

Abstract
A highly ordered array of T7 bacteriophages was created by the electrophoretic capture of phages onto a nanostructured array with wells that accommodated the phages. Electrophoresis of bacteriophages was achieved by applying a positive potential on an indium tin oxide electrode at the bottom of the nanowells. Nanoscale arrays of phages with different surface densities were obtained by changing the electric field applied to the bottom of the nanowells. The applied voltage was shown to be the critical factor in generating a well-ordered phage array. The number of wells occupied by a phage, and hence the concentration of phages in a sample solution, could be quantified by using a DNA intercalating dye that rapidly stains the T7 phage. The fluorescence signal was enhanced by the intrinsic photonic effect made available by the geometry of the platform. It was shown that the quantification of phages on the array was 6 orders of magnitude better than could be obtained with a fluorescent plate reader. The device opens up the possibility that phages can be detected directly without enrichment or culturing, and by detecting phages that specifically infect bacteria of interest, rapid pathogen detection becomes possible.
Keywords: T7 bacteriophages, photonic crystal, capture, electrophoresis, array, pathogen detection
Introduction
Bacteriophages are abundant in the environment and in living systems. They evolved to specifically infect target bacteria and then to multiply within the host. In the production of fermented products that rely upon the action of desirable bacteria to generate products such as biological based therapeutics and foodstuffs such as yoghurt, bacteriophages may be highly undesirable.1 On the other hand, their ability to target particular host bacteria, to multiply (100–1000-fold), and then to lyse the host upon the completion of the lytic cycle has been explored in antimicrobial and bio-sensing applications.2,3
Traditionally, the following technologies have been used for quantifying phages: a plate culture assay for the quantification of plaque forming units (PFU),4 a polymerase chain reaction (PCR) assay,5,6 or an enzyme-linked immunosorbent assay (ELISA)4 with a plate reader. However, these methods are time-consuming, complex, and, in some cases, may lack adequate sensitivity.
The detection of phages typically involves labeling the phages with a fluorescent dye followed by optical interrogation. Usually, the limit of detection of these methods is restricted by the sensitivity of the optical system. However, it is possible to make use of simple devices fabricated on a chip to significantly enhance the intensity of the signal, thereby lowering detection limits to a level that makes on-chip direct detection of phages feasible. The first hurdle to realizing the potential of so-called photonic crystals for phage detection involves the capture and organization of the phages into a periodic array which will be excited by incident light. The organization of phages on substrates has been studied with respect to other emerging applications such as energy storage7,8 and advanced bio-inorganic functional materials.9−11 As an example, Chung et al.12 made use of self-templating and interfacial forces to assemble highly ordered arrays of M13 phages on a surface with attendant photonic properties.
We propose that electrophoresis may be used to capture phages into the features of a photonic crystal. The use of electrophoresis to capture bacteriophages is attractive because many phages have a net negative charge13 and furthermore, the phage DNA can be modified to give rise to a desired zeta potential by modifying the capsid composition. For example, Yoo et al.14 assembled disordered M13 bacteriophages on polyelectrolyte films based on the electrostatic charge of the phage. However, little is known in terms of ordering phages electrophoretically, particularly into a very precise photonic structure. Here we demonstrate a facile electrophoretic technique that can be used to position phages with nanoscale precision on a surface to make use of nanophotonic effects to produce a new generation of assay for phages.
A photonic crystal (PC)15 is created when the dielectric constant of a material is modulated on the nanoscale. This allows selective coupling of light with the PC with increased electric field strength, enabling enhanced excitation and emission from fluorophores trapped within the PC. The DNA in the phages was stained using a DNA intercalating dye. Electrophoretic trapping into the wells exposed this reporting dye to a locally enhanced electric field that was produced by the nanophotonic interaction of light at the excitation wavelength of the dye with the nanostructure.
The bacteriophage T7 was chosen for several reasons: for its rapid lytic life cycle, significant amplification (approximately 200-fold) upon lysis of Escherichia coli bacteria, and its thorough characterization as a result of its use in evolutionary genetics.16 Thus, it is a good model system to demonstrate both the organization of phages in nanowells and the quantification of phages based on fluorescence measurements using a sensitive PC platform technology.
Experimental Section
Phage detection proceeded via the following steps: (1) the phages were labeled with a DNA intercalating dye, (2) labeled phages were trapped electrophoretically in the nanophotonic wells using a DC electric field, and (3) fluorescent emission from the labeled phages in the wells was enhanced through the nanophotonic interaction of excitation light with the nanostructure and efficient coupling to the detector. This nanophotonic interaction generates a locally enhanced electric field that amplifies the fluorescence emission from the DNA dye molecules bound to the trapped phages.
Preparation of T7 Bacteriophage
T7 phages were purchased from ATCC (#BAA-1025-B2) and propagated according to recommend methods by ATCC. Prior to use in experiments, a small amount of previously frozen phage was added to a log phase culture of E. coli BL21 (ATCC #BAA-1025) grown in Luria Broth3 and allowed to incubate in a shaking incubator at 37 °C and 250 rpm for 4 h to allow several phage lifecycles to complete. After 4 h, the culture was centrifuged for 10 min at 5800 rpm to remove bacterial debris. The supernatant containing the phage was decanted and passed through a 0.45 μm filter. A 20% v/v PEG buffer (40% w/v polyethylene glycol (PEG) MW 8000 and 2.5 M NaCl) to phage filtrate was added. The solution was chilled on ice overnight at 4 °C prior to centrifugation at 11,000g for 15 minutes. The supernatant was removed, and the pellet was suspended in sterile water. Phage activity was measured prior to use.
Phage activity was quantified using the standard soft agar overlay plating assay and reported in plaque forming units (PFU mL–1). Briefly, top agar (0.7% agar) was melted, and 3 mL aliquots were kept at 45 °C in a water bath until used. Phage samples were serially diluted into sterile water, and 100 μL of each sample was combined with 250 μL of fresh log phase bacterial culture with an optical density of 1.5 measured at 600 nm. The samples were incubated for 10 min and then combined with an aliquot of molten agar. The molten agar mixture was poured onto a prewarmed LB agar plate and allowed to solidify. Once the plates solidified, they were inverted and incubated at room temperature (22 °C) overnight. After incubation, the plaques were then counted.
The phages were fluorescently labeled with either SYBR Green or SYTOX Orange (Invitrogen, USA). To each 1 mL of quantified phage sample, 2.5 μL of dye was added. The sample was then wrapped to prevent excess light exposure and shaken gently on a platform for 15 min at room temperature. The solution was then passed through a Zeba desalt column (Fisher Scientific) equilibrated with sterile water to remove excess dye. To confirm phage labeling, the sample was tested for absorbance peaks at 497 and 532 nm for SYBR Green and SYTOX Orange, respectively, against an unlabeled phage sample as the blank. After the labeling was confirmed, the phage sample was used further experiments.
Electrophoretic Trapping Bacteriophages into an Array
To establish the condition for facile trapping of phages into an ordered array, chips with 200 nm wide wells spaced 2 μm apart were fabricated (chip 1). The larger separation between the wells permitted the visualization of labeled phages with a fluorescence microscope. T7 bacteriophages were labeled using a DNA intercalating SYBR Green dye (intercalating fluorescent dye: excitation, 497 nm; emission, 520 nm) and were excited at a wavelength of 488 nm. This dye was chosen for the visualization studies in order to be compatible with the standard excitation/emission filter sets in the fluorescent microscope that was used.
Voltage, time, and concentration were varied. Ten microliters of labeled phages at a given concentration (PFU mL–1) was dropped onto a chip that contained nine discrete PC structures. These structures were fabricated in a 3 × 3 format, with each structure measuring 50 × 50 μm. The separation between each PC structure was 250 μm, rendering each array independent of the others. The chip was maintained at a positive potential for trapping of phages. After trapping, the solution was removed by imposing a Couette flow17 that was established by moving the top plate over stationary bottom plate (chip) of the electrophoretic entrapment system (EPES) (Figure 1, top). The positive potential on the chip was maintained during this procedure to avoid the removal of phages from the PC structure. An additional rinsing with 10 μL of deionized (DI) water was performed by the same method. Each individual array on the chip was imaged under a fluorescent microscope. Fluorescent images were analyzed (ImageJ software, http://rsb.info.nih.gov/ij) to measure the amount of T7 phages on the array under different conditions of trapping.
Figure 1.

Schematic of T7 bacteriophage trapping into nanowells using an electrophoretic particle entrapment system. The inherent net negative charge on the phage capsid induces electrophoretic transport of the phages to the bottom of the nanowells of the array along the electric field created between top and bottom indium tin oxide layer.
Anodisc-Captured Using Fluorescent Microscopy
An aliquot (50 μL) of the released secondary T7 phages was captured on a 20 nm Anodisc filter (Whatman) using a gentle vacuum suction. The Anodisc filter was then placed on a glass slide and the DNA of the captured phages was labeled with SYBR Green I dye for 5 min. Then a coverslip was placed over the Anodisc and the sample was visualized on an Olympus IX-7 inverted fluorescence microscope with a 10× oil immersion objective (Olympus PLANApo, NA = 1.4). For the control, only SYBR Green was placed on the Anodisc.
Plate Plaque Assay
Titers of released phages were performed using standard plate plaque assay. Briefly, released phages were serially diluted with sterile water and added to a tube containing 3 mL molten soft agar (3% w/v) with 300 μL of an overnight E. coli BL21 culture. The solution was poured over LB agar plate and incubated overnight at room temperature. The visible plaques were counted the following day.
Fabrication of Photonic Crystal Nanostructured Array
The indium tin oxide5 coated glass wafer was selected for its electrical and optical properties. The high refractive index of ITO (1.8) contributed to the creation of wave-guided modes in the nanoarrays. In addition, its optical transparency aided optic-based detection. The LOL-2000 was spin-coated on the wafer at 6500 rpm for 45 s followed by baking at 180 °C for 300 s. 2% 950 PMMA A2 was spin-coated on the LOL-ITO-glass wafer at 500 rpm for 5 s followed by 3000 rpm for 45 s. The coated wafer was then baked at 180 °C for 80 s. This resulted in a coating thickness of 240 nm on the wafer. The coated wafer was patterned by e-beam lithography. The diameter of the wells was 200 nm, with an inter-well spacing of 2 μm (chip 1) or 350 nm (chip 2). The size of the array was 50 × 50 μm (for assays) or 25 × 25 μm (imaging only). The dimensions and spacing of the features on the array were designed for the specific wavelength of fluorophore emission using the numerical approach that was presented by Han et al.17
Fluorescence Microplate Assay
An aliquot of a DNA binding dye, SYTOX Orange (1:1000 final dilution, Invitrogen) was added to the stock solution (109 PFU mL–1) and incubated for 15 min at room temperature in the dark to allow the dye to bind to the phage DNA. Stained T7 phages were serially diluted with PBS and added to a black wall, clear bottom 96-well plate (Costar 3603, Corning). The fluorescence intensity of the T7 phage was measured using a microplate reader (Molecular Devices SpectraMax M5) at excitation and emission wavelengths of 544 and 572 nm, respectively. Complementary to this approach, we also labeled the phages in each well that had different concentrations of T7 to determine the limit of detection (Supporting information, Figure S5).
Results and Discussion
The T7 bacteriophage has unequal charges on the head (negative) and tail (positive).18,19 The measured zeta potential (−21.1 ± 1.4 mV) of the phages in deionized water confirmed that the charge on the head is in excess, resulting in a net negative charge. Figure 1 shows the schematic of electrophoretic trapping of the T7 phage onto the surface of the positive-charged indium tin oxide5 glass substrate on which a polymeric PC array was fabricated. In the case where a T7 phage is intended for use as a bio-receptor, the head down-trapping facilitates display of the tail-spike protein that is mainly involved in recognizing the target of interest, i.e., pathogen20 or RNA-binding protein21 (the polarity of the trapping electric field oriented the phage capture correctly for this application).
To establish the condition for facile trapping of phages into an ordered array, chips with 200 nm wide wells spaced 2 μm apart were fabricated (chip 1). The larger separation between the wells permitted the visualization of labeled phages with a fluorescence microscope. T7 bacteriophages were labeled using a DNA intercalating SYBR Green dye (intercalating fluorescent dye: excitation, 497 nm; emission, 520 nm) and were excited at a wavelength of 488 nm. This dye was chosen for the visualization studies in order to be compatible with the standard excitation/emission filter sets in the fluorescent microscope that was used.
For a trapping time of 60 min and a concentration of 1011 PFU mL–1, three different voltages (1.5, 2.5, 3 V) were applied to the EPES. At 1.5 V, there was no discernible fluorescent signal from the PC structure. At 2.5 V, pixelated green fluorescent signal from individual wells was identified with some nonspecific binding of the phage to the PMMA between the nanowells. At an applied voltage of 3 V, there was significant agglomeration of the phages on the array with no specific confinement or ordering of phages (Figure 2a). The total fluorescent intensity at each voltage from an array showed an exponential increase with increasing voltage (Figure 2b). To reduce this agglomeration, a shorter time scale of 30 s was tested with no significant improvement towards specifically trapping phages into the nanowells (Figure 2c). This shorter time was calculated based on the electrophoretic mobility of the T7 phage19 applied to our experimental electrophoretic setup that consisted of two ITO slides separated by 490 μm Hence, the concentration of the T7 phage solution was reduced by 100-fold and the trapping of phages was performed at 3 V for 5 min (Figure 2d). This yielded a significant reduction in the agglomeration, but no ordering of phages was observed. From these experiments, it was deduced that the number of the T7 phages migrating from the solution to the array was sensitive to the applied voltage.
Figure 2.

Trapping of T7 phages intercalated with SYBR Green fluorescent dye (excitation, 497 nm; emission, 520 nm) with different voltages, trapping times, and concentrations. (a) 1.5, 2.5, and 3 V for 60 min with 1011 PFU mL–1 T7 phage concentration, (b) Measured intensity from the arrays trapped with T7 phages as a function of voltage: 1.5, 2.5, and 3 V for 60 min or trapping time, (c) 30 s and 5 min for 3 V and 1011 PFU mL–1, (d) 30 s and 5 min for 3 V and 109 PFU mL–1, and (e) 10, 20, 40 and 60 min for 2.133 V and 1011 PFUmL–1. (f) Image analysis of fluorescence intensity corresponding to 30 s and 5 min at 3 V (■), 10, 20, 40, and 60 min trapping of T7 phages at 2.133 V (●). The concentration of the T7 phage was 1011 PFU mL–1.
Neither variation of the trapping time nor the concentration produced satisfactory trapping (Figure 2c,d), leading us to consider voltage as the dominant factor. Because applied voltage of 2.5 V showed trapping of phages within the wells, voltages around 2.5 V were tested for optimization. A spatially ordered nanoarray of phages was obtained from a concentration of 1011 PFU mL–1 of phage solution trapped with 2.133 V. With this voltage, the minimum time necessary for adequate capture of the phages into wells was determined. After 20 min, T7 phages were trapped into all the wells of the array (Figure 2e). With increased time, no significant improvement in the number of phages trapped was observed (Figure 2f), but there was a significant improvement in the arraying of the phages. The fluorescence intensity indicated the saturation of the wells without further agglomeration (Figure 2e). The results showed that the voltage is the critical factor that governs the successful capture of phages into wells. A higher voltage may be useful for trapping of small amounts of the T7 phages within a very short time. The optimized trapping time of 1 h and voltage of 2.133 V were used for the subsequent experiments.
Pixelated images of green fluorescence from the phages in wells in Figure 3a confirmed the electrophoretic trapping of phages into nanowells without obvious attachment of the phages to the PMMA coating between the wells. By using image analysis, fluorescent spots were identified in 476 out of 625 wells per array (Supporting Information, Figure S1) giving 76% occupancy of the array. The low magnification fluorescent image shown in Figure 3b confirms that the trapping of the phages was confined to the PC structure region and that nonspecific binding of the phages onto the surface of the PMMA between the PC structures was absent. Scanning electron microscopy (SEM) further confirmed that T7 phages were confined only to the nanowells (Figure 3a, right); SEM images were taken from two different chips. Figure 3a, right-top shows one of the wells following trapping of phages. The right-bottom image was taken from a similar array without any trapping. Although it is not possible to distinctly resolve the structure of the phage in the well using SEM, comparison to the control image in which no phages were present clearly indicates the difference between the two cases. In addition, the fluorescent intensity of the individual wells, measured by image analysis, showed well-distributed occupation of the phages (Supporting Information, Figure S1).
Figure 3.

PC nanostructure arrayed with T7 bacteriophages intercalated with SYBR Green fluorescent dye (excitation: 497 nm, emission: 520 nm). (a) A fluorescent image of 200 nm wells/2 μm periodicity of the array (size of the array: 50 × 50 μm). On the right side, scanning electron microscope images of the T7 bacteriophage trapped into a well (top), In comparison, no bacteriophage trapped well (bottom). (b) A fluorescent image of the arrays with 200 nm wells/350 nm periodicity (size of the array: 25 × 25 μm). The distance between the arrays is 250 μm.
For reliable quantification of low concentrations of phages on a highly sensitive PC array, it is important to assess the variability in the staining of individual T7 bacteriophages with dye molecules. Therefore, the variation in the staining of T7 DNA was assessed prior to performing the quantification by measuring the fluorescence from phages attached to the surface of an anodisk. T7 phages stained with SYBR Green dye were trapped on an anodisc, resulting in well-dispersed phages. Figure 4a shows the fluorescent microscope image of phages captured on the anodisc. The fluorescence intensity from individual phages was analyzed by ImageJ. The fluorescent distribution plotted in Figure 4b shows that the value of fluorescence, corresponding to an individual phage was ∼300 ± 30 arbitrary units (see also the Supporting Information, Figure S2). The more obvious spots in the image were associated with the anodisc itself, as confirmed by the correlation between the fluorescent (Figure 4c) and white light image (Figure 4d). The fluorescence variation associated with phages that could be attributed to the variation in the amount of staining of the individual phage was less than 10%. This uncertainty will factor into the quantification when only a few phages are captured onto an array.
Figure 4.

Image analysis of the staining of T7 bacteriophage with DNA intercalating dye on an Anodisc. (a) 10× fluorescent image of T7 bacteriophage labeled with SYBR Green on the anodisc membrane after amplification of the phage from 1.0 × 102 PFU mL–1 to 1.4 × 103 PFU mL–1 with 102 CFU bacteria; time for amplification: 30 min. (b) The distribution of the mean fluorescence intensity on the images by image analysis; count means the number of the spots which have specific value of the mean fluorescent intensity on the image. Measured intensities were 303.0 ± 32 (total counts: 950), 265 ± 20 (906), and 297 ± 295 (1281) for Images #1, #2, and #3, respectively. (c) Fluorescent microscope image of only SYBR Green intercalating dye on the anodisc for negative control. (d) White light image corresponding to panel c.
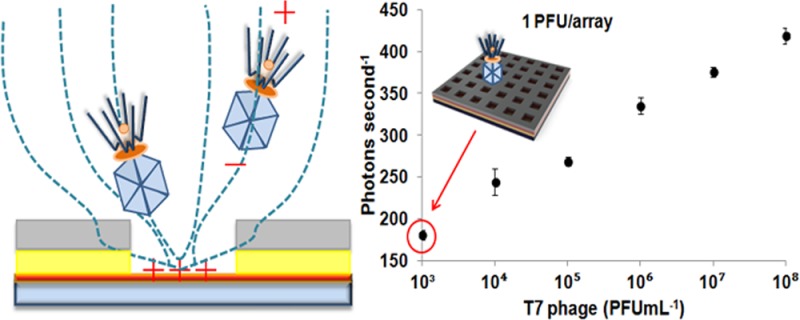
For the quantification of phages on the PC, another chip (chip 2) was designed to optimize the fluorescence enhancement for the emission wavelength of SYTOX Orange.17 The size of the wells was 200 nm, which afforded some degree of freedom for the bacteriophage electrophoresis (diameter of phage head, 60 nm; tail length, 23 nm). Phages that were labeled with SYTOX Orange dye were trapped on chip 2 for fluorescent quantification. Six different concentrations of T7 phages suspended in DI water were measured: 103, 104, 105, 106, 107, and 108 PFU mL–1. The volume of each phage solution and other trapping and rinsing methods were identical to that used for the imaging work. After the T7 phages were trapped, the fluorescent signal from an individual array was detected with a single photon counting detection system17 with excitation from a 532 nm laser and collection of 570 nm emitted fluorescent signal from the T7 SYTOX Orange labeled phages on the array. The photons emitted by SYTOX Orange labeled phages were counted with a high-speed digital oscilloscope. The signal from an individual PC structure (25 × 25 μm) was obtained by averaging the photon count per second over a period of 20 s.
Figure 5 shows the log–linear standard curve (R2: 0.99) for different concentrations of T7-SYTOX Orange in DI water. Each point on the curve was obtained by averaging the signal from an array with four replicates. Background noise for the experiment was established by measuring the laser light that was reflected back from the array in the absence of the phages. The noise was 161 ± 6 photons s–1.
Figure 5.

Quantification of T7 bacteriophages intercalated with SYTOX Orange (excitation: 547 nm, emission: 570 nm) using a photonic crystal nanostructured array. Six different concentrations of T7 SYTOX Orange labeled phages were used: 103, 104, 105, 106, 107, and 108 PFU mL–1 on the PC array (●). For performing control experiment without photonic crystal, the chip with 300 nm periodicity was used, while all other experimental conditions were maintained. A solution of T7-SYTOX Orange at a concentration of 108 PFU mL–1 was serially diluted 10-fold (▲). Dashed line: background signal plus three standard deviation. Error bars: standard deviation over four replicates. Inset: a fluorescence image of the array (200 nm wells with 350 periodicity; 25 × 25 μm) trapped with T7 bacteriophage-SYTOX Orange. Used concentration was 1011 PFU mL–1.
The measured limit of detection (LOD) of 103 PFU mL–1 corresponded to 1 PFU/array, given that the 10 μL volume of the sample was spread across all nine arrays. The LOD was defined as the concentration of phages that corresponded to a fluorescence signal that was three standard deviations higher than the background noise. In addition, a control experiment was performed to understand the change in the sensitivity of phage detection in to the absence of the photonic enhancement. Changing the periodicity of the nanostructure to 300 nm significantly alters the photonic behavior of the chip, leading to reduced enhancement. The experimental conditions for trapping of phages into this nanostructure were the same as previously mentioned. A solution of T7-SYTOX Orange at a concentration of 108 PFU mL–1 was serially diluted 10-fold. The LOD of the control experiment was 107 PFU mL–1, about the same as obtained using a fluorescence plate reader for detection. The result showed that the photonic enhancement yielded a 104 improvement in the sensitivity of phage detection (Figure 5). The CV of measured signals at all tested concentrations was less than 3%, showing uniformity of the trapping into an array. The LOD obtained from the photonic crystal nanostructured chip corresponded to the detection of 102 CFU mL–1 of E. coli in 30 min based on the phage infection assay described in the Supporting Information. To highlight the sensitivity of the PC structure platform, a fluorescence plate reader was also used to determine the LOD of fluorescently labeled phages in a 96-well format (Figure 6). The LOD using a fluorescence plate reader was 109 PFU mL–1. Thus, the PC structure was 106-fold more sensitive than the fluorescence plate reader.
Figure 6.
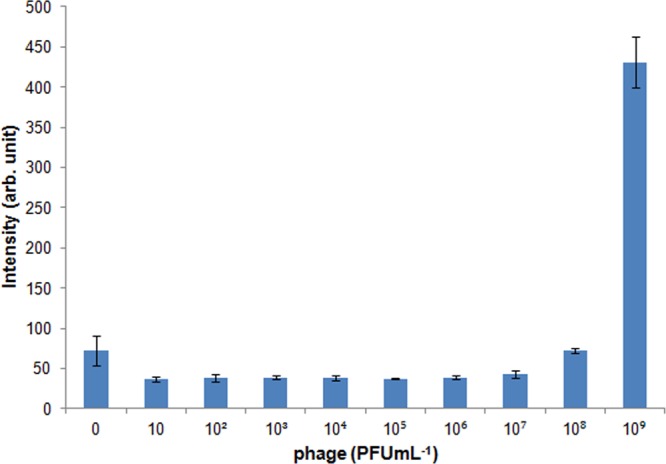
Fluorescent microplate assay using a microplate reader (excitation: 544 nm, emission: 572 nm). Ten different concentrations of T7-SYTOX Orange phages were used: 0, 10, 102, 103, 104, 105, 106, 107, 108, and 109 PFU mL–1. Error bars: standard deviation over three replicates.
The enhancement of the fluorescent signal from the PC structure can significantly improve the sensitivity of phage detection. Lytic phages are typically used as bio-sensing elements for pathogen detection. At the end of the lytic phage lifecycle, the host bacterium is lysed, releasing the secondary phages that were generated within the bacterium. In a typical bio-sensor, the secondary phages indicate the existence of the target pathogen. These secondary phages can be easily labeled and detected using DNA intercalating dyes as demonstrated in this work (see the Supporting Information, Figure S3). The electrophoretic trapping of phages shown here can be used as a rapid bio-sensor for pathogen detection with sufficient sensitivity to detect a single phage per array.
The increased sensitivity of the nanostructured chip, both in terms of detection limits and time required for the assay, are useful for real-world applications. In an industrial setting, two methods for detection of phages are followed. (1) The Heap-Lawrence test,22 which is based on acidification rate of the dairy products such as milk and gives a reliable measure of phage infection. This assay is typically an end stage assay after significant phage amplification (>106 PFU/mL) in a fermentation culture has been achieved. (2) A plaque-forming assay, which allows quantitative analysis of strain-specific phage count. Both methods can take from 1 to 2 days for the detection of phage infection. The limit of detection using the plaque assay is typically around 102 PFU/mL, but it requires culturing of phages in a microbiological laboratory for an extended period of time.
Other methods are available in a laboratory setting. The sensitivity of phage detection using classical one-step PCR ranges between of 104–107 PFU/mL.23,24 The PCR method requires multiple steps of isolation of phage DNA from complex materials such as milk and microbial broth. In addition, a PCR method detects only a narrow class of phages and is both expensive and time consuming (several hours). The limit of detection using a flow cytometry assay using nucleic acid based staining of phage DNA is about 105 to 107 PFU/mL.24 This approach requires a flow cytometer for measurement, which is not feasible in most industrial setups. Impedance spectroscopy requires capture of phages by antibodies or microbial strains on surfaces and its typical detection sensitivity is >104 PFU/mL. In addition, the impedance spectroscopy approach is limited by the lack of diversity of antibodies and the efficiency of absorbing microbes on a sensing platform.25
The direct trapping of phages also eliminates additional steps such as the use of labeled antibodies to detect the phages; this step can result in false positive signals when applied in a sandwich immunoassay. In addition, an intercalating dye offers a simple and effective method to stain the T7 phage without the need for complex and time-consuming conjugation processes and, with a significant bias towards labeling double stranded DNA, not free ssDNA. It is important to note that the LOD of the system was constrained only by the volume of the sample. A larger array with a microfluidic system could significantly improve both the LOD in terms of PFU mL–1 and the dynamic range.
Conclusions
We have demonstrated for the first time that the electrophoretic trapping of phages onto a photonic crystal offers a simple technology for the controlled nanoscale arraying of T7 phages with application as a rapid and sensitive bio-sensor. The direct trapping and quantification of phages has the potential to make a major impact in the bio-pharmaceutical and food industry by significantly shortening the analysis time to quantify either harmful phages themselves or to quantify pathogenic bacteria.
Acknowledgments
We thank the Northern California Nanotechnology Center at the University of California for the facility used in the fabrication of PC-nanowells/array and Professor T. Young for permission to use his zeta potential analyzer. The project was supported by the National Institute of Environmental Health Sciences (NIEHS) Superfund Basic Research Program, award number P42ES004699. The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institute of Environmental Health Sciences or the National Institutes of Health. We acknowledge the support of the W. M. Keck Foundation for a research grant in science and engineering. This work was also supported by a grant from NSF-CAPPS.
Supporting Information Available
Image analysis of the fluorescent intensity of individual wells on the array obtained from ImageJ analysis, phages captured on Anodisc; images #2 and #3 used for image analysis in Figure 4 of the main text, fluorescent microplate assay using a microplate reader. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ali Y.; Kot W.; Atamer Z.; Hinrichs J.; Vogensen F. K.; Heller K. J.; Neve H. Classification of Lytic Bacteriophages Attacking Dairy Leuconostoc Starter Strains. Appl. Environ. Microbiol. 2013, 79123628–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García P.; Martínez B.; Obeso J. M.; Rodriquez A. Bacteriophages and Their Application in Food Safety. Lett. Appl. Microbiol. 2008, 47, 479–485. [DOI] [PubMed] [Google Scholar]
- Minikh O.; Tolba M.; Brovko L. Y.; Griffiths M. W. Bacteriophage-Based Biosorbents Coupled with Bioluminescent ATP Assay for Rapid Concentration and Detection of Escherichia Coli. J. Microbiol. Methods 2010, 82, 177–183. [DOI] [PubMed] [Google Scholar]
- Beaber J. W.; Tam E. M.; Lao L. S.; Rondon I. J. A New Helper Phage for Improved Monovalent Display of Fab Molecules. J. Immunol. Methods 2012, 376, 46–54. [DOI] [PubMed] [Google Scholar]
- Morohashi K.; Arai T.; Saito S.; Watanabe M.; Sakaguchi K.; Sugawara F. A High-Throughput Phage Display Screening Method Using a Combination of Real-Time PCR and Affinity Chromatography. Comb. Chem. High Throughput Screening 2006, 9, 55–61. [DOI] [PubMed] [Google Scholar]
- Imamovic L.; Ballesté E.; Jofre J.; Muniesa M. Quantification of Shiga Toxin-Converting Bacteriophages in Wastewater and in Fecal Samples by Real-Time Quantitative PCR. Appl. Environ. Microbiol. 2010, 76, 5693–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y.; Kim S. N.; Jones S. E.; Wissler L. L.; Naik R. R.; McAlpine M. C. Chemical Functionalization of Graphene Enabled by Phage Displayed Peptides. Nano Lett. 2010, 10, 4559–4565. [DOI] [PubMed] [Google Scholar]
- Neltner B.; Peddie B.; Xu A.; Doenlen W.; Durand K.; Yun D. S.; Speakman S.; Peterson A.; Belcher A. Production of Hydrogen Using Nanocrystalline Protein-Templated Catalysts on M13 Phage. ACS Nano 2010, 4, 3227–3235. [DOI] [PubMed] [Google Scholar]
- Lee S. W.; Mao C. B.; Flynn C. E.; Belcher A. M. Ordering of Quantum Dots Using Genetically Engineered Viruses. Science 2002, 2965569892–895. [DOI] [PubMed] [Google Scholar]
- Mao C. B.; Flynn C. E.; Hayhurst A.; Sweeney R.; Qi J. F.; Georgiou G.; Iverson B.; Belcher A. M. Viral Assembly of Oriented Quantum Dot Nanowires. Proc. Natl. Acad. Sci. U. S. A. 2003, 100126946–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C. B.; Wang F. K.; Cao B. R. Controlling Nanostructures of Mesoporous Silica Fibers by Supramolecular Assembly of Genetically Modifiable Bacteriophages. Angew. Chem., Int. Ed. 2012, 51266411–6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W. J.; Oh J. W.; Kwak K.; Lee B. Y.; Meyer J.; Wang E.; Hexemer A.; Lee S. W. Biomimetic Self-Templating Supramolecular Structures. Nature 2011, 478, 364–368. [DOI] [PubMed] [Google Scholar]
- Anany H.; Chen W.; Pelton R.; Griffiths M. W. Biocontrol of Listeria Monocytogenes and Escherichia Coli O157:H7 in Meat by Using Phages Immobilized on Modified Cellulose Membranes. Appl. Environ. Microbiol. 2011, 77186379–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S. Y.; Oh J.-W.; Lee S.-W. Phage-Chips for Novel Optically Readable Tissue Engineering Assays. Langmuir 2012, 28, 2166–2172. [DOI] [PubMed] [Google Scholar]
- Joannopoulos J. D.; Johnson S. G.; Winn J. N.; Meade R. D.. Photonic Crystals: Molding the Flow of Light, 2nd ed.; Princeton University Press: Princeton, NJ, 1996. [Google Scholar]
- Bull J. J.; Molineux I. J. Predicting Evolution from Genomics: Experimental Evolution of Bacteriophage T7. Heredity 2008, 100, 453–463. [DOI] [PubMed] [Google Scholar]
- Han J.-H.; Sudheendra L.; Kim H.-J.; Gee S.; Hammock B. D.; Kennedy I. M. Ultrasensitive on-Chip Immunoassays with a Nanoparticle-Assembled Photonic Crystal. ACS Nano 2012, 6108570–8582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwer F.; Barott K. Viral Information. Biol. Philos. 2013, 28, 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serwer P.; Pichler M. E. Electrophoresis of Bacteriophage T7 and T7 Capsids in Agarose Gels. J. Virol. 1978, 28, 917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees C. E. D.; Loessner M. J.. Bacteriophages: Biology and Applications; CRC Press: Boca Raton, FL, 2004. [Google Scholar]
- Danner S.; Belasco J. G. T7 Phage Display: A Novel Genetic Selection System for Cloning Rna-Binding Proteins from cDNA Libraries. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 12954–12959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heap H. A. L.; Lawrence R. C. The Selection of Starter Strains for Cheesemaking. N. Z. J. Dairy Sci. Technol. 1976, 2, 16–20. [Google Scholar]
- Zago M.; Rossetti L.; Reinheimer J.; Carminati D.; Giraffa G. Detection and Identification of Lactobacillus Helveticus Bacteriophages by PCR. J. Dairy Res. 2008, 752196–201.18474137 [Google Scholar]
- Marco M. B.; Moineau S.; Quiberoni A. Bacteriophages and Dairy Fermentations. Bacteriophage 2012, 23149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Aljaro C.; Munoz-Berbel X.; Munoz F. J. On-Chip Impedimetric Detection of Bacteriophages in Dairy Samples. Biosens. Bioelectron. 2009, 2461712–1716. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.