

Abstract

Protein and peptide aggregation is an important issue both in vivo and in vitro. Herein, we examine the aggregation behaviors of two well-studied β-hairpins, Trpzip1 and Trpzip2. Previous studies suggested that Trpzip2 remains monomeric up to a concentration of ∼15 mM whereas Trpzip1 readily aggregates at micromolar concentrations at acidic or neutral pH. This disparity is puzzling considering that these two peptides differ only in their turn sequences (i.e., GN vs NG). We hypothesize that these peptides can aggregate from their folded states via native edge-to-edge interactions and that the Lys8 residue in Trpzip2 is a more effective aggregation gatekeeper, because of a more favorable orientation. In support of this hypothesis, we find that increasing the pH to 13 or replacing Lys8 with a hydrophobic and photolabile Lys analogue, Lys(nvoc), leads to a significant increase in the aggregation propensity of Trpzip2, and that the aggregation of this Trpzip2 mutant can be reversed upon restoring the native Lys side chain via photocleavage of the nvoc moiety. In addition, we find that while both Trpzip1 and Trpzip2 form parallel β-sheet aggregates, the Lys(nvoc) Trpzip2 mutant forms antiparallel β-sheets and more stable fibrils. Taken together, these findings provide another example showing how sensitive peptide and protein aggregation is to minor sequence variation and that it is possible to use a photolabile non-natural amino acid, such as Lys(nvoc), to tune the rate of peptide aggregation and to control fibrillar structure.
Protein and peptide aggregation and amyloid formation are commonly associated with various pathological disorders1−3 and, thus, have been the subject of many studies. In addition, aggregation poses a major obstacle in de novo protein design and also in mechanistic studies of protein folding where relatively high protein or peptide concentrations are required. While it is easily recognized that many factors can come into play in determining the aggregation propensity of a given protein or peptide system, in practice the most commonly used strategy in protein design is to incorporate a certain number of charged residues to prevent or alleviate aggregation.4−10 For example, Marqusee and Baldwin have shown that the solubility of alanine-based α-helical peptides in aqueous solution can be significantly increased by dispersing either lysine or glutamate residues in the peptide sequence of interest.11,12 For β-sheet systems, however, the situation can be much more complicated, as the edge strands are often poised for further intermolecular strand–strand association,7,13−18 and as a result, only a small difference in peptide sequence could lead to a significant difference in aggregation propensity.19−22 One distinctive example, which is also the focus of this study, is that two designed, closely related β-hairpins, Trpzip1 and Trpzip2,23 show very different aggregation behaviors. Previous studies23−27 indicated that Trpzip2 remains completely monomeric in the concentration range of 5–12 mM at acidic pH, whereas Trpzip1 has previously shown measurable aggregation at concentrations of >500 μM.28,29 For this reason, Trpzip2 has been extensively used as a model to study the mechanism of β-hairpin folding,24,25,27,30−38 whereas the stronger aggregation propensity of Trpzip1 has made it a less attractive system. As shown (Table 1), these two β-hairpins differ only in the order of the two amino acids in the turn region (i.e., NG vs GN). Considering the fact that both peptides adopt a stable β-hairpin conformation in solution at room temperature and their sequences are almost identical, this difference is surprising.
Table 1. Sequences and Thermal Melting Temperatures (Tm) of the Peptides Studied Herein.
| peptide | sequencea | Tm (°C) |
|---|---|---|
| Trpzip1 | SWTWEGNKWTWK | 49.8 ± 0.3b |
| Trpzip2 | SWTWENGKWTWK | 71.9 ± 0.1b |
| Trpzip2-K | SWTWENG(K*)WTWK | 56.8 ± 0.5c |
| Trpzip2-W4A | SWTAENGKWTWK | 23.7 ± 2.1c |
| Trpzip2-KK | SWTWENG(K*)WTW(K‡) | – |
K* represents lysine-4,5-dimethoxy-2-nitrobenzyloxycarbonyl, and K‡ represents the acylated lysine (Scheme 1).
From ref (23).
Figure S1 of the Supporting Information.
As shown (Figure 1), a comparison of the averaged nuclear magnetic resonance (NMR) structures23 of Trpzip1 and Trpzip2 indicates that the major structural variation between these two β-hairpins is in the relative orientation of the Lys8 side chain. Specifically, in Trpzip1, the side chain of Lys8 is oriented orthogonal to the β-hairpin axis so that it points straight outward, and away from the Trp hydrophobic cluster, whereas in Trpzip2, the same side chain points in an upright parallel direction with respect to the β-hairpin axis. It is well-known that a solvent-exposed Lys side chain is relatively flexible and can fluctuate among several rotamer conformations. Therefore, to obtain a more quantitative assessment of the difference in the Lys8 orientations of these two peptides, we measured the dihedral angles of Lys8 in an ensemble of 20 NMR structures obtained from the Protein Data Bank (PDB)23 using built-in functions in Visual Molecular Dynamics (VMD).39 The results indicate that the greatest angle disparity arises from the difference in χ2, which describes the angle between the Cα–Cβ and Cγ–Cδ planes. For Trpzip1, the Lys8 side chain always adopts a trans configuration along χ2 (171 ± 11°). However, for Trpzip2, χ2 fluctuates between a trans and gauche(+) configuration. When the averaged structure of all frames of Trpzip2 was evaluated, χ2 of Lys8 is ∼70 ± 10°, indicative of a gauche(+) rotamer along the Cβ–Cγ bond.40,41 Thus, we hypothesize that the difference in the aggregation propensities of Trpzip1 and Trpzip2 can be explained by this variation in χ2. In other words, Lys8 in Trpzip2 is an effective aggregation gatekeeper,5−7 which prevents edge-to-edge β-hairpin association through unfavorable electrostatic interactions between neighboring Lys8 contacts. To this end, we further point out that our hypothesis is based on the assumption that the β-hairpin unit in the peptide aggregates possesses a native or nativelike turn structure, which, in conjunction with the confinement effect induced by peptide association, would place Lys8 in a specific configuration that could disfavor aggregation.
Figure 1.
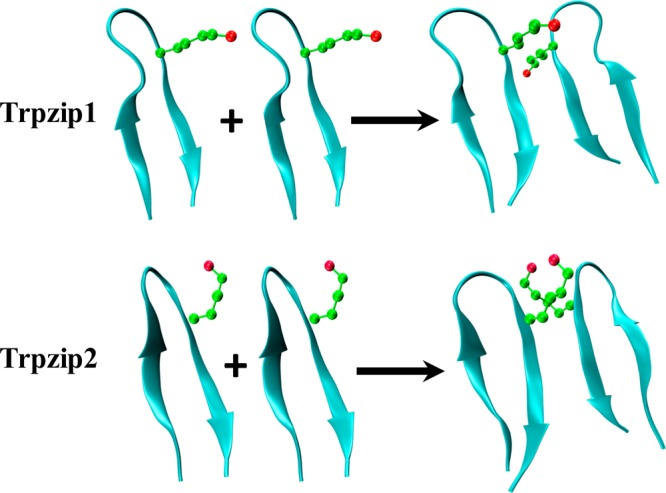
NMR structures of Trpzip1 (PDB entry 1LE0) and Trpzip2 (PDB entry 1LE1), as indicated, and the proposed dimerization scheme, showing the difference in the orientations of the Lys8 side chains.
To test this hypothesis, we examined the aggregation kinetics of both peptides under different concentration and pH conditions. In addition, for Trpzip2, we also used a chemical approach to eliminate the positive charge of Lys8 by replacing it with Lys(4,5-dimethoxy-2-nitrobenzyloxycarbonyl).42,43 The latter is a lysine analogue [hereafter termed Lys(nvoc)] with a photolabile hydrophobic moiety and has been used to control the disassembly of peptide aggregates and hydrogels via illumination.44,45 Should Lys8 indeed serve as an aggregation gatekeeper of Trpzip2, we expect that this Trpzip2 mutant (hereafter termed Trpzip2-K) will exhibit a significantly stronger aggregation propensity.
Materials and Methods
Materials and Sample Preparation
D2O (D, 99.96%) and deuterium chloride (D, 99.5%) were purchased from Cambridge Isotope Laboratories (Andover, MA). Fmoc-Lys(4,5-dimethoxy-2-nitrobenzyloxycarbonyl)-OH [Fmoc-Lys(nvoc)-OH] was purchased from Anaspec, Inc. (Fremont, CA), and used without further purification. Fmoc-protected amino acids were purchased from Advanced Chem Tech (Louisville, KY). All peptides were synthesized on a PS3 peptide synthesizer (Protein Technologies, Woburn, MA) and purified by reverse-phase high-performance liquid chromatography (HPLC). The identity of each peptide was further verified by matrix-assisted laser desorption ionization (MALDI) mass spectrometry. Residual trifluoroacetic acid (TFA) from peptide synthesis was removed by multiple rounds of lyophilization against a 0.1 M DCl solution. All peptide samples were prepared by directly dissolving the lyophilized peptide solid in D2O, and the pH of the peptide samples was approximately 3, unless explicitly indicated separately. The peptide concentration was determined optically using the absorbance at 280 nm, with an ε280 of 22760 cm–1 M–1.
Acylation of Lys12
Lyophilized peptide was first dissolved in 100 mM phosphate buffer (pH 7.4) to a final peptide concentration of 250 μM. This peptide solution was then mixed with a 30 mM N-acryloxysuccinimide (Sigma-Aldrich) solution prepared in 100 mM phosphate buffer (pH 7.4) containing 10% dimethyl sulfoxide with a final N-acryloxysuccinimide:peptide concentration ratio of 8:1. The reaction mixture was stirred for 9 h at 4 °C. The resulting peptide product was purified by HPLC and verified by MALDI mass spectrometry.
Circular Dichroism (CD) and Fourier Transform Infrared (FTIR) Measurements
CD data were collected on an Aviv 62A DS spectrometer (Aviv Associates) using a 1 mm sample cuvette. FTIR spectra were collected on a Magna-IR 860 spectrometer (Nicolet) at 2 cm–1 resolution using a temperature-regulated, 52 μm CaF2 sample cell.46
Photocleavage Experiments
Irradiation of samples was conducted by placing the sample in the optical path of a FluoroLog fluorometer (HORIBA Jobin Yvon), at room temperature. The excitation wavelength was set to 355 nm with a slit width of 0.75 cm. The excitation intensity is approximately 8.8 mW cm–1, estimated on the basis of the measured power of the excitation light and the beam diameter. The nvoc moiety has an extinction coefficient44 (ε350) of 5485 M–1 cm–1 and a photochemical yield (Φ365) of 0.023.47−49
Atomic Force Microscopy (AFM) Measurements
AFM experiments were performed in air at room temperature, using a multimode atomic force microscope (model 5500, Agilent, Santa Clara, CA), equipped with a 90 μm closed loop piezoscanner. Five microliters of a sample solution was applied to a freshly cleaved mica surface and allowed to sit for ∼10 s, rinsed with 100 μL of Millipore water, and subsequently dried with a slow stream of N2 gas. Tapping-mode imaging was conducted with a silicon cantilever, where the tip radius was <10 μm and the force constant was 40 N/m (Ted Pella, Redding, CA). Height and deflection images were obtained with a scan rate of 1.6 Hz and a tapping frequency of 285 kHz. Multiple images were obtained for each sample at different locations on the mica substrate to confirm the presence of fibrils.
Results and Discussion
Aggregation Kinetics of Trpzip1 and Trpzip2
The aggregation kinetics of Trpzip1 and Trpzip2 were examined using FTIR and CD spectroscopy. In particular, the amide I′ band (amide I band in D2O) of the peptide was used as an IR probe of the aggregation process, as this band has proven to be sensitive to intermolecular β-sheet association. For example, the development of a narrow amide I′ band at approximately 1615 cm–1 is indicative of peptide aggregation to form parallel β-sheets,50 whereas the appearance of a pair of narrow bands, i.e., a strong one at ∼1618 cm–1 and a weak one at ∼1685 cm–1, signifies the formation of antiparallel β-sheets.51,52
As shown (Figure 2), the amide I′ bands of Trpzip1 obtained at different concentrations indicate that its aggregation rate is concentration-dependent, as expected. For example, at ∼2 mM and 25 °C, the peptide sample becomes almost completely aggregated after 24 h, as judged by the full development of the 1616 cm–1 band, whereas at ∼350 μM, aggregate formation becomes detectable after just 1 day. In comparison, even at a much higher concentration (i.e., ∼10 mM), Trpzip2 does not show any signs of aggregation under the same conditions (Figure 2, inset). As shown (Figure 3), further time-dependent measurements indicate that the aggregation kinetics of Trpzip1 at a concentration of 1.2 mM, determined by the growth of the 1616 cm–1 band, follow a biexponential function; the magnitude of the signal increases quickly within the first 10 h and then slowly reaches a plateau over ∼150 h. Repeating this measurement at a higher peptide concentration, which results in a faster overall aggregation rate, reproduces this biphasic kinetic pattern (Figure S3, Supporting Information). Similar biphasic growth kinetics have been observed in other peptide aggregation studies,53−55 which were attributed to a separation in the time scales of the fibril nucleation and elongation processes.
Figure 2.
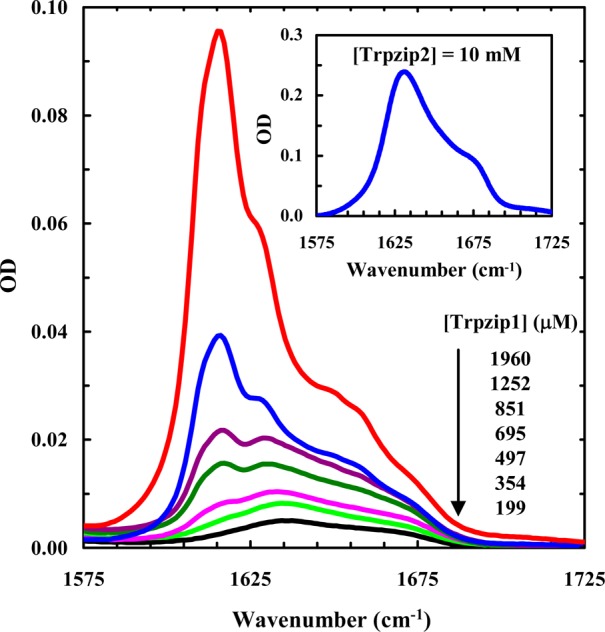
Amide I′ spectra of Trpzip1 at different concentrations, as indicated. Shown in the inset is the amide I′ band of Trpzip2 at 10 mM. These data were collected after the peptide samples had been incubated for 24 h at 25 °C.
Figure 3.
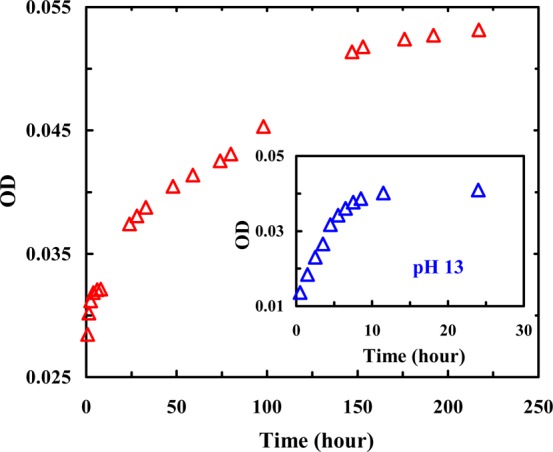
Intensity of the 1616 cm–1 band of Trpzip1 (1.2 mM, pH 3) as a function of incubation time, showing the aggregation kinetics of this peptide at acidic pH. For comparison, the aggregation data of Trpzip1 (0.7 mM) obtained at pH 13 are shown in the inset. The corresponding FTIR spectra are presented in Figure S2 of the Supporting Information.
Aggregation Mechanism and Gatekeeper
In principle, the initial aggregation step can occur through interactions between two folded β-hairpins or two unfolded peptides. However, a simple calculation, based on the thermal stabilities of Trpzip1 and Trpzip2,23 indicates that at 25 °C the unfolded concentrations of Trpzip1 (at a total concentration of 350 μM) and Trpzip2 (at a total concentration of 10 mM) are 89 and 900 μM, respectively. Thus, these results strongly suggest that the aggregation of these β-hairpins is not initiated by association of two unfolded peptides; instead, it is triggered by dimerization of two folded β-hairpins. This notion is consistent with that put forward by Richardson and Richardson,5 who showed that naturally occurring β-sheets can cause aggregation via edge-to-edge β-sheet interactions and that an effective strategy used by nature to defend against this is to place a charged side chain (or gatekeeper residue) on the hydrophobic face of an edge β-strand to mask the aggregation-prone regions with a solvent favorable interaction. In the current case, we hypothesize that Lys8 in both peptides acts as an aggregation gatekeeper residue; however, it is more effective to prevent Trpzip2 from aggregating. Because Lys8 in Trpzip1 and Trpzip2 appears on the opposite face of the hydrophobic core (i.e., the four Trp residues), we note that the aggregation gatekeeper role of this charged residue is somewhat different from that discussed by Richardson and Richardson, but the overall idea remains the same: a charge is used to disfavor the process of intermolecular association. As proposed in Figure 1, in the early stages of the aggregation process, should two Trpzip2 β-hairpins stack to form a dimer in a parallel fashion, the positively charged Lys8 side chains can lead to a more unfavorable intermolecular electrostatic interaction, because of their upward-pointing rotamer geometry and thus greater proximity. As a result, the aggregation propensity of Trpzip2 is weaker than that of Trpzip1. To test this hypothesis, we employed two strategies to eliminate the charge of Lys8 and then investigated how this change affects the aggregation propensities of these β-hairpins. In the first case, we used pH to neutralize the charge, whereas in the second case, we replaced Lys8 with a neutral Lys derivative, Lys(nvoc). The added advantage of using Lys(nvoc) is that the nvoc group can be removed via light, converting the mutant back to its parent sequence. In other words, we expect that the Lys(nvoc) modification in Trpzip2 not only will enhance the aggregation propensity of the peptide considerably but also can render the aggregates thus formed photodissociable, a feature that may find important applications in bioengineering.
As shown (Figure 3, inset), at a concentration of 0.7 mM and pH 13, the aggregation process of Trpzip1, as judged by the intensity of the 1616 cm–1 band, is complete within the first 10 h of dissolution, which is faster than the aggregation rate of Trpzip1 at acidic pH. Similarly, the aggregation of Trpzip2 can also be induced by increasing the pH to 13 (Figure 4). Taken together, these results provide strong evidence supporting the gatekeeper role of Lys8 mentioned above. However, unlike that of Trpzip1, the aggregation process of Trpzip2 does not seem to be complete even after incubation for 6 days, indicating that there are other factors that also play a role in determining the aggregation kinetics.
Figure 4.
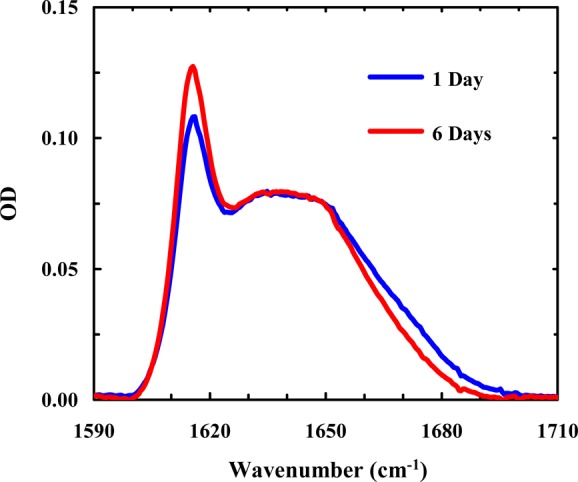
Amide I′ spectra of Trpzip2 (∼6 mM) at pH 13 measured after the peptide sample had been incubated for 1 and 6 days, as indicated.
It is well-documented that the β-hairpin structure in peptide fibrils tends to have extended and flat β-strands as opposed to the twisted conformations characteristically observed in native monomeric β-sheet proteins.56−61 Thus, for the initially formed peptide dimer or oligomers consisting of native or nativelike β-hairpins to further propagate to produce well-ordered and stacked parallel β-sheets, many native side chain–side chain interactions need to be broken to facilitate new intermolecular interactions, such as hydrogen bonding among neighboring monomers. In other words, the native β-hairpin must partially unfold to relax into a flat β-sheet unit, a structure required for the growth of the aggregate nucleus into long fibrils. Indeed, as shown (Figure S4, Supporting Information), the CD spectrum of the aggregated Trpzip1 lacks the distinctive positive band at 227 nm observed for the folded Trpzip1,62,63 indicating that the native edge-to-face Trp–Trp packing is disrupted upon the formation of aggregates.64 This observation is consistent with an aggregation mechanism that requires flattening of the native β-hairpin structure upon incorporation of the peptide into a tightly packed and well-organized fibrillar matrix. While this study does not allow us to describe further structural details, it is reasonable to assume that the Trp residues play an important role in aggregate formation, presumably via non-native hydrophobic stacking. This requirement of native structural change or relaxation would argue that the higher the β-hairpin stability, the more difficult it becomes for the aggregate nucleus to propagate to form mature aggregates or fibrils. Thus, we attribute the slow aggregation growth rate of Trpzip2 at pH 13 to its high stability (Tm = 72 °C).
Because there is another Lys residue (i.e., Lys12) in the peptide sequence, the results obtained at pH 13 may not entirely reflect the effect of Lys8. Therefore, in the second study, we examined the aggregation kinetics of a Trpzip2 mutant wherein Lys8 is replaced with a photolabile lysine analogue, Lys(nvoc).43−45 As shown (Figure 5), the amide I′ band of this mutant (Trpzip2-K) indicates that it aggregates quickly at very low peptide concentrations. Interestingly, the aggregates thus formed adopt an antiparallel β-sheet structure, as judged by the pair of bands centered at 1616 and 1685 cm–1. Because both Trpzip1 and Trpzip2 form parallel β-sheet aggregates, these results not only support the notion that Lys8 is an effective aggregation gatekeeper in Trpzip2 but also indicate that this non-natural lysine residue, which is strongly hydrophobic,65 can alter the aggregation pathway. In addition, the CD spectrum of the aggregated Trpzip2-K sample shows clearly the presence of a positive band at 228 nm (Figure 6), indicating that the native Trp–Trp packing is preserved to a certain extent in the aggregates. This is an interesting finding considering that the aggregates formed by the wild-type peptide do not support native Trp–Trp interactions and hence corroborates the aforementioned notion that Lys(nvoc), because of its higher hydrophobicity, can play a key role in determining the aggregation rate and pathway. This result further substantiates our initial hypothesis. Previously, the vertically pointing Lys8 side chains served as an aggregation deterrent because of the repulsive electrostatic interaction, but in the mutant case, having Lys(nvoc) in this orientation provides another or possibly stronger avenue for aggregation-prone hydrophobes to associate. Moreover, as shown (Figure S6, Supporting Information), upon removal of the nvoc group using light,44,45 which converts Lys(nvoc) to Lys, the aggregates formed by Trpzip2-K spontaneously disassemble to yield Trpzip2 monomers. Thus, this result provides additional evidence supporting the aggregation gatekeeper role of Lys8 in Trpzip2.
Figure 5.
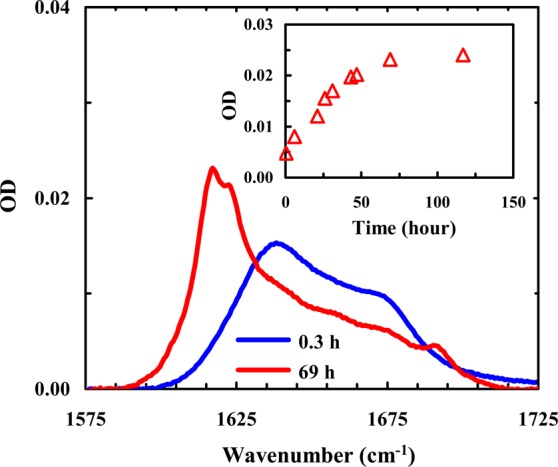
Representative amide I′ spectra of Trpzip2-K (0.5 mM, pH 3) obtained after different sample incubation times, as indicated. Shown in the inset is the intensity of the 1616 cm–1 band as a function of incubation time. The band intensities were obtained from the FTIR spectra shown in Figure S5 of the Supporting Information.
Figure 6.
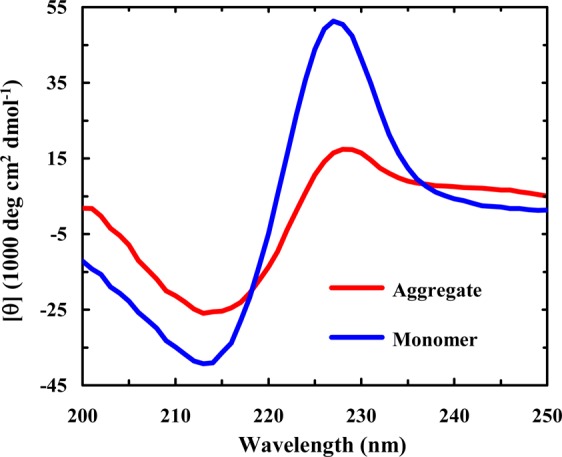
CD spectra of monomeric and aggregated Trpzip2-K samples (40 μM, pH 3), as indicated. The aggregated sample was prepared by diluting a more concentrated peptide sample (0.5 mM) that had been incubated for 14 days to allow aggregate formation.
To further verify the notion that it is Lys8, not Ly12, that plays a key role in mediating the aggregation process, we conducted another photocleavage experiment on an aggregate sample formed by a Tripzip2-K derivative wherein the charged Lys12 side chain was converted to a neutral species. Specifically, the amine group of Lys12 in Trpzip2-K was allowed to react with a common acylation agent, N-hydroxysuccinimide (NHS) ester, to form an amide bond (Scheme 1),66,67 and the resulting peptide is termed Trpzip2-KK. Similar to Trpzip2-K, this peptide readily aggregates at low concentrations, as expected. As shown (Figure 7), however, when the native side chain of Lys8 is recovered via photocleaveage of the nvoc group, the aggregates formed by Trpzip2-KK disassemble. Because the resulting peptide contains only one charged Lys side chain at position 8, this result thus substantiates our hypothesis that Lys12 does not play a significant role in preventing Trpzip hairpins from aggregating.
Scheme 1. Acylation of the Primary Amine of Lys12 in Trpzip2-K.

The resulting peptide is termed Trpzip2-KK.
Figure 7.
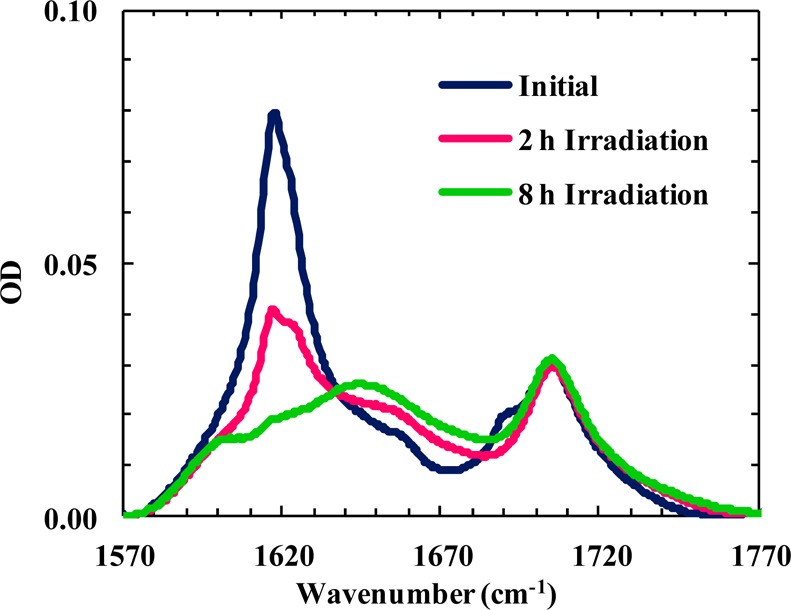
Amide I′ bands of an aggregated Trpzip2-KK sample (2.2 mM, D2O) obtained under different conditions, as indicated. These spectra show that photocleavage of the nvoc group on Lys8 results in aggregate disassembly. The band located near 1700 cm–1 arises from the C=O stretching vibration of the Lys12 side chain acrylamide.
Finally, to rule out the possibility that the weaker aggregation propensity of Trpzip2 is a direct outcome of its higher thermal stability, we tested a second variant in which Trp4 was mutated to Ala (hereafter termed Trpzip2-W4A). As expected (Figure S1, Supporting Information), this mutation significantly decreases the thermal stability of the β-hairpin (Tm of ∼23 °C). However, as indicated (Figure 8), Trpzip2-W4A (10 mM) does not show any detectable aggregation even after incubation for 5 days. Thus, this result further corroborates the proposed notion that the difference in the aggregation propensities of Trpzip1 and Trpzip2 stems from the difference in the Lys8 orientations and is not due to their difference in stability. Furthermore, this result suggests that any interactions between Lys8 and Trp4 are not critical in preventing the β-hairpin from aggregating.
Figure 8.
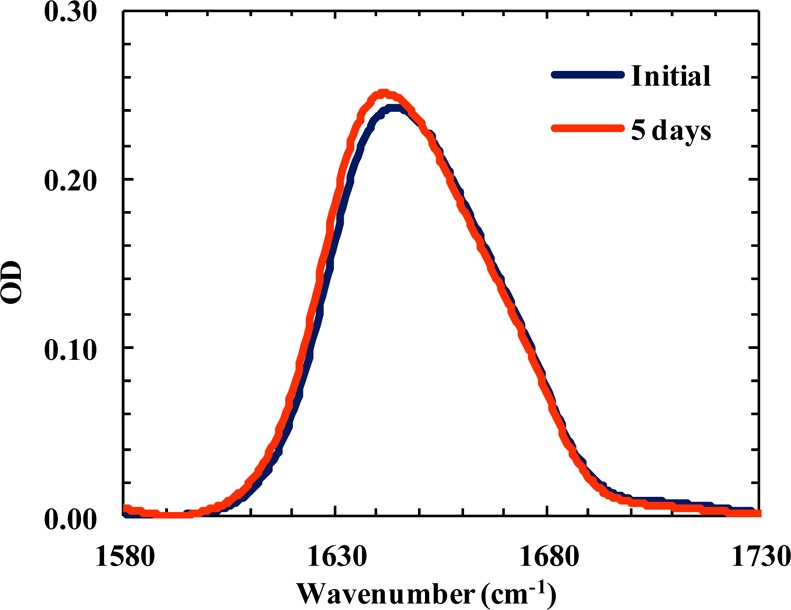
Amide I′ bands of Trpzip2-W4A (10 mM, pH 3) obtained at two incubation times, as indicated.
It is well-known that a stronger turn-promoting sequence can increase the stability of β-hairpins. However, the effect of turn sequence on the aggregation propensity of β-hairpins has not been systematically examined. On the basis of results obtained from this study, we can begin to think of several possible scenarios. If aggregation proceeds from an unfolded conformation, and the native turn structure is not preserved in the aggregates, the effect of a specific turn sequence on aggregation would be directly correlated with its effect on the β-hairpin stability. On the other hand, if aggregation is initiated by association of folded or partially folded β-hairpins, then the effect of a specific turn sequence on aggregation becomes more subtle. Depending on how it directs the distribution of key charged residues, a turn sequence could prevent or retard β-hairpin aggregation by creating unfavorable intermolecular electrostatic interactions or facilitate aggregate formation by weakening any repulsive interactions. To this end, the aggregation gatekeeper notion used in this study should not be simply interpreted as an independent action of one amino acid; rather, it should be discussed in the context of the underlying aggregation mechanism and aggregate structures.
Aggregate Stability and Morphology
Because both the strength of hydrophobic interactions and the unfolded population increase with an increase in temperature, many proteins and peptides show a stronger tendency to aggregate at higher temperatures.68−71 Interestingly, temperature has the opposite effect on Trpzip1 aggregation. As shown (Figure 9), Trpzip1 aggregates readily dissociate at higher temperatures. On the other hand, the aggregates formed by Trpzip2-K do not show any detectable heat-induced dissociation (Figure 10). These results suggest that the aggregates formed by Trpzip1 are less stable and less rigid than those formed by Trpzip2-K, which is corroborated by AFM measurements. As shown (Figure 11), the AFM image of a Trpzip2-K aggregate sample shows a well-defined fibrillar network, with a homogeneous distribution of fibrils approximately 3.8 nm wide, consistent with previously engineered β-hairpin aggregates.72 In comparison, the AFM image of a Trpzip1 aggregate sample reveals a more heterogeneous morphology, with the presence of variously sized fibrils and amorphous aggregates (Figure 12). Thus, taken together, the FTIR and AFM results indicate that the nvoc moiety in Lys(nvoc) not only significantly increases the aggregation rate of the peptide by eliminating the native aggregation gatekeeper in Trpzip2 but also guides the β-hairpins in the fibrils to stack in an antiparallel fashion. In other words, these results suggest that intermolecular Lys(nvoc) interactions can provide a strong driving force for peptide association and thus a new avenue for fibril formation. We believe this is an important finding as it suggests that it is possible to use Lys(nvoc) to control the fibrillization rate, as well as the fibrillar architecture, of the peptide of interest. In addition, another advantage of using Lys(nvoc) is that it makes the fibrils photoresponsive, a feature that could be exceedingly useful in certain bioengineering applications.73−76
Figure 9.
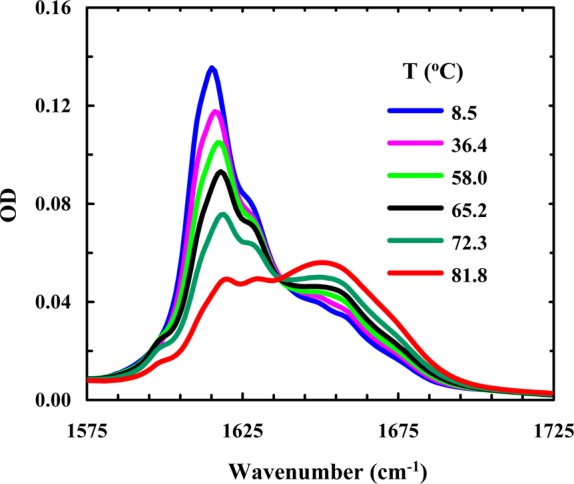
Amide I′ band of Trpzip1 (2.4 mM, pH 3) as a function of temperature.
Figure 10.
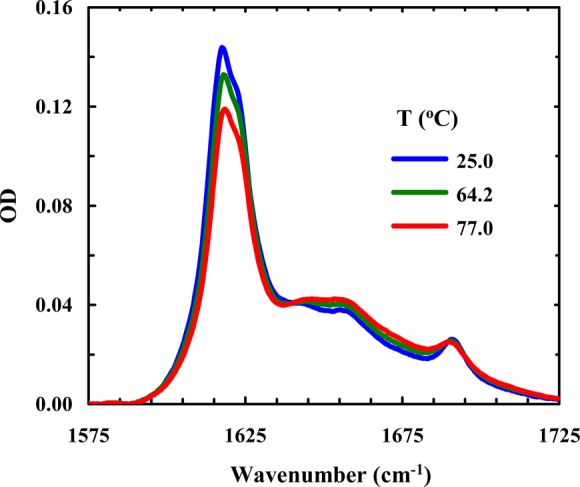
Amide I′ band of Trpzip2-K (4 mM, pH 3) as a function of temperature.
Figure 11.
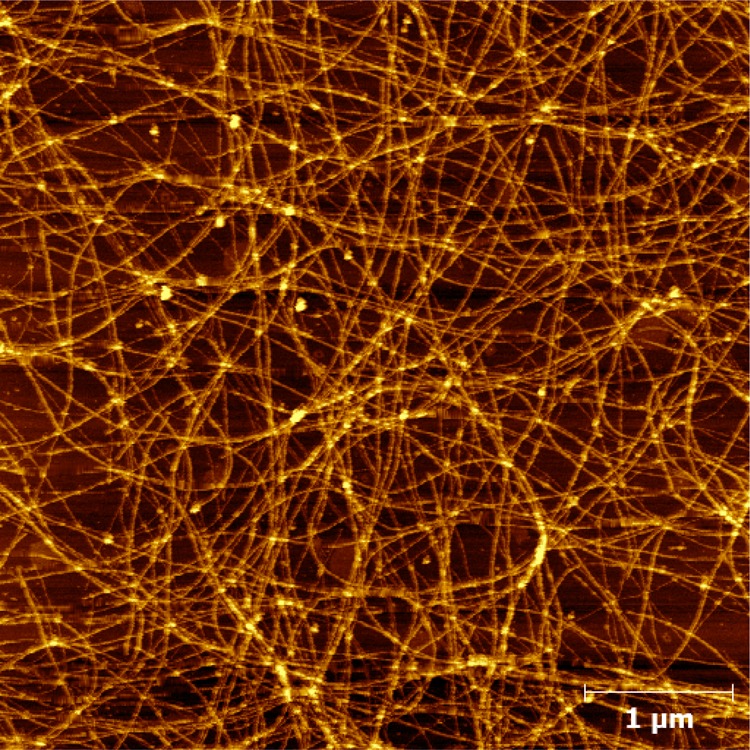
Representative AFM image of the peptide fibrils formed by Trpzip2-K after an incubation period of 14 days.
Figure 12.

Representative AFM image of the peptide fibrils and aggregates formed by Trpzip1 after an incubation period of 14 days.
Conclusion
Protein and peptide aggregation can have dire biological consequences. For example, it may lead to degenerative diseases in vivo and dysfunction of protein and peptide therapeutics in vitro. Therefore, many studies have been conducted in the past, with the aim of understanding the important factors that control protein and peptide aggregation and devising strategies to prevent it from happening. Herein, we study the aggregation properties of two closely related β-hairpins, Trpzip1 and Trpzip2, seeking to gain further insight into the mechanism of this phenomenon. Despite the minor difference in their turn sequences (i.e., NG vs GN), these two peptides exhibit totally different aggregation propensities; at acidic pH, Trpzip1 readily aggregates at micromolar concentrations, while under the same conditions, Trpzip2 does not show detectable aggregation even at concentrations of tens of millimolar. On the basis of the difference in their NMR structures and the fact that both peptides form aggregates rich in parallel β-sheets, we propose that (1) aggregation is initiated by association of two folded β-hairpins via edge-to-edge interactions and (2) Lys8 acts as an aggregation gatekeeper in both cases and its higher efficiency in preventing Trpzip2 from aggregating arises from the vertically pointing side chain rotamer preference. To test this hypothesis, we utilized two strategies, one by increasing the pH and the other by mutating Lys8 to a non-natural amino acid, Lys(nvoc), to examine how elimination of the positive charge on Lys8 affects the aggregation kinetics. We found that at pH 13 both Trpzip1 and Trpzip2 aggregate faster, which is consistent with the notion that Lys8 behaves as an aggregation gatekeeper. Further evidence in support of this hypothesis is that the Lys(nvoc) Trpzip2 mutant aggregates quickly, even at submillimolar concentrations, to form antiparallel amyloid-like fibrils that can be disassembled via photocleavage of the nvoc group. Moreover, our findings are consistent with an aggregation mechanism in which folded β-hairpins first associate to form a nucleus and the subsequent growth of this nucleus requires partial unfolding of the native structure. Finally, our results indicate that Lys(nvoc), because of its high hydrophobicity, can alter the aggregation mechanism and, hence, can be used to control, in conjunction with light, the morphology and structure of peptide fibrils.
Acknowledgments
We thank Yue Zhang, Ethan Glor, and Prof. Zahra Fakhraai at the University of Pennsylvania for assistance with AFM measurements.
Supporting Information Available
Additional CD and FTIR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
We gratefully acknowledge the financial support from the National Institutes of Health (GM-065978).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kelly J. W. (1996) Alternative conformations of amyloidogenic proteins govern their behavior. Curr. Opin. Struct. Biol. 6, 11–17. [DOI] [PubMed] [Google Scholar]
- Dobson C. M. (2003) Protein folding and misfolding. Nature 426, 884–890. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2003) Folding proteins in fatal ways. Nature 426, 900–904. [DOI] [PubMed] [Google Scholar]
- RamirezAlvarado M.; Blanco F. J.; Serrano L. (1996) De novo design and structural analysis of a model β-hairpin peptide system. Nat. Struct. Biol. 3, 604–612. [DOI] [PubMed] [Google Scholar]
- Richardson J. S.; Richardson D. C. (2002) Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. U.S.A. 99, 2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Hecht M. H. (2002) Rationally designed mutations convert de novo amyloid-like fibrils into monomeric β-sheet proteins. Proc. Natl. Acad. Sci. U.S.A. 99, 2760–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht M. H.; Das A.; Go A.; Bradley L. H.; Wei Y. N. (2004) De novo proteins from designed combinatorial libraries. Protein Sci. 13, 1711–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura S.; Zurdo J.; Narayanan S.; Parreño M.; Mangues R.; Reif B.; Chiti F.; Giannoni E.; Dobson C. M.; Aviles F. X.; Serrano L. (2004) Short amino acid stretches can mediate amyloid formation in globular proteins: The Src homology 3 (SH3) case. Proc. Natl. Acad. Sci. U.S.A. 101, 7258–7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokala N.; Handel T. M. (2005) Energy functions for protein design: Adjustment with protein–protein complex affinities, models for the unfolded state, and negative design of solubility and specificity. J. Mol. Biol. 347, 203–227. [DOI] [PubMed] [Google Scholar]
- Monsellier E.; Chiti F. (2007) Prevention of amyloid-like aggregation as a driving force of protein evolution. EMBO Rep. 8, 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marqusee S.; Baldwin R. L. (1987) Helix stabilization by Glu–···Lys+ salt bridges in short peptides of de novo design. Proc. Natl. Acad. Sci. U.S.A. 84, 8898–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marqusee S.; Robbins V. H.; Baldwin R. L. (1989) Unusually stable helix formatoin in short alanine-based peptides. Proc. Natl. Acad. Sci. U.S.A. 86, 5286–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serag A. A.; Altenbach C.; Gingery M.; Hubbell W. L.; Yeates T. O. (2002) Arrangement of subunits and ordering of β-strands in an amyloid sheet. Nat. Struct. Biol. 9, 734–739. [DOI] [PubMed] [Google Scholar]
- Thirumalai D.; Klimov D. K.; Dima R. I. (2003) Emerging ideas on the molecular basis of protein and peptide aggregation. Curr. Opin. Struct. Biol. 13, 146–159. [DOI] [PubMed] [Google Scholar]
- Pawar A. P.; Dubay K. F.; Zurdo J.; Chiti F.; Vendruscolo M.; Dobson C. M. (2005) Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 350, 379–392. [DOI] [PubMed] [Google Scholar]
- Munoz V.; Ghirlando R.; Blanco F. J.; Jas G. S.; Hofrichter J.; Eaton W. A. (2006) Folding and aggregation kinetics of a β-hairpin. Biochemistry 45, 7023–7035. [DOI] [PubMed] [Google Scholar]
- Nagarkar R. P.; Hule R. A.; Pochan D. J.; Schneider J. P. (2008) De novo design of strand-swapped β-hairpin hydrogels. J. Am. Chem. Soc. 130, 4466–4474. [DOI] [PubMed] [Google Scholar]
- Smith M. H.; Miles T. F.; Sheehan M.; Alfieri K. N.; Kokona B.; Fairman R. (2010) Polyglutamine fibrils are formed using a simple designed β-hairpin model. Proteins: Struct., Funct., Bioinf. 78, 1971–1979. [DOI] [PubMed] [Google Scholar]
- Azriel R.; Gazit E. (2001) Analysis of the minimal amyloid-forming fragment of the islet amyloid polypeptide: An experimental support for the key role of the phenylalanine residue in amyloid formation. J. Biol. Chem. 276, 34156–34161. [DOI] [PubMed] [Google Scholar]
- Williams A. D.; Portelius E.; Kheterpal I.; Guo J.-t.; Cook K. D.; Xu Y.; Wetzel R. (2004) Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335, 833–842. [DOI] [PubMed] [Google Scholar]
- Senguen F. T.; Doran T. M.; Anderson E. A.; Nilsson B. L. (2011) Clarifying the influence of core amino acid hydrophobicity, secondary structure propensity, and molecular volume on amyloid-β 16–22 self-assembly. Mol. BioSyst. 7, 497–510. [DOI] [PubMed] [Google Scholar]
- Lakshmanan A.; Cheong D. W.; Accardo A.; Di Fabrizio E.; Riekel C.; Hauser C. A. E. (2013) Aliphatic peptides show similar self-assembly to amyloid core sequences, challenging the importance of aromatic interactions in amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 110, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran A. G.; Skelton N. J.; Starovasnik M. A. (2001) Tryptophan zippers: Stable, monomeric β-hairpins. Proc. Natl. Acad. Sci. U.S.A. 98, 5578–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du D.; Zhu Y.; Huang C. Y.; Gai F. (2004) Understanding the key factors that control the rate of β-hairpin folding. Proc. Natl. Acad. Sci. U.S.A. 101, 15915–15920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.; Wu L.; McElheny D.; Bour P.; Roy A.; Keiderling T. A. (2009) Cross-strand coupling and site-specific unfolding thermodynamics of a trpzip β-hairpin peptide using C-13 isotopic labeling and IR spectroscopy. J. Phys. Chem. B 113, 5661–5674. [DOI] [PubMed] [Google Scholar]
- Popp A.; Wu L.; Keiderling T. A.; Hauser K. (2012) Impact of β-turn sequence on β-hairpin dynamics studied with infrared-detected temperature jump. Spectroscopy 27, 557–564. [Google Scholar]
- Jones K. C.; Peng C. S.; Tokmakoff A. (2013) Folding of a heterogeneous β-hairpin peptide from temperature-jump 2D IR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 110, 2828–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow C. D.; Qiu L.; Du D.; Gai F.; Hagen S. J.; Pande V. S. (2004) Trp zipper folding kinetics by molecular dynamics and temperature-jump spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 101, 4077–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekiyo T.; Wu L.; Yoshimura Y.; Shimizu A.; Keiderling T. A. (2009) Relationship between hydrophobic interactions and secondary structure stability for trpzip β-hairpin peptides. Biochemistry 48, 1543–1552. [DOI] [PubMed] [Google Scholar]
- Yang W. Y.; Pitera J. W.; Swope W. C.; Gruebele M. (2004) Heterogeneous folding of the trpzip hairpin: Full atom simulation and experiment. J. Mol. Biol. 336, 241–251. [DOI] [PubMed] [Google Scholar]
- Guvench O.; Brooks C. L. (2005) Tryptophan side chain electrostatic interactions determine edge-to-face vs parallel-displaced tryptophan side chain geometries in the designed β-hairpin “trpzip2”. J. Am. Chem. Soc. 127, 4668–4674. [DOI] [PubMed] [Google Scholar]
- Smith A. W.; Chung H. S.; Ganim Z.; Tokmakoff A. (2005) Residual native structure in a thermally denatured β-hairpin. J. Phys. Chem. B 109, 17025–17027. [DOI] [PubMed] [Google Scholar]
- Krejtschi C.; Huang R.; Keiderling T. A.; Hauser K. (2008) Time-resolved temperature-jump infrared spectroscopy of peptides with well-defined secondary structure: A trpzip β-hairpin variant as an example. Vib. Spectrosc. 48, 1–7. [Google Scholar]
- Narayanan R.; Pelakh L.; Hagen S. J. (2009) Solvent friction changes the folding pathway of the tryptophan zipper TZ2. J. Mol. Biol. 390, 538–546. [DOI] [PubMed] [Google Scholar]
- Nymeyer H. (2009) Energy landscape of the trpzip2 peptide. J. Phys. Chem. B 113, 8288–8295. [DOI] [PubMed] [Google Scholar]
- Yang L. J.; Shao Q.; Gao Y. Q. (2009) Thermodynamics and folding pathways of trpzip2: An accelerated molecular dynamics simulation study. J. Phys. Chem. B 113, 803–808. [DOI] [PubMed] [Google Scholar]
- Roy S.; Jansen T. L. C.; Knoester J. (2010) Structural classification of the amide I sites of a β-hairpin with isotope label 2DIR spectroscopy. Phys. Chem. Chem. Phys. 12, 9347–9357. [DOI] [PubMed] [Google Scholar]
- Culik R. M.; Jo H.; DeGrado W. F.; Gai F. (2012) Using thioamides to site-specifically interrogate the dynamics of hydrogen bond formation in β-sheet folding. J. Am. Chem. Soc. 134, 8026–8029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: Visual molecular dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Dunbrack R. L.; Cohen F. E. (1997) Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 6, 1661–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel J. S.; Sturtevant J. M.; Regan L. (1999) Sidechain interactions in parallel β sheets: The energetics of cross-strand pairings. Structure 7, 1333–1343. [DOI] [PubMed] [Google Scholar]
- Patchorn A.; Amit B.; Woodward R. B. (1970) Photosensitive protecting groups. J. Am. Chem. Soc. 92, 6333–6335. [Google Scholar]
- Rusiecki V. K.; Warne S. A. (1993) Synthesis of Nα-Fmoc-Nε-Nvoc-lysine and use in the preparation of selectively functionalized peptides. Bioorg. Med. Chem. Lett. 3, 707–710. [Google Scholar]
- Measey T. J.; Gai F. (2012) Light-triggered disassembly of amyloid fibrils. Langmuir 28, 12588–12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Measey T. J.; Markiewicz B. N.; Gai F. (2013) Amide I band and photoinduced disassembly of a peptide hydrogel. Chem. Phys. Lett. 580, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.-Y.; Getahun Z.; Zhu Y.; Klemke J. W.; DeGrado W. F.; Gai F. (2002) Helix formation via conformation diffusion search. Proc. Natl. Acad. Sci. U.S.A. 99, 2788–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood J. S.; Koszelak M.; Liu J.; Lawrence D. S. (1998) A Caged Protein Kinase Inhibitor. J. Am. Chem. Soc. 120, 7145–7146. [Google Scholar]
- Loudwig S., and Bayley H. (2005) Light-Actived Proteins: An Overview. In Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules (Goeldner M., and Givens R., Eds.) pp 253–303, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany. [Google Scholar]
- Solomek T.; Mercier S.; Bally T.; Bochet C. G. (2012) Photolysis of ortho-nitrobenzylic derivatives: The importance of the leaving group. Photochem. Photobiol. Sci. 11, 548–555. [DOI] [PubMed] [Google Scholar]
- Kubelka J.; Keiderling T. A. (2001) Differentiation of β-sheet-forming structures: Ab initio-based simulations of IR absorption and vibrational CD for model peptide and protein β-sheets. J. Am. Chem. Soc. 123, 12048–12058. [DOI] [PubMed] [Google Scholar]
- Lansbury P. T. Jr.; Costa P. R.; Griffiths J. M.; Simon E. J.; Auger M.; Halverson K. J.; Kocisko D. A.; Hendsch Z. S.; Ashburn T. T.; Spencer R. G.; et al. (1995) Structural model for the β-amyloid fibril based on interstrand alignment of an antiparallel-sheet comprising a C-terminal peptide. Nat. Struct. Biol. 2, 990–998. [DOI] [PubMed] [Google Scholar]
- Lee C.; Cho M. (2004) Local amide I mode frequencies and coupling constants in multiple-stranded antiparallel β-sheet polypeptides. J. Phys. Chem. B 108, 20397–20407. [Google Scholar]
- Lomakin A.; Chung D. S.; Benedek G. B.; Kirschner D. A.; Teplow D. B. (1996) On the nucleation and growth of amyloid β-protein fibrils: Detection of nuclei and quantitation of rate constants. Proc. Natl. Acad. Sci. U.S.A. 93, 1125–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty S. A.; Decatur S. M. (2005) Intersheet rearrangement of polypeptides during nucleation of β-sheet aggregates. Proc. Natl. Acad. Sci. U.S.A. 102, 14272–14277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perálvarez-Marín A.; Barth A.; Gräslund A. (2008) Time-resolved infrared spectroscopy of pH-induced aggregation of the alzheimer Aβ1–28 peptide. J. Mol. Biol. 379, 589–596. [DOI] [PubMed] [Google Scholar]
- Jimenez J. L.; Nettleton E. J.; Bouchard M.; Robinson C. V.; Dobson C. M.; Saibil H. R. (2002) The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 99, 9196–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Ishii Y.; Balbach J. J.; Antzutkin O. N.; Leapman R. D.; Delaglio F.; Tycko R. (2002) A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandomeneghi G.; Krebs M. R.; McCammon M. G.; Fandrich M. (2004) FTIR reveals structural differences between native β-sheet proteins and amyloid fibrils. Protein Sci. 13, 3314–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S.; Gawlak G.; Makabe K.; Tereshko V.; Koide A.; Koide S. (2007) Hydrophobic surface burial is the major stability determinant of a flat, single-layer β-sheet. J. Mol. Biol. 368, 230–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larini L.; Shea J.-E. (2012) Role of β-hairpin formation in aggregation: The self-assembly of the amyloid-β(25–35) peptide. Biophys. J. 103, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W.; Yau W.-M.; Luo Y.; Mattson M. P.; Tycko R. (2012) Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 4443–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishina I. B.; Woody R. W. (1994) Contributions of tryptophan side chains to the circular dichroism of globular proteins: Exciton couplets and coupled oscillators. Faraday Discuss. 245–262. [DOI] [PubMed] [Google Scholar]
- Wu L.; McElheny D.; Huang R.; Keiderling T. A. (2009) Role of tryptophan–tryptophan interactions in trpzip β-hairpin formation, structure, and stability. Biochemistry 48, 10362–10371. [DOI] [PubMed] [Google Scholar]
- Barrow C. J.; Yasuda A.; Kenny P. T. M.; Zagorski M. G. (1992) Solution conformations and aggregational properties of synthetic amyloid β-peptides of Alzheimer’s disease: Analysis of circular-dichroism spectra. J. Mol. Biol. 225, 1075–1093. [DOI] [PubMed] [Google Scholar]
- Nasir S.; Ramirez P.; Ali M.; Ahmed I.; Fruk L.; Mafe S.; Ensinger W. (2013) Nernst-Planck model of photo-triggered, pH-tunable ionic transport through nanopores functionalized with “caged” lysine chains. J. Chem. Phys. 138, 034709–034711. [DOI] [PubMed] [Google Scholar]
- Abello N.; Kerstjens H. A. M.; Postma D. S.; Bischoff R. (2007) Selective acylation of primary amines in peptides and proteins. J. Proteome Res. 6, 4770–4776. [DOI] [PubMed] [Google Scholar]
- Basle E.; Joubert N.; Pucheault M. (2010) Protein chemical modification on endogenous amino acids. Chem. Biol. 17, 213–227. [DOI] [PubMed] [Google Scholar]
- Clark A. H.; Saunderson D. H.; Suggett A. (1981) Infrared and laser-Raman spectroscopic studies of thermally-induced globular protein gels. Int. J. Pept. Protein Res. 17, 353–364. [DOI] [PubMed] [Google Scholar]
- Casal H. L.; Kohler U.; Mantsch H. H. (1988) Structural and conformational changes of β-lactoglobulin B: An infrared spectroscopic study of the effect of pH and temperature. Biochim. Biophys. Acta 957, 11–20. [DOI] [PubMed] [Google Scholar]
- Naumann D.; Schultz C.; Gorne-Tschelnokow U.; Hucho F. (1993) Secondary structure and temperature behavior of the acetylcholine receptor by Fourier transform infrared spectroscopy. Biochemistry 32, 3162–3168. [DOI] [PubMed] [Google Scholar]
- Fabian H.; Schultz C.; Naumann D.; Landt O.; Hahn U.; Saenger W. (1993) Secondary structure and temperature-induced unfolding and refolding of ribonuclease T1 in aqueous solution. A Fourier transform infrared spectroscopic study. J. Mol. Biol. 232, 967–981. [DOI] [PubMed] [Google Scholar]
- Rughani R. V.; Schneider J. P. (2008) Molecular design of β-hairpin peptides for material construction. MRS Bull. 33, 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines L. A.; Rajagopal K.; Ozbas B.; Salick D. A.; Pochan D. J.; Schneider J. P. (2005) Light-activated hydrogel formation via the triggered folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 127, 17025–17029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer G.; Heckel A. (2006) Biologically active molecules with a “light switch”. Angew. Chem., Int. Ed. 45, 4900–4921. [DOI] [PubMed] [Google Scholar]
- Peng K.; Tomatsu I.; Kros A. (2010) Light controlled protein release from a supramolecular hydrogel. Chem. Commun. 46, 4094–4096. [DOI] [PubMed] [Google Scholar]
- Knowles T. P. J.; Buehler M. J. (2011) Nanomechanics of functional and pathological amyloid materials. Nat. Nanotechnol. 6, 469–479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.