

Abstract
CYP19A1, or aromatase, a cytochrome P450 responsible for estrogen biosynthesis in humans, is an important therapeutic target for the treatment of breast cancer. There is still controversy surrounding the identity of reaction intermediate that catalyzes carbon–carbon scission in this key enzyme. Probing the oxy-complexes of CYP19A1 poised for hydroxylase and lyase chemistries using resonance Raman spectroscopy and drawing a comparison with CYP17A1, we have found no significant difference in the frequencies or isotopic shifts for these two steps in CYP19A1. Our experiments implicate the involvement of Compound I in the terminal lyase step of CYP19A1 catalysis.
The mammalian Cytochrome P450 enzymes participate in a vast array of important physiological functions through the involvement of potent oxidizing intermediates capable of epoxidation or hydroxylation of even relatively inert substrates.1−3 Defining the structural dynamics of reactions involving these enzymatic intermediates, identified in Figure 1,3 is one of the most challenging and important goals of heme protein research. While the Compound I species is considered to be the most common active oxidant, mainly effecting hydroxylation of substrates,3−5 the ferric peroxo- and hydroperoxo- intermediates are also powerful oxidants that have been suggested to participate in physiologically important processes.6,7 The availability of these multiple potential oxidants leads to various possible outcomes for a given cytochrome P450 under different conditions. The peroxo- intermediate formed upon delivery of an electron to the ferrous-dioxygen adduct can react in the presence of a susceptible (electrophilic) substrate or be converted to the subsequent hydroperoxo-form with a rate depending on the efficiency of proton delivery. If delivery of a second proton to the terminal (distal) oxygen of the hydroperoxo-intermediate is restricted, O–O bond cleavage is impeded and the system either “uncouples” by loss of H2O2 or, if a susceptible substrate such as an alkene is present, a rather efficient epoxidation reaction can occur.7 In the absence of conditions which permit interception of these peroxo- and hydroperoxo- intermediates, Compound I is formed, the efficiency of which also depends on the extent of the “decoupling” reactions that releases superoxide and hydrogen peroxide. Based on this abbreviated scheme, it is clear that the catalytic behavior of a given Cytochrome P450 depends on numerous factors, including the structure and reactivity of a particular substrate, the influence of the partner reductant, and the environmental conditions.
Figure 1.

Catalytic cycle for cytochrome P450.3
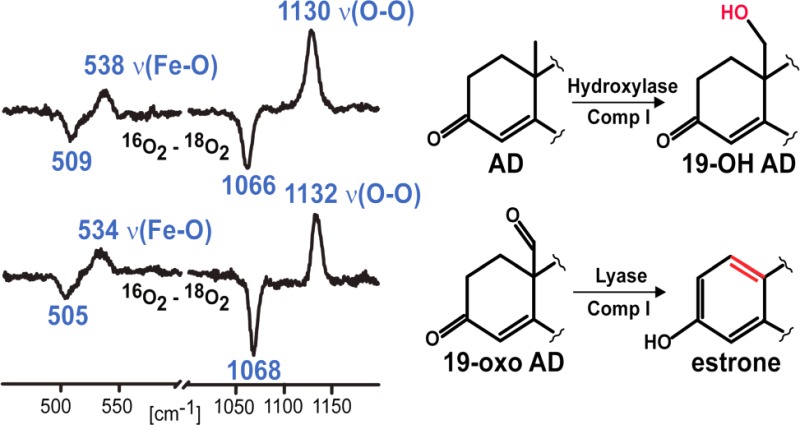
Aromatase (CYP19A1) plays a crucial role in the biosynthesis of steroid hormones, being responsible for the conversion of androgens to estrogens.8 The conversion of androstenedione (AD) to an aromatic C18 estrogen, estrone, involves two consecutive oxidations at the C19 methyl group and a third “lyase” step that culminates in cleavage of the C10–C19 bond of the C19-aldehyde and A ring aromatization (Figure 2).9,10 The first and second oxidative steps are generally accepted to proceed as classic hydroxylations at the C19 of androgens mediated by the Compound I species, producing the C19 primary alcohol, and then, in a second hydroxylation reaction, a gem-diol intermediate is formed that spontaneously dehydrates to the C19 aldehyde.9−11 The operative mechanism associated with the lyase step has been the subject of much debate during the past two decades.9,12−16 In agreement with an early proposal by Fishman and Raju,12 Korzekwa et al. favored involvement of Compound I as the active oxidant.13 However, it has also been suggested that the nucleophilic ferric peroxo- intermediate attacks the substrate’s aldehyde group yielding the observed product via decomposition of a transient peroxo hemiacetal,9 a C–C bond cleavage reaction similar to that which has been proposed to occur for CYP2B4, CYP51, and CYP17 in their processing of substrates bearing aldehyde or ketone functionalities.14−16 However, Hackett et al. applied density functional theory to a minimal CYP19A1 active site model, finding support for an energetic preference for a Compound I mediated hydrogen abstraction from the C1 carbon of 19-oxo-AD rather than direct attack of the ferric peroxo-species on the C19 aldehyde carbon.17 Until recently, definitive experimental evidence to identify the reactive intermediates involved in this third stage of the C10–C19 bond cleavage/aromatization process has been lacking.
Figure 2.

Proposed pathway for aromatization reaction of androstenedione by human CYP19A1.
To address this issue one of our groups recently exploited the kinetic solvent isotope effect (KSIE) technique, which had earlier been shown to be useful in quantifying consecutive proton-transfer processes involved in generating high-valent iron-oxo intermediates,18 to investigate the bond cleavage reactions involved in CYP17A1 and CYP19A1 enzymatic cycles.16,19 Briefly, the effect of deuterium substitution on the enzymatic rates reveal information regarding the number of protons involved in O–O bond heterolysis,20 with Compound I formation, which requires at least 2 protons, leading to lowered rates in 2H2O, by a factor of ∼1.3, whereas direct reaction of the nucleophilic ferric peroxo intermediate involves no proton transfer step. KSIE studies on CYP17A1 showed that while the hydroxylase reaction, proceeding through a Compound I intermediate, revealed an expected kH/kD ratio of 1.3, the stage wherein a C20–C17 lyase reaction occurs exhibited an inverse KSIE of 0.39, a value consistent with involvement of a ferric peroxo intermediate.16 In contrast to this substrate-dependent difference in mechanism for CYP17A1, similar KSIE studies for CYP19A1 showed comparably slower rates in 2H2O vs H2O for conversion of AD to 19-OH-AD and for conversion of oxo-AD to estrone, with a KSIE of ≥2.5 in both cases, suggesting the involvement of the same intermediate during catalysis and therefore implicating the involvement of Compound I species in the controversial C10–C19 lyase step for CYP19A1.12,13,17,19
Such KSIE studies suggest the involvement of a Compound I intermediate in the lyase reaction of CYP19A1. However, a lingering question to be addressed is what specific active site structural element(s), including substrate fragments, enable the lyase reaction to proceed via the peroxo- intermediate for CYP17A1/17-OH PREG, but are apparently absent in the case of CYP19A1 bound with 19-oxo AD, thereby relegating the task of C–C bond cleavage to the later Compound I intermediate? Obviously, one potentially important difference between the CYP17A1 and CYP19A1 systems is that the CYP17A1 case houses a substrate bearing a hydroxyl group juxtaposed to the Fe–O–O fragment, while the relevant substrate in the CYP19A1 system possesses no such H-bond donor fragment. However, other potential H-bond donors, including active site water molecules, are conceivably present to interact with the Fe–O–O fragment of CYP19A1/AD and CYP19A1/19-oxo-AD, so the possibility of differentially directed H-bonding interactions cannot be ruled out a priori and prompts further investigation. Gaining insight into this issue requires a method for direct interrogation of active site structure. While many spectroscopic methods are available for documenting structural parameters of the heme, its associated axial ligands, and its immediate protein environment, resonance Raman (rR) spectroscopy has been shown to be especially powerful for these in their stable terminal states as well as for trapped intermediates.22−25 In fact, in a recent work focused on the dioxygen adducts of CYP17A1 in the presence of 17-OH-progesterone (17-OH PROG) or 17-OH-pregnenolone (17-OH PREG),26 we were able to show that distinctive patterns of vibrational modes for the Fe–O–O fragments arise for these two situations. In the case of 17-OH PREG, which shows a 50-fold increase in its tendency to undergo a lyase reaction,27 compared to 17-OH PROG and proceeding via a peroxo- intermediate,16 the dioxygen adduct exhibits a vibrational spectral pattern consistent with H-bonding to the proximal oxygen atom of the Fe–O–O fragment, an interaction expected to stabilize a subsequent ferric peroxo-fragment.28 Conversely, the dioxygen adduct of the 17-OH PROG-bound CYP17A1 system exhibits a vibrational mode pattern associated with H-bonding to the terminal oxygen in the Fe–O–O fragment, an arrangement that promotes O–O bond cleavage and follows the Compound I pathway to effect hydroxylation.29
In the present work rR spectroscopy is used to characterize the active site structures of dioxygen adducts of CYP19A1 bound with the first (AD) or third (19-oxo-AD) substrates encountered in the enzymatic cycle so as to assess whether or not there is definitive evidence for functionally significant structural differences of the Fe–O–O fragments in the two cases. As in our earlier works with CYP17A1 and CYP19A1,16,19,26 the Nanodisc sampling system was used, which effectively mimics the natural membrane environment, yielding well-behaved functional properties, as evidenced by the expected distributions of spin-state populations and enhanced stability of the dioxygen adducts.30−32
As shown in Figure 3 (trace A), a rather strong feature is observed at 1130 cm–1 in the rR spectrum of the 16O2 adduct of CYP19A1 when bound with AD, which is reasonably assigned to the ν(16O–16O) stretching mode based on the fact that, as illustrated in the difference spectrum shown in trace B, it disappears and is replaced by a new feature located at 1066 cm–1 for the corresponding 18O2 adduct, the 64 cm–1 shift being entirely consistent with that expected in the harmonic oscillator approximation.33 The difference spectrum shown in trace C, obtained from solutions prepared from deuterated solvents, shows a possible, though barely significant, shift of the ν(16O–16O) and ν(18O–18O) modes to slightly higher frequencies. Shown in the inset are the 16O2–18O2 difference spectra acquired in the low frequency region indicating that the ν(Fe–16O) stretching mode occurs at 538 cm–1, exhibiting an apparent16O2–18O2 shift of ∼29 cm–1, a value that is somewhat higher than that expected in the harmonic oscillator approximation, which is between 20 and 24 cm–1, depending on whether the oxygen masses are assumed to be 16 and 18 amu vs 32 and 36 amu.33 Such slightly large shifts observed for dioxygen adducts of cytochromes P450 have been observed previously and can be explained in terms of an effective “three body oscillator” model, (Fe–O–O).34 Shown in Figure 4 are the corresponding rR spectra obtained for the dioxygen adducts of CYP19A1 having the third substrate, 19-oxo-AD, bound in its active site. There it is seen that the observed frequencies and isotopic shifts obtained are quite similar to the data acquired for the AD-bound enzyme, with only the values for the ν(Fe–O) modes differing, but even then only by a few wavenumbers, at most. It is noted that, while in our experience alteration of substrate structure within the P450 distal pocket is not expected to significantly affect the ν(Fe–S) mode,35,36 we nevertheless confirmed that the values of ν(Fe–S) are identical for CYP19A1 bound with these two substrates (see Supporting Information), reflecting the validity of the point made above.
Figure 3.

High frequency rR spectra of AD-bound oxy CYP19A1 (A) and the difference traces, 16O2–18O2 in H2O buffer (B) and 16O2–18O2 in D2O buffer (C). Inset shows corresponding difference traces in low frequency region.
Figure 4.

High frequency rR spectra of 19-oxo-AD-bound oxy CYP19A1 (A) and the difference traces, 16O2–18O2 in H2O buffer (B) and 16O2–18O2 in D2O buffer (C). Inset shows corresponding difference traces in low frequency region.
It is now possible and instructive to compare the vibrational data for the dioxygen adducts of the CYP17A1 and CYP19A1 systems, bound with various substrates, in light of the functional properties as reflected in the extracted KSIE data.16,19 In the case of CYP17A1 oxidation of 17-OH-PREG, the KSIE data support involvement of the peroxo- species and, as is shown in a recent spectroscopic study,26 the ν(Fe–O) frequency for this form was observed at 526 cm–1, which signals an H-bonding interaction with the proximal O-atom of the Fe–O–O fragment, whereas the ν(Fe–O) mode for the dioxygen adduct of 17-OH PROG, as well as the data for dioxygen adducts of PROG and PREG, which undergo hydroxylation reactions, appear near 540 cm–1 and are consistent with H-bond donation to the terminal oxygen of the Fe–O–O fragment.24,25,37−40 The essential point is that the internal modes of Fe–O–O fragments, especially the ν(Fe–O) modes, differ considerably for systems poised for the lyase and hydroxylase reaction pathways. Consequently, the quite similar vibrational mode patterns observed here for dioxygen adducts with the first and third substrates of CYP19A1 are consistent with the similar KSIE values obtained for the associated reactions and provide convincing evidence for an H-bonding interaction with the terminal oxygen atom of the Fe–O–O fragment that would facilitate O–O bond cleavage and imply the involvement of Compound I.
Returning to the question posed above regarding functionally important active site structural elements, the collection of rR data acquired for the dioxygen adducts of CYP19A1 with two different substrates and, previously for CYP17A1 with four different substrates,26 provides evidence indicating that CYP17A1 housing the “lyase promoting substrate”, 17-OH PREG, represents a special case in which the substrate hydroxyl group is uniquely positioned to provide an H-bonding interaction with the proximal oxygen atom of the Fe–O–O fragment, an active site structural arrangement that is not duplicated even with the structurally very similar 17-OH PROG,26 confirming the importance of a finely tuned active site architecture in orchestrating the enzymatic cycles of these enzymes. The spectroscopic data acquired here for oxy-CYP19A1 bound with AD and 19-oxo-AD suggest that neither of these systems gives rise to the unusual H-bonding interaction with the proximal oxygen of the Fe–O–O fragment. Instead they exhibit a vibrational spectral pattern consistent with H-bond donation to the terminal oxygen and therefore implicate Compound I in both hydroxylation and aromatization steps of CYP19A1 catalysis. Finally, it is interesting consider the necessity for divergent mechanisms operating in C–C bond cleavage in terms of the demands imposed by the substrate structure. The lyase reaction proceeds through a more commonly encountered Compound I pathway for CYP19A1 via attack on the susceptible C1–H bond. Inasmuch as no such reaction is possible with the lyase substrates of CYP17A1, the system has evolved a “substrate-assisted” catalysis, facilitating the cleavage by employing a ferric peroxo- intermediate stabilized by H-bonding from the substrate hydroxyl fragment.
Acknowledgments
This work was supported by National Institutes of Health Grants GM31756 and GM33775 to S.G.S. and GM96117 to J.R.K.
Supporting Information Available
Materials and Methods sections, experimental details of rR measurements, Figure S1 showing the ν(Fe–S) modes. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cytochrome P450: Structure, Mechanism, and Biochemistry; Ortiz de Montellano P. R., Ed.; Kluwer Academic/Plenum Publisher: New York, 2005. [Google Scholar]
- Metal Ions in Life Sciences; Sigel A., Sigel H., Sigel R. K. O., Eds.; John Wiley & Sons, Ltd.: 2007,. Vol 3. [Google Scholar]
- Denisov I. G.; Makris T. M.; Sligar S. G.; Schlichting I. Chem. Rev. 2005, 105, 2253. [DOI] [PubMed] [Google Scholar]
- Groves J. T. J. Inorg. Biochem. 2006, 100, 434. [DOI] [PubMed] [Google Scholar]
- Rittle J.; Green M. T. Science 2010, 330, 933. [DOI] [PubMed] [Google Scholar]
- Akhtar M.; Corina D. L.; Miller S. L.; Shyadehi A. Z.; Wright J. N. J. Chem. Soc., Perkin Trans. 1 1994, 263. [Google Scholar]
- Vaz A. D. N.; McGinnity D. F.; Coon M. J. Proc. Natl. Acad. Sci. U.S.A. 1988, 95, 3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D.; Griswold J.; Erman M.; Pangborn W. Nature 2009, 457, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar M.; Calder M. R.; Corina D. L.; Wright J. N. Biochem. J. 1982, 201, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar M.; Njar V. V.; Wright J. N. J. Steroid. Biochem. Mol. Biol. 1993, 44, 375. [DOI] [PubMed] [Google Scholar]
- Bernhardt R.; Waterman M. R. Metal Ions in Life Sciences; Sigel A., Sigel H., Sigel R. K. O., Eds.; John Wiley & Sons, Ltd.: 2007; Vol. 3, pp 361–396. [Google Scholar]
- Fishman J.; Raju M. S. J. Biol. Chem. 1981, 256, 4472. [PubMed] [Google Scholar]
- Korzekwa K. R.; Trager W. F.; Mancewicz J.; Osawa Y. J. Steroid Biochem. Mol. Biol. 1993, 44, 367. [DOI] [PubMed] [Google Scholar]
- Vaz D. N.; Pernecky S. J.; Raner G. M.; Coon M. J. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyadehi Z.; Lamb D. C.; Kelly S. L.; Kelly D. E; Schunck W. H.; Wright J. N.; Corina D.; Akhtar M. J. Biol. Chem. 1996, 271, 12445. [DOI] [PubMed] [Google Scholar]
- Gregory M. C.; Denisov I. G.; Grinkova Y. V.; Khatri Y.; Sligar S. G. J. Am. Chem. Soc. 2013, 135, 16245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett J. C.; Brueggemeier R. W.; Hadad C. M. J. Am. Chem. Soc. 2005, 127, 5224. [DOI] [PubMed] [Google Scholar]
- Aikens J.; Sligar S. G. J. Am. Chem. Soc. 1994, 116, 1143. [Google Scholar]
- Khatri Y.; Luthra A.; Duggal R.; Sligar S. G., to be submitted.
- Vidakovic M.; Sligar S. G.; Li H.; Poulos T. L. Biochemistry 1998, 37, 9211. [DOI] [PubMed] [Google Scholar]
- Biological Applicfations of Raman Spectroscopy; Spiro T. G., Ed.; John Wiley & Sons: New York, 1988. [Google Scholar]
- Kincaid J. R. In The Porphyrin Handbook; Kadish K. M., Smith K. M., Guilard R., Eds.; Academic Press: 2000; Vol. 7, pp 225–291. [Google Scholar]
- Terner J.; Palaniappan V.; Gold A.; Weiss R.; Fitzgerald M. M.; Sullivan A. M.; Hosten C. M. J. Inorg. Biochem. 2006, 100, 480. [DOI] [PubMed] [Google Scholar]
- Spiro T. G.; Soldatova A. V.; Balakrishnan G. Coord. Chem. Rev. 2013, 257, 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov I. G.; Mak P. J.; Makris T. M.; Sligar S. G.; Kincaid J. R. J. Phys. Chem. A 2008, 112, 13172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory M.; Mak P. J.; Sligar S. G.; Kincaid J. R. Angew. Chem., Int. Ed. 2013, 52, 5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usanov S. A.; Gilep A. A.; Sushko T. A. Biochim. Biophys. Acta 2011, 1814, 200. [DOI] [PubMed] [Google Scholar]
- Pant K.; Crane B. R. Biochemistry 2006, 45, 2537. [DOI] [PubMed] [Google Scholar]
- Ogliaro F. O.; de Visser S. P.; Cohen S.; Sharma P. K.; Shaik S. J. Am. Chem. Soc. 2002, 124, 2806. [DOI] [PubMed] [Google Scholar]
- Denisov I. G.; Sligar S. G. Biochim. Biophys. Acta 2011, 1814, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler M. A.; Denisov I. G.; Sligar S. G. Methods Mol. Biol. 2013, 74, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra A.; Gregory M.; Grinkova Y. V.; Denisov I. G.; Sligar S. G. Methods Mol. Biol. 2013, 987, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto K.Infrared and Raman Spectra of Inorganic and Coordination Compounds; John Wiley & Sons, Inc.: 2009; Part A, p 10. [Google Scholar]
- Macdonald I. D. G.; Sligar S. G.; Christian J. F.; Unno M.; Champion P. M. J. Am. Chem. Soc. 1999, 121, 376. [Google Scholar]
- Mak P. J.; Im S.-C.; Zhang H.; Waskell L. A.; Kincaid J. R. Biochemistry 2008, 47, 3950. [DOI] [PubMed] [Google Scholar]
- Mak P. J.; Gregory M. C.; Sligar S. G.; Kincaid J. R. Biochemistry 2013, 53, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.; Egawa T.; Wainwright L. M.; Poole R. K.; Yeh S.-R. J. Biol. Chem. 2007, 282, 13627. [DOI] [PubMed] [Google Scholar]
- Chartier F. J. M.; Couture M. J. Biol. Chem. 2007, 282, 20877. [DOI] [PubMed] [Google Scholar]
- Tosha T.; Kagawa N.; Arase M.; Waterman M. R.; Kitagawa T. J. Biol. Chem. 2008, 283, 3708. [DOI] [PubMed] [Google Scholar]
- Li D.; Kabir M.; Stuehr D. J.; Rousseau D. L.; Yeh S.-R. J. Am. Chem. Soc. 2007, 129, 6943. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.