

Abstract
Cationic Antimicrobial Peptides (CAMPs) represent a first line of defense against bacterial colonization. When fighting Gram-negative bacteria, CAMPs initially interact electrostatically with the negatively charged phosphate groups in lipid A and are thought to kill bacteria by disrupting their membrane integrity. However, many human pathogens, including Salmonella and Pseudomonas, have evolved lipid A modification mechanisms that result in resistance to CAMPs and related antibiotics such as Colistin. The addition of 4-amino-4-deoxy-l-Arabinose (Ara4N) to a phosphate group in lipid A is one such modification, frequently found in Pseudomonas isolated from cystic fibrosis patients. The pathway for biosynthesis of Ara4N-lipid A requires conversion of UDP-Glucuronic acid into UDP-Ara4N and subsequent transfer of the amino-sugar to lipid A. ArnB is a pyridoxal-phosphate (PLP) dependent transaminase that catalyzes a crucial step in the pathway: synthesis of UDP-Ara4N from UDP-4-keto-pentose. Here we present the 2.3 Å resolution crystal structure of an active site mutant of ArnB (K188A) in complex with the reaction intermediate aldimine formed by UDP-Ara4N and PLP. The sugar–nucleotide binding site is in a cleft between the subunits of the ArnB dimer with the uracil buried at the interface and the UDP ribose and phosphate groups exposed to the solvent. The Ara4N moiety is found in the 4C1 conformation and its positioning, stabilized by interactions with both the protein and cofactor, is compatible with catalysis. The structure suggests strategies for the development of specific inhibitors that may prove useful in the treatment of resistant bacteria such as Pseudomonas found in cystic fibrosis patients.
The envelope of Gram-negative bacteria is characterized by an outer membrane that consists of phospholipids in the inner leaflet and lipopolysaccharide (LPS) in the outer leaflet.1 Lipid A is the conserved lipid anchor portion of LPS and is recognized by several branches of the host innate immune system. However, Gram-negative bacteria, including the human pathogens Escherichia coli, Yersinia pestis, Salmonella typhimurium, and Pseudomonas aeruginosa, have evolved mechanisms to modify the structure of lipid A and resist the antimicrobial response mounted by the host. Such modifications include both acylation and deacylation of the lipid core, as well as addition of phosphoethanolamine and 4-amino-4-deoxy-arabinose (Ara4N) to phosphate groups among other mechanisms.2,3
Cationic Antimicrobial Peptides (CAMPs) constitute a branch of the innate immune system. They are small, positively charged amphipathic peptides that destroy microorganisms primarily by membrane disruption.4−6 CAMPs initially bind to the Gram-negative bacterial cell surface through electrostatic interactions with the negatively charged phosphate groups of lipid A and the lipopolysaccharide core. There are several proposed models for the mechanism of membrane disruption by CAMPs, and the specific mechanism may depend on the features of the CAMP.7 In general, they are thought to coalesce and form a pore that permeabilizes the membrane. In Gram-negative bacteria, breaching of the outer membrane allows additional peptides to gain access to the periplasmic space where they can disrupt the inner membrane causing bacterial death.8−10 In addition to membrane disruption mechanisms, CAMPs may have intracellular targets that contribute to their antimicrobial activity. Peptidoglycan cell wall synthesis inhibition has been well documented,11,12 whereas inhibition of nucleic acid and protein synthesis has also been reported.7
The modification of a phosphate group in lipid A with 4-amino-4-deoxy-Arnabinose (Ara4N) leads to reduced density of negatively charged groups on the bacterial outer membrane, resulting in reduced CAMP binding. This mechanism mediates resistance to both CAMPs of the host immune system as well as related antibiotics such as Colistin and other polymyxins.13−18 The pathway specific proteins responsible for the biosynthesis of Ara4N and its attachment to lipid A are encoded in pmrHFIJKLM operon (pmr stands for polymyxin resistance, now renamed arn for AraN synthesis) under the control of the PmrA/PmrB and PhoP/PhoQ two-component systems.3,19 The biosynthesis of UDP-Ara4N from UDP-Glucuronic acid (UDP-GlcA) is carried out by two enzymes (Figure 1). ArnA is a bifunctional enzyme whose C-terminal domain catalyzes the oxidative decarboxylation of UDP-GlcA to yield UDP-4-keto-pentose (UDP-Ara4O). ArnB uses glutamate as an amino group donor and catalyzes a transamination reaction to yield UDP-4-amino-4-deoxy-arabinose (UDP-Ara4N). This is a reversible reaction with an equilibrium constant of approximately 0.1 in the forward direction of the pathway.20 In subsequent steps, Ara4N is transiently formylated, transferred to the lipid carrier undecaprenyl-phosphate, and flipped to the periplasmic side of the membrane where it is transferred to lipid A.21−23
Figure 1.

Initial steps in the biosynthesis of lipid A-Ara4N. The pathway starts with UDP-glucose that is oxidized to UDP-glucuronic acid (UDP-GlcA) by UDP-glucose dehydrogenase (Ugd). UDP-GlcA is then oxidatively decarboxylated to UDP-4-ketopentose (UDP-Ara4O) by the C-terminal domain of ArnA. UDP-Ara4O is transaminated by ArnB to form UDP-Ara4N that, after transient formylation, sugar transfer to undecaprenyl-phosphate, and flipping to the periplasmic side of the inner membrane is finally transferred to lipid A.
All the proteins encoded by the arn operon are essential for lipid A modification with Ara4N and resistance to polymyxins.3,19 Therefore, they represent attractive targets for development of inhibitors that would abolish resistance to CAMPs of the host immune system and last-resort antibiotics such as Colistin. Such inhibitors would be particularly useful in the treatment of chronic Pseudomonas infections in cystic fibrosis (CF) patients. Analysis of prevalence has showed that 33% of the Pseudomonas isolates from CF patients, and as much as 44% of the isolates from infants with CF display Lipid A modified with Ara4N.13 It is striking that the unique environment encountered by Pseudomonas in the CF airway appears to induce the pathway for Ara4N Lipid A modification, hence making the bacteria resistant to the host innate immune response (CAMPs). Using cultured epithelial cells, it has been shown that normal, but not CF airway, epithelia can efficiently kill Pseudomonas aeruginosa.24 The bactericidal activity was traced to CAMPs synthesized by the epithelium, underscoring the importance of these compounds preventing bacterial colonization.24 In addition, aerosolized polymyxins are clinically used for the treatment of Pseudomonas infections in CF patients.25−29 Therefore, inhibitors of the Ara4N Lipid A modification pathway would not only enhance the ability of the host immune system to clear the infection but could also be administered together with clinically useful CAMPs such as polymyxins to increase their effectiveness.
In this study, we focus on ArnB, a pyridoxal-phosphate (PLP) dependent transaminase. Previous crystal structures of ArnB revealed the overall type I fold of the enzyme as well as the residues involved in binding the cofactor, α-keto-glutarate, and the inhibitor cycloserine.30 However, the basis for specificity toward the substrate sugar–nucleotide remained elusive. Here, we enzymatically synthesized UDP-Ara4N and expressed in E. coli a mutant of Salmonella typhimurium ArnB to prepare, in vitro, a ternary complex of the enzyme with UDP-Ara4N forming the external aldimine with PLP in the active site. We present the crystal structure of the complex refined to 2.3 Å resolution, define the sugar nucleotide binding determinants, and discuss the implications for the design of specific inhibitors.
Materials and Methods
Cloning, Expression, and Purification of Wild Type and Mutant ArnB
The arnB gene of S. typhimurium was PCR amplified from genomic DNA (forward primer AGC GGC GTC ATA TGG CTG AAG GAA AAA TGA TG, reverse primer CGT CTA GAG CTC TTA TTG TCC TGC TAT CTG ATG) incorporating NdeI and SacI restriction sites at the N- and C-terminus of the coding region, respectively. The gene was cloned into pMS122 (a derivative of pET28(+) modified to include an N-terminal His-tag followed by a tobacco etch virus (TEV) protease cleavage site) digested with the same restriction enzymes. The correct cloning was confirmed by sequencing and the plasmid designated pMS206. E. coli BL21(DE3) cells were transformed with pMS206 and a single colony used to inoculate 60 mL of LB broth containing 50 μg/mL kanamycin and grown at 37 °C overnight with shaking at 225 rpm. The starter culture was diluted 1/100 into fresh LB medium containing 50 μg/mL of kanamycin and incubated at 37 °C with shaking at 225 rpm until OD600 reached about 0.6. The culture was cooled down in ice for 30 min and overexpression of the protein was induced by adding isopropyl-1-thio-β-d-galactopyranoside (IPTG) to a final concentration of 0.5 mM, and incubation overnight at room temperature with shaking at 225 rpm. All the purification steps were carried out at 4 °C. The cells were harvested by centrifugation at 6000g for 20 min in a Beckman JLA 8.1000 rotor, resuspended in 60 mL of lysis buffer containing 25 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10% glycerol, 5 mM β-mercaptoethanol, and protease inhibitor cocktail, and broken by sonication (20 cycles of 10 s sonication and 30 s rest). The crude cell lysate was centrifuged at 40 000g for 30 min in a Beckman JA20 rotor to remove cell debris and unbroken cells, and the supernatant loaded on a 10 mL Ni-NTA column pre-equilibrated with equilibration buffer containing 25 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10% glycerol, and 5 mM β-mercaptoethanol. The column was washed with 50 mL of washing buffer (equilibration buffer supplemented with 25 mM imidazole) and the bound protein eluted with 50 mL of elution buffer (equilibration buffer containing 300 mM imidazole). The eluted protein solution was concentrated in a Vivaspin 20 centrifugal concentrator (MWCO 10K) and the His-tag cleaved by incubation with His-tagged-TEV protease while dialyzing against equilibration buffer containing 10 mM DTT. The sample was again loaded on a Ni-NTA column equilibrated as before to remove the cleaved 6-His tag as well as the His-tagged-TEV protease. The flow-through was concentrated to 4 mL and loaded on Superdex 200 gel filtration column pre-equilibrated with buffer containing 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% glycerol, and 5 mM β-mercaptoethanol. Fractions containing highly purified ArnB, as judged by SDS-PAGE, were pooled and concentrated with Vivaspin 20 centrifugal concentrator (MWCO 10K) to 26 mg/mL. The K188A mutant of ArnB was prepared using the QuickChange kit (Agilent) and primers (forward primer TCC TTC CAC GCC ATT GCC AAC ATT ACC TGC GCT, reverse primer AGC GCA GGT AAT GTT GGC AAT GGC GTG GAA GGA). The mutation was confirmed by sequencing and the protein expressed and purified as described above for the wild-type protein.
Synthesis and Purification of UDP-Ara-4N
The ArnA dehydrogenase domain (ArnA_DH) required for the production of UDP-Ara-4O was prepared as previously described.31 UDP-Ara-4N was enzymatically synthesized in two steps. First, a reaction containing 25 mM Tris-HCl (pH 8.0), 5 mM β-mercaptoethanol, 0.2 mg/mL BSA, 10% glycerol, 100 mM KCl, 100 mM NAD+, 50 mM UDP-glucuronic acid, and 200 nM ArnA_DH was incubated at room temperature overnight. To test the reaction progress, 1 μL of the reaction was diluted 50-fold in 25 mM Tris-HCl (pH 8.0), filtered through an Amicon ultrafilter (MWCO 5K) to remove the protein and 10 μL injected into an ion-pair HPLC column (Hypersil Gold 250H × 3D mm, Thermo Scientific) pre-equilibrated 5 mM TPA (pH 7.0). The column was developed with a gradient of acetonitrile in 5 mM TPA pH 7.0 (0–10% in 20 min, flow rate 1 mL/min). Elution of the UDP-Ara-4O, UDP-glucuronic acid, and NAD+ was monitored at 254 nm and elution of NADH was monitored at 340 nm (SI Figure S1). After confirming complete conversion of UDP-glucuronic acid to UDP-Ara-4O by ion-pair HPLC, the reaction was filtered to remove protein as described above. In a second step, the reaction mixture was supplemented with l-glutamic acid to a final concentration of 500 mM and incubated with 200 nM ArnB at room temperature overnight. Reaction progress was monitored by ion-pair HPLC as described above, confirming complete conversion of UDP-Ara-4O to UDP-Ara-4N (SI Figure S1). The reaction mixture was ultrafiltered to remove ArnB and loaded onto a semipreparative C18 HPLC column (Hypersil Gold 250H × 21.2D mm, Thermo Scientific) for purification. The column was pre-equilibrated with 5 mM TPA (pH 7.0) and the sample eluted with a two step gradient of acetonitrile in 5 mM TPA pH 7.0 at a 5 mL/min flow rate (0–1.6% acetonitrile in 80 min, and 1.6–10% acetonitrile in 100 min). Elution of the UDP-ArnB-4N was monitored at 254 nm and the eluted product collected and lyophilized. The identity of the product was confirmed by mass spectrometry and NMR (SI Figure S2).
Electrospray Ionization Mass Spectrometry (ESI-MS) and Nuclear Magnetic Resonance (NMR)
ESI-MS was conducted with a Synapt G2 High Definition Mass Spectrometer (Waters Company) at the Central Analytical Laboratory of the University of Colorado at Boulder. The ESI-MS settings were as follows: capillary voltage 2.2 kV (ESI-), sampling cone voltage 30 V, extraction cone 4.0 V, source temperature 80 °C, desolvation temperature 150 °C, and desolvation gas (N2) 600 L/h. The flow rate for the infusing sample solution (in 100% MeOH) was 5 μL/min. For NMR data collection, 1 mg of lyophilized UDP-Ara4N was dissolved in 0.5 mL D2O and the proton NMR spectrum was collected at 25 °C on a Varian Inova 500 MHz spectrometer as described in ref (20).
Protein Crystallization and X-ray Data Collection
Purified ArnB (K188A) was incubated with 5 mM PLP on ice for 4 h. Then, UDP-Ara-4N was added to a final concentration of 5 mM and the mixture incubated for 16 h on ice prior to crystallization. The initial search for suitable crystallization conditions for ArnB/PLP/UDP-Ara-4N ternary complex was conducted with numerous commercial and homemade crystal screen solutions in sitting drop 96-well plates set up with the Phoenix drop setter (Art Robbins Instruments) and incubated at 20 °C. Conditions that yielded crystals in the presence of UDP-Ara4N and PLP, but not with PLP alone, were refined using the hanging drop vapor diffusion method at 20 °C, with a precipitant volume of 500 μL and drops initially containing 1.5 μL of protein sample (12 mg/mL) and 1.5 μL of reservoir solution. A precipitant formulation consisting of 0.1 M sodium citrate (pH 4.5), 19% PEG 20 000 yielded well-formed crystals, which were harvested and frozen directly in mother liquor by immersion in liquid nitrogen. After initial evaluation using our home X-ray source, the crystals were analyzed using synchrotron radiation at the Beamline 8.2.1 of the Advanced Light Source, Lawrence Berkeley National Laboratory. Initial diffraction and data collection strategy were evaluated using iMosfilm.32 Data were collected at a 1 Å wavelength using 0.5° oscillations. The diffraction data were indexed, integrated, and scaled with DENZO and SCALEPACK incorporated into the HKL2000 package.33 Data processing statistics are summarized in Table 1.
Table 1. Data Collection and Refinement Statisticsa.
| data collection statistics | K188A |
|---|---|
| Wavelength (Å) | 1.0 |
| Resolution (Å) | 37.18–2.3 (2.174–2.099) |
| Space Group | P43212 |
| Cell Dimensions (Å) | a = b = 90.952, c = 129.123, α = β = γ = 90° |
| Unique Reflections | 29611 (3067) |
| Completeness (%) | 91.53 (96.78) |
| I/σ | 17.75 (2.81) |
| Wilson B-factor | 35.67 |
| refinement statistics | |
|---|---|
| Rwork (%) | 22.64 (34.44) |
| Rfree (%) | 26.08 (34.92) |
| Number of non-hydrogen atoms | 2948 |
| Number of macromolecule atoms | 2768 |
| Number of ligands atoms | 49 |
| Number of water molecules | 131 |
| Number of amino acid residues | 363 |
| Mean B-value (Å2) | 40.20 |
| Mean B-value protein (Å2) | 40.30 |
| Mean B-value ligands (Å2) | 39.20 |
| Mean B-value solvent molecules (Å2) | 38.60 |
| RMS deviation from ideal values: | |
| Bond lengths (Å) | 0.003 |
| Bond angles (deg.) | 0.692 |
| Residues in Ramachandran plot: | |
| Most favored regions (%) | 98 |
| Generously allowed regions (%) | 2.0 |
| Outliers (%) | 0.0 |
Statistics for the highest-resolution shell are shown in parentheses.
Structure Determination and Refinement
All crystallographic calculations were carried out with the PHENIX software suit.34 The structure was determined by molecular replacement using the published ArnB/PLP/α-ketoglutarate ternary complex structure (Protein Data Bank code 1MDX)30 as a search model. The PLP and α-ketoglutarate ligands and water molecules were removed from the search model and residue K188 was mutated to alanine before calculations. Five percent of the unique reflections were reserved for the calculation of free R for validation.35 Unambiguous solutions to the rotation and translation functions were found. An electron density map calculated with phases from the model after a single round of positional and B-factor refinement revealed clear density for the UDP-Ara4N-PLP aldimine in the active site. A set of coordinates and restraints for the ligand were obtained using a SMILES string and the program ELBOW.36 The ligand was then built into the electron density using the program COOT37 and the model subjected to alternating cycles of refinement and rebuilding. After the Rfree had dropped to 0.29, water molecules were added to the model and additional rounds of refinement and manual rebuilding were carried out until no further improvement of the Rfree was observed. Final refinement statistics are summarized in Table 1. Atomic coordinates and structure factors have been deposited with the RCSB Protein Data Bank under accession code 4OCA.
Results and Discussion
Synthesis and Purification of UDP-Ara-4N
The reaction catalyzed by ArnB is a reversible transamination. In the forward direction of the pathway, glutamic acid donates the amino group to UDP-L-Ara4O yielding UDP-L-Ara4N and α-ketoglutarate.20 Whereas the ArnB binding site for α-ketoglutarate has been defined crystallographically,30 the UDP-sugar binding site has remained unknown and co-crystallization efforts have been hampered by the lack of a commercial source for these sugar–nucleotides. To address this problem, UDP-Ara4N was targeted for enzymatic synthesis and purification (UDP-Ara4O has been described as unstable20). First, UDP-Ara4O was synthesized from UDP-glucuronic acid (UDP-GlcA) by NAD+ dependent oxidative decarboxylation catalyzed by the dehydrogenase domain of ArnA31,38,39 (Figure 1). The reaction progress was examined by ion-pair reversed-phase HPLC monitored at 256 and 340 nm (to detect NADH) (SI Figure S1). In a second step, ArnA decarboxylase domain was removed by ultrafiltration and the reaction supplemented with ArnB and a large excess of glutamate to drive the synthesis of UDP-Ara4N. Analysis of the reaction products by HPLC showed disappearance of the UDP-Ara4O substrate with concomitant appearance of an early eluting peak (SI Figure S1). Mass spectrometry analysis of this early eluting material indicated a molecular mass of 534.1 Da consistent with UDP-Ara4N (theoretical molecular weight 535.29) (SI Figure S2A). NMR analysis of this material showed a spectrum essentially identical to that of UDP-Ara4N reported by Raetz and co-workers20 confirming the identity of the purified nucleotide (SI Figure S2B).
Structure of ArnB in Complex with PLP and UDP-Ara4N
Initial attempts to determine the structure of an ArnB/PLP/UDP-Ara-4N ternary complex were conducted by incubating wild-type ArnB with purified UDP-Ara4N in crystallization screens. Analysis of the resulting crystals indicated the presence of the cofactor in the active site, but no additional electron density could be ascribed to UDP-Ara4N (data not shown). At the available resolution (3.5 Å), it was not clear if the cofactor was PLP or PMP. However, further analysis of the structure showed that the side chain of the catalytic residue K188 was in a position previously reported for the ArnB/PMP structure.30 Furthermore, the plane of the cofactor was tilted close to the PMP position in the ArnB/PMP structure. This was interpreted as an indication that, (i) during crystallization incubation, UDP-Ara4N reacted with PLP to produce PMP and UDP-Ara4O, and (ii) under the conditions of the experiment, this PMP-bound form of the enzyme did not bind the sugar nucleotides tightly enough to result in a stable ternary complex.
In a second approach to crystallize an ArnB/PLP/UDP-Ara-4N ternary complex, the catalytic lysine 188 was mutated to alanine. In the proposed catalytic mechanism (SI Figure S3) K188 acts as a general base and abstracts a proton from the 4′ carbon of the UDP-Ara4N-PLP aldimine to generate a quinonoid intermediate. Thus, it was reasoned that the K188A mutant might trap the UDP-Ara4N-PLP aldimine in the enzyme active site. To this end, a binary complex was prepared first, by incubating ArnB K188A with PLP to saturate the cofactor binding site. This step was necessary, because in ArnB K188A, the PLP is not stably linked to the enzyme by an internal aldimine with K188. The protein was then incubated with an excess of purified UDP-Ara-4N in crystallization screens. Conditions that yielded crystals in the presence of UDP-Ara-4N, but not in its absence, were refined. Large diamond-shaped crystals obtained in 0.1 M Sodium acetate (pH 4.5), 19% PEG20000 were cryoprotected and used to collect a data set to 2.3 Å resolution. The crystals belonged to space group P43212 with unit cell dimensions: a = b = 90.952 and c = 129.123 Å, which suggested one protomer per asymmetric unit. The structure was determined by molecular replacement using S. typhimurium ArnB (PDB ID 1MDX) stripped of its ligands as a search model. After an initial round of positional and B-factor refinement, clear electron density for the UDP-Ara-4N-PLP aldimine was observed confirming the achievement of a ternary complex. The final model (Rwork 0.23; Rfree 0.26) contains residues 9 to 384 except a short stretch of polypeptide spanning residues 219 to 232, which could not be unambiguously modeled presumably due to conformational flexibility. This region was also missing in the structures of ArnB with other ligands.30 The final model also contains the UDP-Ara-4N-PLP aldimine and 131 water molecules. Data collection and refinement statistics are summarized in Table 1.
The ArnBK188A/PLP/UDP-Ara4N ternary complex superimposes with wild-type ArnB with an RMS deviation of 0.3 Å for all Cα atoms, indicating that the K188A mutation does not alter the overall conformation. Furthermore, ligand binding does not appear to induce any significant conformational changes in the protein. ArnB is a dimer in solution as defined by its size exclusion chromatography elution profile (data not show). Although some type-I aminotransferases assemble into larger oligomers, members of this family are known to be catalytically active as dimers.40 The dimer twofold symmetry axis is incorporated into the crystal symmetry, which was used to generate a model of the ArnB dimer (Figure 2A). We and others have proposed that ArnB may oligomerize with other enzymes in the pathway to form a multiprotein complex in vivo where the substrates are channeled between active sites for protection of labile intermediates and efficient synthesis of product.21,41,42 However, no evidence for such an assembly has been reported, and we expect the ArnB dimer to be the functional unit in vivo.
Figure 2.

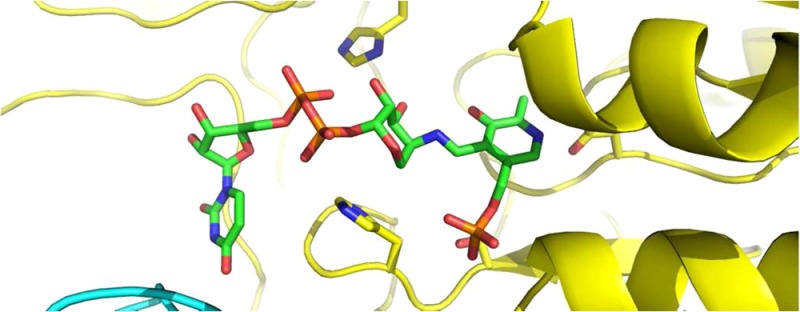
ArnB ligand binding Site. (A) Molecular surface representation of the dimeric structure of ArnB showing the UDP-Ara4N-PLP binding pocket. Subunits 1 and 2 are colored in yellow and cyan, respectively. UDP-Ara4N-PLP from subunit 1 is shown as a stick model. The insert is a close up view of the binding site showing the exposed UDP moiety. (B) Stereoview of a simulated annealing Fo-Fc omit map (contour level 3.0 σ) where the ligand UDP-Ara4N-PLP was omitted. The structure of the refined ligand is superimposed on the map for reference.
Substrate and Cofactor Binding Sites
The cofactor and substrate binding sites are at the interface between the two subunits. Pyridoxal-phosphate binds in a deep pocket within one subunit (Figure 2A), with D160 interacting with the pyridine ring nitrogen (Figure 3A) and increasing its electron-sink character as is typical in this family of aminotransferases.40,43 The PLP phosphate group interacts with T64, S183, the main chain amides from A63 and T64 (Figure 3A). In addition, a well-defined water molecule (wat1) mediates binding of the phosphate group of PLP to the side chains of K241 and H215 from the opposite subunit. Another water molecule (wat2) also mediates binding of the phosphate group of PLP to the side chain of E194 and the main chain amide and carbonyl oxygen of G195 as observed previously in the cofactor bound structures of ArnB.30 The UDP-Ara4N binding site is more solvent exposed than the PLP site (Figure 2A). The uracil ring is sandwiched between the side chains of P16, A186, and I187 from one subunit and the indole ring of W34 from the opposite subunit (Figure 3B). Binding is also stabilized by a hydrogen bond between the 4-oxygen of uracil and the main chain amide of I35 from the second subunit. The ribose adopts a 3′ endo conformation and makes no direct hydrogen bonds with the protein. The phosphate groups of UDP-Ara4N make only two hydrogen bonds with ArnB (Figure 3B). The side chain of His185 contacts the β-phosphate of the UDP moiety, whereas the α-phosphate forms a hydrogen bond with the side chain of H329 (Figure 3B). Remarkably, H329 forms a nonproline cis peptide bond with F330 stabilized by a hydrogen bond between the side chain of Y136 and the carbonyl oxygen of H329. Nonproline cis peptide bonds are observed with a frequency of only 0.03%44 and are typically found in active sites. Curiously, they appear to be enriched in carbohydrate binding or processing enzymes.44 The unusual cis peptide bond between H329 and F330 and its stabilization by Y136 was noted in the previous structures of ArnB without UDP-Ara4N, but its role was undefined.30 The function of H329 in binding the sugar nucleotide substrate is now established.
Figure 3.

Close up views of the active site of S. typhimurium ArnB. (A) Binding site of PLP moiety. (B) Binding site of UDP moiety. (C) Stereoview of the binding site of Ara4N moiety. Residues from subunit 1 and subunit 2 are colored in yellow and cyan, respectively. Possible hydrogen bonds lying within ∼3.2 Å are indicated by the blue dashed lines. Water molecules are shown in red sphere.
The Ara4N pyranose ring adopts a 4C1 chair conformation in the ArnB ternary complex structure (Figure 3C). Raetz and co-workers identified this as the most stable conformation for the UPD-Ara4N pyranose ring based on the characteristic H1–H2 and H2–H3 J coupling constants as well as the diagnostic chemical shift of the C1 carbon linked to an axial glycosidic oxygen.20 In this orientation the 4″ proton is ideally positioned for abstraction by the catalytic K188 (A188 in the mutant structure). This suggests that UDP-Ara4N is bound in a physiologically relevant, catalytically competent orientation in the structure. The pyranose ring is packed against the indole group of W89 and its binding stabilized by several hydrogen bond interactions (Figure 3C). The 5-hydroxyl group of PLP interacts with the 3″OH of Ara4N while the 2″OH interacts with the carbonyl oxygen of L328 through a water molecule (wat3). The water molecule (wat1) that links the PLP phosphate with H215 and K241 from the opposite subunit (Figure 3A) is also within hydrogen bonding distance of the pyranose oxygen and may contribute to the sugar binding, although the geometry is not ideal for a hydrogen bond.
Comparison to Other Aminotransferases and Implications for Inhibitor Design
ArnB is most closely related to bacterial PLP-dependent aminotransferases that act on sugar nucleotides and are involved in cell envelope synthesis or modification such as PseC and perosamine synthase. PseC catalyzes amino transfer to the 4″-keto group of UDP-2-acetamido-2,6-dideoxy-4-hexulose yielding UDP-4-amino-4,6-dideoxy-AltNAc, a crucial intermediate in the synthesis of pseudaminic acid, which glycosylates flagelins in Helicobacter and Campylobacter strains.35,45 Similarly, GDP-perosamine synthase aminates the 4-keto group of GDP-4-keto-6deoxymannose to yield GDP-perosamine, an unusual sugar found in the O-antigen of Vibrio and Caulobacter strains.46,47 Despite low sequence identity (on the order of 30%), ArnB shares with these enzymes both the overall fold and the sugar-nucleotide binding site (RMS deviations of 1.5 Å for 356 Cα superimposed on PseC (PDB ID 2FNU(48)); and 1.4 Å for 359 Cα superimposed on perosamine synthase (PDB ID 3DR4(49)) as calculated with Dali45). The ArnB active site is most similar to that of PseC, which also binds a UDP-sugar nucleotide. As shown in Figure 4, most active site residues are conserved despite the relatively low overall sequence identity, and the substrate is bound in the same orientation with the pyranose ring retaining the 4C1 chair conformation. In fact, it appears possible that both these enzymes may be able to catalyze transamination of both substrates. However, this structure-based hypothesis awaits experimental confirmation.
Figure 4.
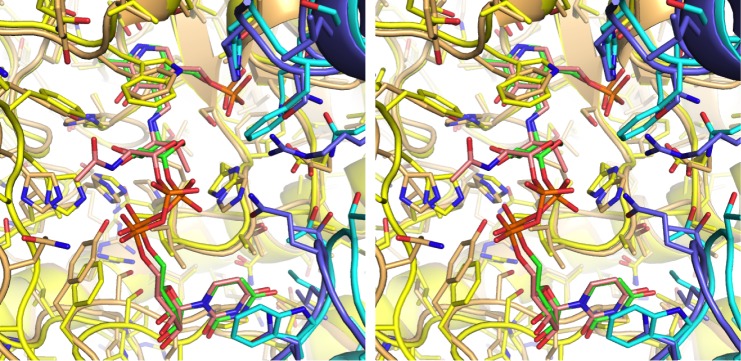
Comparison of ligand binding site in ArnB and PseC. Stereo representation of the active site of ArnB (subunits colored yellow and cyan) superimposed on PseC (subunits colored light orange and slate blue). The position and conformation of the ArnB and PseC ligands, UDP-Ara4N and UDP-4-amino-4,6-dideoxy-AltNAc, respectively, are very similar. Carbon atoms in the ArnB and PseC ligands are colored green and pink, respectively, whereas oxygen is red, nitrogen is blue, and phosphorus is orange.
The similarity of ArnB to human amino transferases is more limited. Sequence similarity is barely detectable, with the closest homologue, δ-aminolevulinate synthase, sharing only 20% identity and 29% similarity (aligned using DELTA-BLAST50). Despite poor sequence conservation, many PLP dependent human enzymes retain the type I aminotransferase fold. The closest structural homologues of ArnB in humans, as detected by Dali,45 include cystathionine-γ-lyase51 and kynurenine aminotransferase.52 However, these enzymes bind substrates totally different from those of ArnB and the geometry of their active sites is incompatible with binding UDP-Arabinose sugar nucleotides (Figure 5). Furthermore, whereas plants and lower eukaryotes such as leishmania synthesize UDP or GDP-Ara, to the best of our knowledge no arabinose-nucleotides have been described in mammals. This provides a rationale for the development of small molecule inhibitors that would target bacterial but not human PLP-dependent enzymes.
Figure 5.
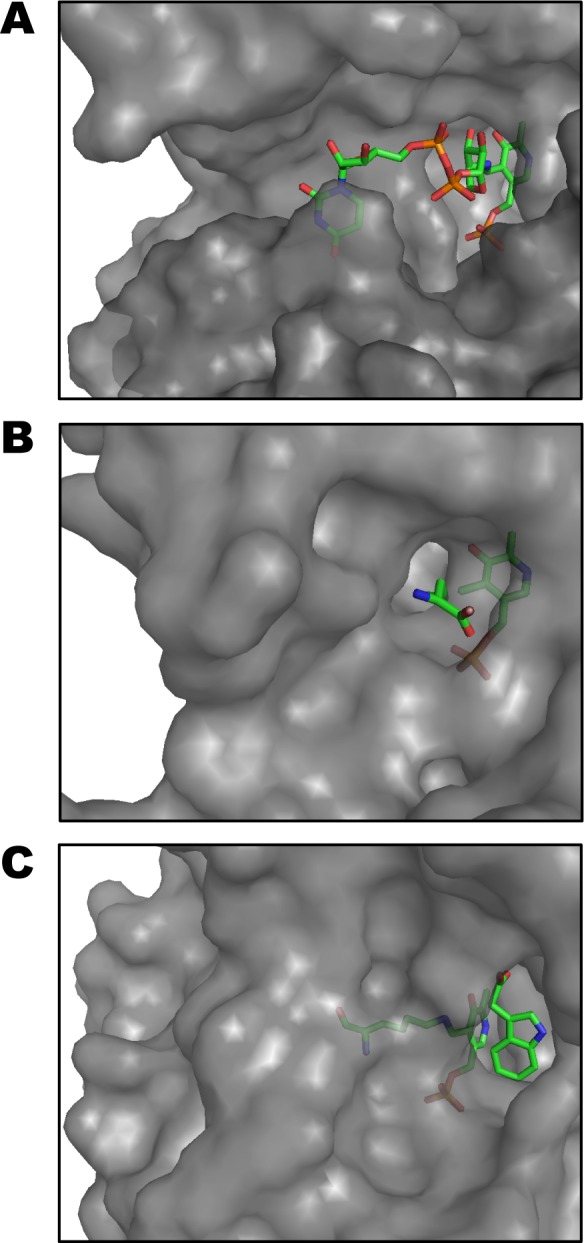
Comparison of ligand binding site in ArnB and its human structural homologues. Surface representations of the active sites of S. typhimurium ArnB (A); Human cystathion-γlyase (B); and Human kynurenine aminotransferase I (C). The proteins were superimposed using Dali and they are displayed in the same orientation with their respective ligands shown as stick figures. Whereas the PLP cofactor is bound in approximately similar conformations, the substrates are accommodated in completely different binding sites.
We propose that ArnB is an excellent target for development of selective inhibitors that would abolish lipid A modification and render bacteria sensitive to host CAMPs and antibiotics that act by similar mechanisms such as Colistin. Molecules designed to occupy the UDP-Ara4N binding site should provide specificity for bacterial targets. In addition, analogs designed to incorporate cycloserine or aminodyhydroquinolone functionalities in place of the arabinose moiety may result in irreversible inhibitors where the external aldimine formed with PLP tautomerizes to a stable enamine. This strategy has been used successfully to develop specific, irreversible inhibitors of kynurenine aminotransferase for the treatment of schizophrenia.53−55 It is also worth highlighting that ArnB makes only two hydrogen bonds with the phosphate groups of UDP-Arn4N. This suggests that it may be possible to develop analogs that replace the phosphates with noncharged functionalities capable of satisfying the hydrogen bonding requirements. This would improve the membrane permeability characteristics of the inhibitor as well as facilitate the synthesis of analogs.
Acknowledgments
We are indebted to Danielle Castagneri for assistance with cloning, and to Dr. David McKay for X-ray facility support. We also thank Sabrina Hunt and Dr. Arthur Pardi for assistance with NMR experiments identifying UDP-Ara4N.
Glossary
Abbreviations:
- CAMPs
cationic antimicrobial peptides
- PLP
pyridoxal-phosphate
- PMP
pyridoxamine-phosphate
- LPS
lipopolysaccharide
- CF
cystic fibrosis
- UDP-Ara4N
uridine 5′-diphospho-β-(4-amino-4-deoxy-l-arabinose)
- UDP-Ara4O
uridine 5′-diphospho-β-(l-threopentapyranosyl-4″-ulose)
- IPTG
iso-propyl-1-thio-β-d-galactopyranoside
Supporting Information Available
Chromatography traces tracking the synthesis of UDP-Ara4N (Figure S1); the mass spectrometry and NMR spectra identifying UDP-Ara4N (Figure S2); and a scheme of the proposed reaction mechanism of ArnB. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported in part by NIH grant AI060841 (Sousa). Structural biology research at the University of Colorado, Boulder is supported in part by the William M. Keck Foundation. Part of the work presented in this manuscript was carried out at The Advanced Light Source, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02−05CH11231.
Accession Codes
Coordinate and structure factors for ArnB/PLP/UDP-Ara4N have been deposited in the Protein Databank with ID 4OCA.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Raetz C. R. (1990) Biochemistry of endotoxins. Annu. Rev. Biochem. 59, 129–170. [DOI] [PubMed] [Google Scholar]
- Needham B. D.; Trent M. S. (2013) Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 11, 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn J. S.; Ryan S. S.; Van Velkinburgh J. C.; Ernst R. K.; Miller S. I. (2000) Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect. Immun. 68, 6139–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott M. G.; Hancock R. E. (2000) Cationic antimicrobial peptides and their multifunctional role in the immune system. Crit. Rev. Immunol. 20, 407–431. [PubMed] [Google Scholar]
- Zasloff M. (2002) Antimicrobial peptides of multicellular organisms. Nature 415, 389–395. [DOI] [PubMed] [Google Scholar]
- Zasloff M. (1992) Antibiotic peptides as mediators of innate immunity. Curr. Opin. Immunol. 4, 3–7. [DOI] [PubMed] [Google Scholar]
- Guilhelmelli F.; Vilela N.; Albuquerque P.; Derengowski L. D.; Silva-Pereira I.; Kyaw C. M. (2013) Antibiotic development challenges: the various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 4, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K. (1999) Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1462, 1–10. [DOI] [PubMed] [Google Scholar]
- Shai Y. (1999) Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1462, 55–70. [DOI] [PubMed] [Google Scholar]
- Yang L.; Weiss T. M.; Lehrer R. I.; Huang H. W. (2000) Crystallization of antimicrobial pores in membranes: magainin and protegrin. Biophys. J. 79, 2002–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount N. Y.; Yeaman M. R. (2013) Peptide antimicrobials: cell wall as a bacterial target. Ann. N.Y. Acad. Sci. 1277, 127–138. [DOI] [PubMed] [Google Scholar]
- Schneider T.; Kruse T.; Wimmer R.; Wiedemann I.; Sass V.; Pag U.; Jansen A.; Nielsen A. K.; Mygind P. H.; Raventos D. S.; Neve S.; Ravn B.; Bonvin A. M.; De Maria L.; Andersen A. S.; Gammelgaard L. K.; Sahl H. G.; Kristensen H. H. (2010) Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science 328, 1168–1172. [DOI] [PubMed] [Google Scholar]
- Ernst R. K.; Moskowitz S. M.; Emerson J. C.; Kraig G. M.; Adams K. N.; Harvey M. D.; Ramsey B.; Speert D. P.; Burns J. L.; Miller S. I. (2007) Unique lipid a modifications in Pseudomonas aeruginosa isolated from the airways of patients with cystic fibrosis. J. Infect. Dis. 196, 1088–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R. K.; Yi E. C.; Guo L.; Lim K. B.; Burns J. L.; Hackett M.; Miller S. I. (1999) Specific lipopolysaccharide found in cystic fibrosis airway Pseudomonas aeruginosa. Science 286, 1561–1565. [DOI] [PubMed] [Google Scholar]
- Haagensen J. A.; Klausen M.; Ernst R. K.; Miller S. I.; Folkesson A.; Tolker-Nielsen T.; Molin S. (2007) Differentiation and distribution of colistin- and sodium dodecyl sulfate-tolerant cells in Pseudomonas aeruginosa biofilms. J. Bacteriol. 189, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamp S. J.; Gjermansen M.; Johansen H. K.; Tolker-Nielsen T. (2008) Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol. Microbiol. 68, 223–240. [DOI] [PubMed] [Google Scholar]
- Roland K. L.; Martin L. E.; Esther C. R.; Spitznagel J. K. (1993) Spontaneous pmrA mutants of Salmonella typhimurium LT2 define a new two-component regulatory system with a possible role in virulence. J. Bacteriol. 175, 4154–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer W. M.; Casey S. G.; Spitznagel J. K. (1984) Lipid A and resistance of Salmonella typhimurium to antimicrobial granule proteins of human neutrophil granulocytes. Infect. Immun. 43, 834–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn J. S.; Lim K. B.; Krueger J.; Kim K.; Guo L.; Hackett M.; Miller S. I. (1998) PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol. Microbiol. 27, 1171–1182. [DOI] [PubMed] [Google Scholar]
- Breazeale S. D.; Ribeiro A. A.; Raetz C. R. H. (2003) Origin of lipid A species modified with 4-amino-4-deoxy-L-arabinose in polymyxin-resistant mutants of Escherichia coli. An aminotransferase (ArnB) that generates UDP-4-deoxy-L-arabinose. J. Biol. Chem. 278, 24731–24739. [DOI] [PubMed] [Google Scholar]
- Breazeale S. D.; Ribeiro A. A.; McClerren A. L.; Raetz C. R. (2005) A formyltransferase required for polymyxin resistance in Escherichia coli and the modification of lipid A with 4-Amino-4-deoxy-L-arabinose. Identification and function oF UDP-4-deoxy-4-formamido-L-arabinose. J. Biol. Chem. 280, 14154–14167. [DOI] [PubMed] [Google Scholar]
- Yan A.; Guan Z.; Raetz C. R. (2007) An undecaprenyl phosphate-aminoarabinose flippase required for polymyxin resistance in Escherichia coli. J. Biol. Chem. 282, 36077–36089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trent M. S.; Ribeiro A. A.; Lin S.; Cotter R. J.; Raetz C. R. H. (2001) An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J. Biol. Chem. 276, 43122–43131. [DOI] [PubMed] [Google Scholar]
- Smith J. J.; Travis S. M.; Greenberg E. P.; Welsh M. J. (1996) Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 85, 229–236. [DOI] [PubMed] [Google Scholar]
- Driscoll J. A.; Brody S. L.; Kollef M. H. (2007) The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67, 351–368. [DOI] [PubMed] [Google Scholar]
- Griese M.; Muller I.; Reinhardt D. (2002) Eradication of initial Pseudomonas aeruginosa colonization in patients with cystic fibrosis. Eur. J. Med. Res. 7, 79–80. [PubMed] [Google Scholar]
- Hodson M. E.; Gallagher C. G.; Govan J. R. (2002) A randomised clinical trial of nebulised tobramycin or colistin in cystic fibrosis. Eur. Respir. J. 20, 658–664. [DOI] [PubMed] [Google Scholar]
- Marchetti F.; Candusso M.; Faraguna D.; Assael B. M. (2002) Early Pseudomonas aeruginosa colonisation in cystic fibrosis patients. Lancet 359, 626–627. [DOI] [PubMed] [Google Scholar]
- Taccetti G.; Campana S.; Neri A. S.; Boni V.; Festini F. (2008) Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis. J. Chemother. 20, 166–169. [DOI] [PubMed] [Google Scholar]
- Noland B. W.; Newman J. M.; Hendle J.; Badger J.; Christopher J. A.; Tresser J.; Buchanan M. D.; Wright T. A.; Rutter M. E.; Sanderson W. E.; Muller-Dieckmann H. J.; Gajiwala K. S.; Buchanan S. G. (2002) Structural studies of Salmonella typhimurium ArnB (PmrH) aminotransferase: a 4-amino-4-deoxy-L-arabinose lipopolysaccharide-modifying enzyme. Structure (Cambridge, MA, U.S.) 10, 1569–1580. [DOI] [PubMed] [Google Scholar]
- Gatzeva-Topalova P. Z.; May A. P.; Sousa M. C. (2004) Crystal structure of Escherichia coli ArnA (PmrI) decarboxylase domain. A key enzyme for lipid A modification with 4-amino-4-deoxy-L-arabinose and polymyxin resistance. Biochemistry 43, 13370–13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye T. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirm M.; Soo E. C.; Aubry A. J.; Austin J.; Thibault P.; Logan S. M. (2003) Structural, genetic and functional characterization of the flagellin glycosylation process in Helicobacter pylori. Mol. Microbiol. 48, 1579–1592. [DOI] [PubMed] [Google Scholar]
- Moriarty N. W.; Grosse-Kunstleve R. W.; Adams P. D. (2009) electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Breazeale S. D.; Ribeiro A. A.; Raetz C. R. H. (2002) Oxidative decarboxylation of UDP-glucuronic acid in extracts of polymyxin-resistant Escherichia coli. Origin of lipid a species modified with 4-amino-4-deoxy-L-arabinose. J. Biol. Chem. 277, 2886–2896. [DOI] [PubMed] [Google Scholar]
- Gatzeva-Topalova P. Z.; May A. P.; Sousa M. C. (2005) Crystal structure and mechanism of the Escherichia coli ArnA (PmrI) transformylase domain. An enzyme for lipid A modification with 4-amino-4-deoxy-L-arabinose and polymyxin resistance. Biochemistry 44, 5328–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G.; Kack H.; Lindqvist Y. (2000) The manifold of vitamin B6 dependent enzymes. Structure 8, R1–6. [DOI] [PubMed] [Google Scholar]
- Gatzeva-Topalova P. Z.; May A. P.; Sousa M. C. (2005) Structure and mechanism of ArnA: conformational change implies ordered dehydrogenase mechanism in key enzyme for polymyxin resistance. Structure 13, 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G. J.; Breazeale S. D.; Raetz C. R.; Naismith J. H. (2005) Structure and function of both domains of ArnA, a dual function decarboxylase and a formyltransferase, involved in 4-amino-4-deoxy-L-arabinose biosynthesis. J. Biol. Chem. 280, 23000–23008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansonius J. N. (1998) Structure, evolution and action of vitamin B6-dependent enzymes. Curr. Opin. Struct. Biol. 8, 759–769. [DOI] [PubMed] [Google Scholar]
- Jabs A.; Weiss M. S.; Hilgenfeld R. (1999) Non-proline cis peptide bonds in proteins. J. Mol. Biol. 286, 291–304. [DOI] [PubMed] [Google Scholar]
- Szymanski C. M.; Logan S. M.; Linton D.; Wren B. W. (2003) Campylobacter--a tale of two protein glycosylation systems. Trends Microbiol. 11, 233–238. [DOI] [PubMed] [Google Scholar]
- Cook P. D.; Holden H. M. (2008) GDP-perosamine synthase: structural analysis and production of a novel trideoxysugar. Biochemistry 47, 2833–2840. [DOI] [PubMed] [Google Scholar]
- Redmond J. W. (1975) 4-Amino-4,6-dideoxy-D-mannose (D-perosamine): a component of the lipopolysaccharide of Vibrio cholerae 569B (Inaba). FEBS Lett. 50, 147–149. [DOI] [PubMed] [Google Scholar]
- Schoenhofen I. C.; Lunin V. V.; Julien J. P.; Li Y.; Ajamian E.; Matte A.; Cygler M.; Brisson J. R.; Aubry A.; Logan S. M.; Bhatia S.; Wakarchuk W. W.; Young N. M. (2006) Structural and functional characterization of PseC, an aminotransferase involved in the biosynthesis of pseudaminic acid, an essential flagellar modification in Helicobacter pylori. J. Biol. Chem. 281, 8907–8916. [DOI] [PubMed] [Google Scholar]
- Cook P. D.; Carney A. E.; Holden H. M. (2008) Accommodation of GDP-linked sugars in the active site of GDP-perosamine synthase. Biochemistry 47, 10685–10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boratyn G. M.; Schaffer A. A.; Agarwala R.; Altschul S. F.; Lipman D. J.; Madden T. L. (2012) Domain enhanced lookup time accelerated BLAST. Biol. Direct 7, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q.; Collins R.; Huang S.; Holmberg-Schiavone L.; Anand G. S.; Tan C. H.; van-den-Berg S.; Deng L. W.; Moore P. K.; Karlberg T.; Sivaraman J. (2009) Structural basis for the inhibition mechanism of human cystathionine gamma-lyase, an enzyme responsible for the production of H(2)S. J. Biol. Chem. 284, 3076–3085. [DOI] [PubMed] [Google Scholar]
- Han Q.; Robinson H.; Cai T.; Tagle D. A.; Li J. (2009) Structural insight into the inhibition of human kynurenine aminotransferase I/glutamine transaminase K. J. Med. Chem. 52, 2786–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dounay A. B.; Anderson M.; Bechle B. M.; Campbell B. M.; Claffey M. M.; Evdokimov A.; Evrard E.; Fonseca K. R.; Gan X. M.; Ghosh S.; Hayward M. M.; Horner W.; Kim J. Y.; McAllister L. A.; Pandit J.; Paradis V.; Parikh V. D.; Reese M. R.; Rong S.; Salafia M. A.; Schuyten K.; Strick C. A.; Tuttle J. B.; Valentine J.; Wang H.; Zawadzke L. E.; Verhoest P. R. (2012) Discovery of brain-penetrant, irreversible kynurenine aminotransferase II inhibitors for schizophrenia. ACS Med. Chem. Lett. 3, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dounay A. B.; Anderson M.; Bechle B. M.; Evrard E.; Gan X. M.; Kim J. Y.; McAllister L. A.; Pandit J.; Rong S. B.; Salafia M. A.; Tuttle J. B.; Zawadzke L. E.; Verhoest P. R. (2013) PF-04859989 as a template for structure-based drug design: Identification of new pyrazole series of irreversible KAT II inhibitors with improved lipophilic efficiency. Bioorg. Med. Chem. Lett. 23, 1961–1966. [DOI] [PubMed] [Google Scholar]
- Tuttle J. B.; Anderson M.; Bechle B. M.; Campbell B. M.; Chang C.; Dounay A. B.; Evrard E.; Fonseca K. R.; Gan X. M.; Ghosh S.; Horner W.; James L. C.; Kim J. Y.; McAllister L. A.; Pandit J.; Parikh V. D.; Rago B. J.; Salafia M. A.; Strick C. A.; Zawadzke L. E.; Verhoest P. R. (2013) Structure-based design of irreversible human KAT II inhibitors: discovery of new potency-enhancing interactions. ACS Med. Chem. Lett. 4, 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.