL by addition of deionized water. Elemental analysis was performed using ICP-OES (Spectro Genesis ICP) at the Department of Earth and Environmental Science, University of Pennsylvania. For ICP-OES analysis, bismuth analytical standards were purchased from Fisher Scientific (Pittsburgh, U.S.A.). The concentration of bismuth was determined for each sample and then multiplied by the dilution factor. The concentrations thus obtained were averaged to give the final bismuth concentration of the samples diluted for quantitative CT measurements and cell viability assays. ICP–mass spectrometry (ICP-MS) was performed by Elemental Analysis Inc. (Lexington, KY).
Quantitative CT Measurements
Solutions of bismuth or iodine ranging from 0 to 100 mM in concentration were prepared in triplicate in 1.5 mL microcentrifuge tubes. The samples were BiNPs, Bi(NO3)3 or iopamidol, a commercially available iodine-based contrast agent. The BiNP and iopamidol were diluted in PBS, whereas Bi(NO3)3 was diluted in ethylene glycol due to poor water solubility. The concentration of the BiNP samples was determined from ICP-OES measurements, as described above. The samples, each at several different concentrations, were placed in a plastic rack, and the rack was wrapped in parafilm. The rack was then placed on top of another rack and subsequently taped to the (inside) bottom of a 24 cm wide plastic container. Racks inside the container were covered with water to 21 cm in height to simulate attenuation effects of a patient. The preparation of these body phantoms has been described previously.11
Each set of samples was scanned in the container using a 64-slice clinical CT scanner (Definition DS, Siemens Medical Solutions, Malvern, PA) in single source acquisition mode at 80, 100, 120, or 140 kV and 300 mA with a matrix of 512 × 512, field of view of 37 × 37 cm, and slice thickness of 0.6 cm. The reconstruction kernel used was B30f. Images were analyzed using Osirix 64 bit (v3.7.1). The attenuation value in Hounsfield Units (HU) for each sample tube was recorded from three different slices and averaged for each concentration. Attenuation rates (HU/mM) reported herein are the gradients calculated from graphs of attenuation (HU) versus concentration (mM).13,28
In Vitro Experiments
Cell Culture
J774A.1 murine macrophage cells and HeLa human cervix adenocarcinoma cells were purchased from ATCC, Manassas, VA, U.S.A. J774A.1 cells were maintained in a culture medium of DMEM (Dulbecco’s Modified Eagle’s Medium), while HeLa cells were maintained in EMEM (Eagle’s Minimum Essential Medium). In each case the medium was supplemented with 10% FBS, 45 IU mL–1 penicillin, and 45 IU mL–1 streptomycin. The cells were incubated at 37 °C in 5% CO2.
Analysis of CT Contrast in Cell Pellets
Incubations with BiNPs were performed as previously described.29 Briefly, both types of cells were seeded from confluent flasks at a 1:1 dilution into six well plates (2 mL/well) and allowed to adhere and normalize for 24 h. The resulting cell monolayers were then washed with sterile phosphate buffered saline (PBS). BiNPs dispersed at concentrations of 0, 0.10, 0.25, or 0.50 mg/mL in the appropriate cell medium were then incubated with the cells. After 1 or 24 h incubation, the medium was removed and the cells were gently washed three times with PBS to remove noninternalized nanoparticles. Following incubation and washing, HeLa cells were trypsinized, and the J774A.1 cells were collected using cell scrapers. The cells were centrifuged at 800 rpm, the supernatant removed, and the cell pellets dispersed in 2% (v/v) aqueous glutaraldehyde. The cells were allowed to settle into loosely packed pellets for further quantitative CT attenuation measurements. Each experiment was done in triplicate. Cells were scanned using the same scanner and analyzed using the same software as detailed in the previous section. The scanning parameters used were 140 kV and 160 mA with a matrix size of 512 × 512 cm, field of view at 25 × 25 cm, and a slice thickness of 0.4 cm. The reconstruction kernel used was U30u. These experiments were done with a clinical scanner using parameters that could be used for patients.
TEM of Cells
After CT scanning, the cells were prepared for TEM using typical methods.30 In short, the cell pellets were incubated in 2.5% glutaraldehyde and 2.0% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, overnight at 4 °C. After subsequent buffer washes, samples were postfixed with 2% aqueous osmium tetraoxide for 1 h, en bloc stained with 2% uranyl acetate, and dehydrated in a graded ethanol series. After two changes of propylene oxide, samples were infiltrated and embedded in EMBed-812 (Electron Microscopy Sciences). Thin (∼80 nm) sections of these samples were taken and counter stained with uranyl acetate and lead citrate prior to imaging. TEM imaging was performed on the sections using a JEOL 1010 electron microscope fitted with a Hamamatsu digital camera and using AMT Advantage image capture software.
MTS ((3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) Cell Viability Assay
Cytotoxicity assays were performed in 96-well microplates (Nunc, Denmark) seeded with 100 μL/well of 1 × 105 cells/mL HeLa or J774A.1 cell suspensions. After 24 h, cells were washed with PBS and treated with solutions of BiNPs dispersed in complete culture medium and incubated at 37 °C in a 5% CO2 humidified atmosphere. Aqueous DMSO (10% v/v) was used as the positive control for both cell lines.
Assays were carried out according to the manufacturer’s instructions. Briefly, media were removed after 1 or 24 h and the cells were washed gently with sterile PBS, 100 μL of cell culture medium, and 20 μL of MTS/phenazine methosulfate solution. Six wells were used per condition. After incubation and washing, the absorbance of each well was measured in a microplate reader at 490 nm to assess cellular viability. Three independent experiments were performed for each exposure concentration, and the percentage of viable cells was calculated relative to untreated cells. Data are presented as mean ± standard deviation (n = 3).
Results and Discussion
Synthesis of Size and Morphologically Uniform, Aqueous BiNP X-ray Contrast Agents
We recently reported an aqueous synthesis method that produced dextran-coated BiNPs by NaBH4 reduction of bismuth(III) nitrate, which was suspended in glycine buffer containing 75 kDa dextran polymer.27 When synthesized at pH 10, BiNPs produced using this method have a number of appealing properties for use in XCA applications, including aqueous solubility and stability, dense elemental bismuth cores, and a biocompatible surface-stabilizing polymer coating. These particles had an average bismuth core diameter of 20 nm by TEM and an average hydrodynamic diameter of 60 nm by DLS; using these diameters and modeling the particles as spherical inorganic core/polymer shell NPs, the dextran-coated BiNPs produced by this method contained less than 4% bismuth by volume. We found that it was difficult to concentrate these nanoparticles such that they produced solutions with strong X-ray contrast, and we concluded that the poor X-ray opacity of dextran-coated BiNP solutions was due to the large surfactant shell and low loading of bismuth in these core/shell NPs. We therefore decided to explore small sugar molecules, such as glucose, fructose, maltose, and others, that are chemically similar to dextran and should produce much thinner surface-stabilizing coatings around the bismuth nanocrystal cores (see Figure 1).
Substitution of the dextran polymer with glucose in the previously described aqueous synthesis routinely produced BiNPs with poor and uncontrolled morphology. We attributed this to the generally poor solubility of bismuth(III) salts in aqueous solution, and thus we sought alternate biocompatible solvents with greater ability to solvate bismuth(III) ions. Good solubility should provide better uniformity in particle nucleation and growth and thus result in greater synthetic control over particle size, size distribution, and morphology. We found that polyalcohol solvents, specifically ethylene glycol, glycerol, and PPD, were capable of forming stable solutions of bismuth(III) ions, with concentrations of up to 2 M.
The polyol reaction, in which metal cations are reduced to elemental nanoparticles in a solvothermal process, has previously been used to produce BiNPs in a variety of polyalcohol solvents; these reactions typically use polymeric PVP surfactant to control the particle size and morphology.23,24,31−34 However, in general, we found that PVP-coated BiNPs produced by the polyol method are oxidatively unstable when dispersed in water. Experimentally, we observed that suspending such particles in water resulted in immediate aggregation as well as slow oxidation, which was indicated by the formation of an amorphous white precipitate over time. This aggregation and oxidation behavior in polar solvents has been previously observed by others working with polyol-prepared, PVP-coated BiNPs.31 Under typical polyol reaction conditions (>180 °C), we observed that inclusion of sugar molecules in place of the PVP surfactant to attempt to increase the aqueous stability of the product BiNPs resulted in a highly polymerized solvent/surfactant/particle aggregate, which was difficult to work up further.
To avoid high temperature polymerization/decomposition of the sugar surfactants, we therefore used a chemical reducing agent to produce BiNPs from bismuth(III) nitrate in glucose saturated PPD at lower temperatures (60–100 °C). Borane morpholine was chosen as the reducing agent for its kinetically slower reactivity relative to NaBH4, which should aid in size and morphology control during BiNP growth, and for anticipated ease of removal of the expected decomposition products (morpholine and borate) from the aqueous BiNPs. Of the biocompatible polyalcohol solvents with good bismuth(III) solubility, we optimized the BiNP synthesis method in PPD solvent because of its greater biocompatibility relative to ethylene glycol and lower viscosity relative to glycerol (31 vs 612 mPa·s, respectively).35,36 PPD is known to be safely biologically metabolized and is commonly used in food products.37
In a typical BiNP synthesis (Figure 2A) bismuth(III) nitrate pentahydrate was dissolved in PPD with sonication to produce a fully dissolved bismuth(III) precursor solution. This solution was added to a solution of PPD saturated with glucose surfactant, and the resulting solution was subsequently heated in air to a reaction temperature of 60–120 °C. Addition of borane morpholine solution at the reaction temperature initiated the formation of black BiNP colloids, and after 60 s, the reaction was terminated by rapid dilution in ice water.
Figure 2.

(A) BiNP reaction scheme. (B) BiNP size and morphology dependence on synthetic temperature determined by TEM. Scale the same for each panel.
Qualitatively, the morphology and yield of BiNPs produced by this method were observed to be correlated with synthesis temperature (Figure 2B). In general, BiNPs synthesized at higher temperatures (100 °C) were spherical with a high degree of size polydispersity, while BiNPs synthesized at lower temperatures (60 °C) were highly faceted with a narrower size distribution. However, synthesis at lower temperatures also resulted in lower synthesis yields after purification (gravimetrically determined). At synthetic temperatures below 60 °C no reaction was observed, and at temperatures at or above 120 °C the sugar surfactant polymerized/decomposed. We found that morphology, size, uniformity, and yield trade-offs, on average, were best at 80 °C.
To further probe the role of glucose in particle formation and surface stabilization, the effects of alternate surfactants on nanoparticle morphology were surveyed when the reaction was performed at 80 °C. The nanoparticle synthesis can be visually monitored by observing the formation of a black solution of BiNPs upon addition of reducing agent at the reaction temperature. In the absence of glucose, no color change was observed upon the addition of borane morpholine to the bismuth(III) starting material in PPD. Thus, we can conclude that glucose aids either in the reduction of bismuth(III) or in the nucleation of bismuth nanocrystallites, since the black color characteristic of bismuth(0) nanocrystallites was not observed. Additionally, when sugar alcohols, such as sorbitol or xylitol, replaced glucose in the synthesis, no particle formation was observed. These results indicate that glucose participates in the reduction step and suggest that surfactants bearing carbonyl groups are necessary to initiate low temperature BiNP formation (some fraction of glucose will be its open chain form with an aldehyde functional group). Synthesis of BiNPs at 80 °C in the presence of small molecules with carbonyl groups, such as dihydroxyacetone, ribose (an aldopentose), fructose (a ketohexose), or maltose (a reducing disaccharide), all enabled the formation of BiNPs, albeit with substantially altered morphology relative to the glucose-coated BiNPs (Figure 3). Of the small molecule surfactants that initiated BiNP formation, we found glucose to produce the greatest uniformity in morphology and size, and so CT and cytotoxicity experiments were performed on nanoparticles synthesized at 80 °C coated with this ligand.
Figure 3.

TEM images of BiNPs synthesized using (A) dihydoxyacetone, (B) ribose, (C) fructose, (D) glucose, and (E) maltose as capping ligands. The synthesis was performed in ligand saturated PPD at 80 °C in each case. Scale the same for each panel.
Characterization of Aqueous BiNP Colloids
BiNPs synthesized at 80 °C appear by TEM imaging to be discrete, highly crystalline, and faceted (Figure 4A,B). Because of the anisotropic nanocrystal morphologies observed, a longest diameter (LD) size distribution (Figure 4C) was determined by manually measuring the longest apparent internal distance for >1000 nanoparticles from various sections of the TEM grid. The BiNP core diameter was thus determined to be 74 ± 14 nm. The LD core size distribution determined in this way is shown in Figure 4C overlaid on a corresponding hydrodynamic diameter size distribution measurement obtained by DLS; the average hydrodynamic diameter determined by DLS was 86 ± 35 nm. The TEM and DLS size distributions demonstrate the ultrahigh volumetric loading of bismuth, since only a small portion of the hydrodynamic diameter is due to the organic surface-stabilizing coating. This result can also be seen by comparing corresponding TEM and SEM images (Figure 4A,B, respectively, scale bars 200 nm), where the size observed by SEM after gold sputter-coating is a measurement of the size of the NPs inclusive of their electron-poor organic shells. The 74 nm core to 86 nm HDR ratio, when modeled as spheres, indicates that the BiNPs are ∼64% inorganic bismuth by volume; 74 nm bismuth nanocrystals are estimated to contain ∼6 million atoms/nanocrystal. This is a substantial improvement over our previously synthesized BiNPs, which were calculated to contain 100 000 atoms and be ∼4% inorganic bismuth by volume.27 Furthermore, ICP-MS analysis of samples indicated the BiNP to be composed of 75% bismuth and 25% other material, in reasonable agreement with the values determined from TEM and DLS measurements.
Figure 4.

Size and morphology of BiNPs synthesized in glucose saturated PPD at 80 °C by (A) TEM and (B) SEM. (C) The core diameter (from TEM) and hydrodynamic diameter (measured by DLS).
Elemental bismuth crystallizes in the rhombohedral crystal system (R3̅m) and interestingly we observed multiple nanoparticle morphologies by TEM consistent with preferential face growth of rhombohedral nanocrystals (Figure 4A). The PXRD pattern of the BiNPs matches that reported for elemental bismuth (Figure 5A; JCPDS Card No. 00-044-1246), and no crystalline oxidation products are observed.
Figure 5.

Characterization of BiNP: (A) the powder XRD pattern, (B) the 1H NMR spectrum, and (C) the FT-IR spectrum.
The identity of BiNP surface ligands was determined by 1H NMR and FT-IR spectroscopies. The 1H NMR spectrum of washed and dialyzed BiNPs contains peaks from both PPD and glucose, which indicates that both the synthesis solvent and surfactant are present on the BiNP surfaces (Figure 5B). The 1H NMR spectrum of the BiNPs notably contains a peak at δ = 1.1 ppm, assigned as the methyl protons from PPD, and a peak at δ = 5.2 ppm, assigned as the hydrogen bonded to the anomeric carbon in glucose. Integration of these peaks reveals a 9:2 molar ratio of glucose to PPD in the final formulation. The broad overlapping peaks observed for the BiNPs between δ = 3–4 ppm are difficult to definitively assign since protons from both PPD and glucose are found in this region. Peak broadening in the BiNP spectrum may be indicative of a slower tumbling rate of the organic molecules in solution when adhered to large inorganic nanoparticles.
Consistent with the 1H NMR results, the FT-IR spectrum of the BiNPs has features from both solvent and surfactant, indicating that both are present on the surfaces (Figure 5C). The broad infrared absorbance between 1000 and 1100 cm–1 can be assigned to C–O and C–C bonds, and this region most resembles that of the glucose standard; however, PPD also has sharp peaks in this region that may contribute to the BiNP spectrum. Conversely, the C–H stretching region around 2900 cm–1 most resembles the corresponding feature in the PPD spectrum. Finally, the as-prepared BiNPs show a broad O–H absorbance between ∼3000–3500 cm–1, which is expected of both hydroxylated surfactant ligands.38 However, after lypophilization workup, the BiNPs showed a markedly different FT-IR spectrum, with a decreased number of peaks between 700 and 1500 cm–1, in addition to loss of the O–H vibration. This observation can be attributed to conformational restrictions of the organic surfactants on the BiNP surfaces or perhaps to surface molecule cross-linking, both of which could result from a high degree of lypophilization-induced hydrogen bonding upon removal of water. The degree of hydrogen bonding, indicative of the degree of macromolecular organization, has been previously examined using FT-IR spectroscopy to track the degree of crystallinity during cellulose hydrolysis.39 Here, the simplified FT-IR spectrum of the lyophilized (vs as-prepared) BiNPs suggests restricted movements and vibrations of the organic shell, which indicate increased hydrogen bonding and ordering of small molecules on the BiNP surfaces. Thermogravimetric analysis was attempted on the BiNP; however, distinct plateaus that correlate with the loss of water and organic material were not observed, and only a continuous, gradual weight loss was observed when dried colloids were heated up to 400 °C. Therefore, this method did not yield information on the weight percent of the surface ligands.
X-ray Contrast Evaluation of Ultrahigh Payload BiNPs
The X-ray attenuation of BiNPs was investigated using a clinical computed tomography (CT) scanner. BiNPs were diluted and sealed in microcentrifuge tubes that were submerged in a tank of water to mimic imaging conditions in a patient. Concentrations of bismuth from 0 to 65 mM were scanned at four commonly used X-ray tube voltages, namely, 80, 100, 120, and 140 kV. As an example, the CT image obtained for the BiNP solutions scanned at 140 kV is shown in Figure 6A. The CT attenuation in Hounsfield units (HU) plotted as a function of bismuth concentration (Figure 6B) reveals a linear correlation between bismuth concentration and attenuation as expected.11 The CT attenuation of bismuth in the form of aqueous BiNPs, relative to that of solvated bismuth(III) nitrate ions and an aqueous iodine standard (iopamidol), is also shown (Figure 6C) and was obtained by calculating the slope of the attenuation vs concentration plot for each tube voltage. There is no statistically significant difference in attenuation between the BiNPs and solvated bismuth ions. Both forms of bismuth show significantly higher X-ray attenuation relative to iodine. Interestingly, the iodine attenuation rate decreases with increasing X-ray tube voltage, while the attenuation of bismuth(III) ions and BiNPs is relatively insensitive to tube voltage in the range used in clinical CT scanners, which is advantageous as the same contrast would be produced using any CT imaging protocol.
Figure 6.

CT attenuation evaluation of BiNPs. (A) CT image of solutions of BiNP varying in concentration between 1 and 65 mM bismuth. (B) The attenuation of bismuth nanoparticles as a function of concentration and X-ray tube voltages (80–140 kV). (C) The rates of attenuation for BiNPs, bismuth(III) ions, and iodine at different X-ray tube voltages.
To demonstrate the potential of BiNPs to create contrast in biological tissue, incubations were performed with two commonly used cell lines: HeLa cells and murine macrophages (J774A.1). BiNPs were dispersed in culture media at a range of concentrations (0.1, 0.25, and 0.5 mg/mL) and incubated with the cells for 24 h before collection by centrifugation and washing with PBS. CT imaging of the resulting cell pellets shows clear increases in attenuation for both cell lines when incubated with BiNPs, with more marked increases for the macrophages than for the HeLa cells (Figure 7A), consistent with the high phagocytotic activity of macrophages. Uptake by both cell lines is linear with concentration, indicating a nonspecific uptake process (Figure 7B). We performed electron microscopy on sections of the cells to confirm whether the increase in attenuation was due to BiNP uptake. Nanoparticles could clearly be observed within the HeLa cells and fixed macrophage cells show visible formation of endosomal/lysosomal structures around BiNPs (Figure 7C,D).
Figure 7.

Uptake of BiNPs in model cell lines. (A) CT images of J774A.1 and HeLa cells incubated with BiNPs and formed into pellets. (B) Quantitative analysis of the attenuation in the cell pellets. TEM shows that BiNPs are taken up in endosomal/lysosomal compartments in both J774A.1 (C) and HeLa cells (D).
Cell Viability Experiments
Cell viability was examined using the MTS assay, which yields quantitative information on cellular enzyme activity (Figure 8). After one hour of incubation with BiNPs, no decrease in cellular viability is observed for either cell type. After 24 h of incubation with BiNPs, there was no measured decrease in viability for HeLa cells at any BiNP concentration tested; however, macrophage cell viability drops, with an apparent LD50 of 50 μg/mL (0.2 mM). Decreased macrophage cell viability at such elevated concentrations and long exposure times is unsurprising but will warrant careful toxicity investigations in eventual in vivo experiments. The CT imaging data (Figure 7A,B) indicates that the concentration of bismuth in the macrophages at 24 h must be extremely high, which is likely the cause of the observed toxicity. The sustained viability of HeLa cells in the presence of BiNPs and the viability of the macrophage cells after incubation for one hour is suggestive of good biological compatibility. Future in vivo studies will further examine biocompatibility and BiNP fate (i.e., clearance) in addition to characterization of any BiNP decomposition products.
Figure 8.

Viability of HeLa and J774.A cells incubated with BiNPs for (A) 1 h and (B) 24 h.
Conclusions
Nanoscale XCAs have been advocated for their many potential advantages, including potential alternate biological clearance routes and longer circulation times relative to molecular XCAs, which may enable better blood pool imaging, site-directed imaging, and decreased administration volumes. Relative to previously reported gold nanoparticle XCAs, BiNPs are less oxidatively stable and higher in atomic number, which may decrease bioaccumulation while providing increased X-ray contrast. Here we describe and characterize a novel synthetic strategy for the production of aqueous, colloidally stable BiNPs using PPD as an inexpensive biologically compatible and highly coordinating solvent, coupled with a volumetrically small, inexpensive glucose surfactant layer that protects the BiNP surface from aqueous oxidative decomposition. The nanoparticles have a large inorganic elemental bismuth core relative to the total hydrodynamic size, and 1H NMR and FT-IR both indicate solvent and surfactant are present on the particle surface. The nanoparticles have high X-ray attenuation properties, as demonstrated by CT imaging, and promising biological compatibility. These data suggest this agent could be a highly potent nanoXCA that enables targeted imaging with CT. Future studies will focus on the in vivo application and toxicity assessment of this agent.
Acknowledgments
This work was supported by the NIH (R00 EB012165, D.P.C.), the Burroughs Wellcome Fund (Award Number 1007294.01, A.M.G.), and the Oregon Nanoscience and Microtechnologies Institute (A.M.G) and via start up funds from the University of Pennsylvania (D.P.C.) and Portland State University (A.M.G.). In addition, partial support was provided by the Penn Nanotoxicology Alliance (Nanotechnology Institute, Nano-Biointerface Center, Center for Translational Targeted Therapeutics and Nanomedicine and the Center of Excellence in Environmental Toxicology – P30ES13508) with funds from the Vice Provost of Research (D.P.C.).
We also acknowledge the National Science Foundation for XRD instrumentation (NSF-MRI, Award Number DMR-0923572). We thank the Center for Electron Microscopy and Nanofabrication (CEMN), Portland State University, for microstructural and compositional analysis services. We gratefully acknowledge the assistance of Srikar Rao Darmakkolla with thermogravimetric analysis experiments.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Choi H. S.; Liu W.; Misra P.; Tanaka E.; Zimmer J. P.; Ipe B. I.; Bawendi M. G.; Frangioni J. V. Nat. Biotechnol. 2007, 25, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakhmola A.; Anton N.; Vandamme T. F. Adv. Healthcare Mater. 2012, 1, 413. [DOI] [PubMed] [Google Scholar]
- Cormode D. P.; Naha P.; Fayad Z. A. Contrast Media Mol. Imaging 2014, 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabin O.; Perez J. M.; Grimm J.; Wojtkiewicz G.; Weissleder R. Nat. Mater. 2006, 5, 118. [DOI] [PubMed] [Google Scholar]
- Kinsella J. M.; Jimenez R. E.; Karmali P. P.; Rush A. M.; Kotamraju V. R.; Gianneschi N. C.; Ruoslahti E.; Stupack D.; Sailor M. J. Angew. Chem., Int. Ed. 2011, 50, 12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allijn I. E.; Leong W.; Tang J.; Gianella A.; Mieszawska A. J.; Fay F.; Ma G.; Russell S.; Callo C. B.; Gordon R. E.; Korkmaz E.; Post J. A.; Zhao Y.; Gerritsen H. C.; Storm G.; Thran A.; Proksa R.; Daerr H.; Fuster V.; Fisher E. A.; Fayad Z. A.; Mulder W. J.; Cormode D. P. ACS Nano 2013, 7, 9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonitatibus P. J.; Torres A. S.; Kandapallil B.; Lee B. D.; Goddard G. D.; Colborn R. E.; Marino M. E. ACS Nano 2012, 6, 6650. [DOI] [PubMed] [Google Scholar]
- Torres A. S.; Bonitatibus P. J.; Colborn R. E.; Goddard G. D.; FitzGerald P. F.; Lee B. D.; Marino M. E. Invest. Radiol. 2012, 47, 578. [DOI] [PubMed] [Google Scholar]
- Alric C.; Taleb J.; Le Duc G.; Mandon C.; Billotey C.; Le Meur-Herland A.; Brochard T.; Vocanson F.; Janier M.; Perriat P.; Roux S.; Tillement O. J. Am. Chem. Soc. 2008, 130, 5908. [DOI] [PubMed] [Google Scholar]
- Chou S.-W.; Shau Y.-H.; Wu P.-C.; Yang Y.-S.; Shieh D.-B.; Chen C.-C. J. Am. Chem. Soc. 2010, 132, 13270. [DOI] [PubMed] [Google Scholar]
- Oh M. H.; Lee N.; Kim H.; Park S. P.; Piao Y.; Lee J.; Jun S. W.; Moon W. K.; Choi S. H.; Hyeon T. J. Am. Chem. Soc. 2011, 133, 5508. [DOI] [PubMed] [Google Scholar]
- Lee N.; Cho H. R.; Oh M. H.; Lee S. H.; Kim K.; Kim B. H.; Shin K.; Ahn T. Y.; Choi J. W.; Kim Y. W.; Choi S. H.; Hyeon T. J. Am. Chem. Soc. 2012, 134, 10309. [DOI] [PubMed] [Google Scholar]
- Galper M. W.; Saung M. T.; Fuster V.; Roessl E.; Thran A.; Proksa R.; Fayad Z. A.; Cormode D. P. Invest. Radiol. 2012, 47, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.metalprices.com/metal/bismuth (accessed June 6, 2013). [Google Scholar]
- Briand G. G.; Burford N. Chem. Rev. 1999, 99, 2601. [DOI] [PubMed] [Google Scholar]
- Serfontein W. J.; Mekel R. Res. Commun. Chem. Pathol. 1979, 26, 391. [PubMed] [Google Scholar]
- Pan D.; Roessl E.; Schlomka J.-P.; Caruthers S. D.; Senpan A.; Scott M. J.; Allen J. S.; Zhang H.; Hu G.; Gaffney P. J.; Choi E. T.; Rasche V.; Wickline S. A.; Proksa R.; Lanza G. M. Angew. Chem., Int. Ed. 2010, 49, 9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain M.; Luo Y.; Sun Z. Y.; Wang C. M.; Zhang M. H.; Fu H. Y.; Qiao Y.; Su M. Biosens. Bioelectron. 2012, 38, 348. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Wang C. M.; Hossain M.; Qiao Y.; Ma L. Y.; An J. C.; Su M. Anal. Chem. 2012, 84, 6731. [DOI] [PubMed] [Google Scholar]
- Richards V. N.; Shields S. P.; Buhro W. E. Chem. Mater. 2011, 23, 137. [Google Scholar]
- Wang F. D.; Tang R.; Yu H.; Gibbons P. C.; Buhro W. E. Chem. Mater. 2008, 20, 3656. [Google Scholar]
- Yarema M.; Kovalenko M. V.; Hesser G.; Talapin D. V.; Heiss W. J. Am. Chem. Soc. 2010, 132, 15158. [DOI] [PubMed] [Google Scholar]
- Goia C.; Matijevic E.; Goia D. V. J. Mater. Res. 2005, 20, 1507. [Google Scholar]
- Wang Y. W.; Kim K. S. Nanotechnology 2008, 19, 265303. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhao J. Z.; Zhao X.; Tang L. Q.; Li Y. L.; Wang Z. C. Mater. Res. Bull. 2009, 44, 220. [Google Scholar]
- Ma D. C.; Zhao J. Z.; Zhao Y.; Hao X. L.; Li L. Z.; Zhang L.; Lu Y.; Yu C. Z. Colloids Surf., A 2012, 395, 276. [Google Scholar]
- Brown A. L.; Goforth A. M. Chem. Mater. 2012, 24, 1599. [Google Scholar]
- Cormode D. P.; Fayad Z. A. Eur. Radiol. 2013, 23, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormode D. P.; Skajaa T.; van Schooneveld M. M.; Koole R.; Jarzyna P.; Lobatto M. E.; Calcagno C.; Barazza A.; Gordon R. E.; Zanzonico P.; Fisher E. A.; Fayad Z. A.; Mulder W. J. M. Nano Lett. 2008, 8, 3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak D.; Babic M.; Jendelova P.; Herynek V.; Trchova M.; Pientka Z.; Pollert E.; Hajek M.; Sykova E. Bioconjugate Chem. 2007, 18, 635. [DOI] [PubMed] [Google Scholar]
- Wang Y. L.; Xia Y. N. Nano Lett. 2004, 4, 2047. [Google Scholar]
- Cheng G.; Wu J. L.; Xiao F.; Yu H.; Lu Z.; Yu X. L.; Chen R. Mater. Lett. 2009, 63, 2239. [Google Scholar]
- Wang W. Z.; Poudel B.; Ma Y.; Ren Z. F. J. Phys. Chem. B 2006, 110, 25702. [DOI] [PubMed] [Google Scholar]
- Wang J. W.; Wang X.; Peng Q.; Li Y. D. Inorg. Chem. 2004, 43, 7552. [DOI] [PubMed] [Google Scholar]
- Saleh M. A.; Begum S.; Begum S. K.; Begum B. A. Phys. Chem. Liq. 1999, 37, 785. [Google Scholar]
- Segur J. B.; Oberstar H. E. Ind. Eng. Chem. 1951, 43, 2117. [Google Scholar]
- Huff E. Biochim. Biophys. Acta 1961, 48, 506. [DOI] [PubMed] [Google Scholar]
- Ibrahim M.; Alaam M.; El-Haes H.; Jalbout A. F.; de Leon A. Eclet. Quim. 2006, 31, 15. [Google Scholar]
- Oh S. Y.; Yoo D. I.; Shin Y.; Kim H. C.; Kim H. Y.; Chung Y. S.; Park W. H.; Youk J. H. Carbohydr. Res. 2005, 340, 2376. [DOI] [PubMed] [Google Scholar]