

Abstract
The crystal structure of the flavin mononucleotide (FMN)-containing redox partner to P450cin, cindoxin (Cdx), has been determined to 1.3 Å resolution. The overall structure is similar to that of the FMN domain of human cytochrome P450 reductase. A Brownian dynamics–molecular dynamics docking method was used to produce a model of Cdx with its redox partner, P450cin. This Cdx–P450cin model highlights the potential importance of Cdx Tyr96 in bridging the FMN and heme cofactors as well P450cin Arg102 and Arg346. Each of the single-site Ala mutants exhibits ∼10% of the wild-type activity, thus demonstrating the importance of these residues for binding and/or electron transfer. In the well-studied P450cam system, redox partner binding stabilizes the open low-spin conformation of P450cam and greatly decreases the stability of the oxy complex. In sharp contrast, Cdx does not shift P450cin to a low-spin state, although the stability of oxy-P450cin is decreased 10-fold in the presence of Cdx. This indicates that Cdx may have a modest effect on the open–closed equilibrium in P450cin compared to that in P450cam. It has been postulated that part of the effector role of Pdx on P450cam is to promote a significant structural change that makes available a proton relay network involving Asp251 required for O2 activation. The structure around the corresponding Asp in P450cin, Asp241, provides a possible structural reason for why P450cin is less dependent on its redox partner for functionally important structural changes.
Cytochrome P450 (CYP) enzymes activate molecular dioxygen for hydroxylation of a diverse library of substrates, many of which are saturated alkanes with low reactivity. Almost all P450s must accept electrons from redox partners containing flavin mononucleotide (FMN) or FeS cofactors to initiate and complete monooxygenase activity. P450s have traditionally been divided into two classes depending on the type and organization of their redox partners. Class I enzymes consist of multiple redox partners where NAD(P)H-derived electrons are successively ferried to the P450 heme through a flavin adenine dinucleotide (FAD) flavoprotein and a [2Fe-2S] iron–sulfur cluster ferredoxin. Class I systems are predominately found in prokaryotes, although they also are present in vertebrate mitochondria where the cytosolic ferredoxin, adrenodoxin (Adx), shuttles electrons from membrane-tethered adrenodoxin reductase (Adr) to a number of membrane-bound CYPs. Most mammalian microsomal P450s fall into type II, which are present at the highest concentrations in the liver and adrenal glands, where they are responsible for xenobiotic clearance and steroid biogenesis. These P450s are membrane-bound and accept electrons from membrane-bound cytochrome P450 reductase (CPR) that contains FAD and FMN redox cofactors.
We now know that the class I versus class II definition is too limited, and various combinations have been discovered.1 The P450cin system2 is one of these outliers, having an FMN-containing flavodoxin redox partner instead of a 2Fe-2S ferredoxin. The sequence of cindoxin (Cdx) is 37% homologous to those of both the human CPR-FMN domain3 and bacterial fusion protein XplA.4 To date, there have been only three crystal structures of a P450 in a complex with its redox partner determined: the FMN-heme domains of P450BM3,5 the Adx–CYP11A1 complex,6 and the recent Pdx–P450cam structures.7,8 The Adx–CYP11A1 and P450BM3 FMN-heme crystal structures exhibited few differences compared to the isolated domains. However, a large structural change was observed in the Pdx–P450cam X-ray7 and nuclear magnetic resonance (NMR)8 structures. These two structures are consistent with data gathered over many years9 supporting the role of Pdx in promoting structural changes in P450cam necessary for O2 activation, electron transfer, and product formation. As a step toward elucidating the structural details of the Cdx–P450cin complex, we have determined the structure of Cdx and used computer simulations to develop a hypothetical model of the complex. This model has been used to guide mutagenesis studies. We also have conducted some initial experiments to determine if binding of Cdx to P450cin results in structural changes similar to what has been observed in the P450cam system.
Experimental Procedures
Cdx Six-His Cloning and Site-Directed Mutagenesis
For ease of purification, Cdx was cloned into the Pet22B vector as a C-terminal six-His-tagged fusion protein. Briefly, Cdx was amplified with primers containing NdeI (forward) and XhoI (reverse) restriction sites from the Cdx-PcWori construct, kindly provided by J. DeVoss. The gel-purified polymerase chain reaction (PCR) product was then ligated into Pet28 cut with the same enzymes. The resulting construct, Cdx-Pet28, encoded eight extra residues at the C-termini consisting of a Leu-Glu dipeptide followed by a six-His tag. PIPE mutagenesis10 utilizing overlapping complementary 5′ primer ends was used to amplify mutant constructs for all mutants. PCR proceeded with 28 cycles of denaturation at 95 °C followed by a combined extension and annealing step at 68 °C for 30 s.
Expression, Purification, and Characterization of Protein
Untagged PcWori-Cdx was expressed and purified as described previously.11 The Cdx-Pet28 plasmid was chemically transformed into DE3 BL21 Star (Invitrogen) and plated overnight at 37 °C on Luria-Bertani broth agar plates. A single colony was used to inoculate 100 mL of overnight 2×YT culture, and 5 mL of overnight culture was used to inoculate 1 L of Terrific Broth (7 L total). The cultures were grown at 225 rpm and 37 °C to a final OD600 of 1–1.5. The temperature was decreased to 30 °C and the rate of shaking to 125 rpm. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was not added because small scale trials showed much higher protein yields from noninduced cells. The cells were centrifuged 17–24 h later and pellets stored at −80 °C. Cell pellets were then thawed and resuspended in 300 mL of lysis buffer containing 50 mM sodium phosphate (pH 8), 50 mM KCl, 2 μg/mL apostatin, 2 μg/mL pepstatin, 20 μg/mL trypsin inhibitor, 0.1 mM PMSF, 10 mM MgCl2, 0.7 g of lysozyme, and 10 μg/mL DNase. Cells were spun on a magnetic stirrer for 1 h at 4 °C, sonicated, and centrifuged. The supernatant was loaded over nickel-NTA superflow resin (Clontech). Cdx does not bind very tightly, and the column was washed with 50 mM sodium phosphate (pH 8.0) containing 50 mM KCl and only 5 mM imidazole. Buffer containing 50 mM imidazole was used for elution. Yellow fractions were pooled and diluted 2-fold with sodium phosphate (pH 7.4), 50 mM KCl, and 2 mM DTT and loaded directly onto a DEAE-Sepharose FastFlow (GE Healthcare) drip column. The column was washed in the buffer described above with 200 mM KCl and eluted in 250 mM KCl. Fractions with an A415/A280 ratio of >0.3 were pooled and concentrated in an Amicon 10 kDa molecular mass cutoff filter unit (Millipore). Protein to be used for crystallography was then loaded onto a Superdex 26/60 (GE Healthcare) gel filtration column and run at a rate of 1 mL/min in 50 mM Tris buffer (pH 7.5). Samples consisting of a single band on sodium dodecyl sulfate–polyacrylamide gel electrophoresis and with an A415/A280 ratio of >0.35 were subsequently pooled and concentrated. Untagged Cdx was expressed and purified analogously with the following exceptions. IPTG was added when the temperature was reduced to 30 °C, and the lysate was loaded directly onto DEAE resin followed by a second identical DEAE purification.
Characterization of Cdx
The FMN content was determined by adding 20 μL of 10% SDS to 10 μM Cdx in 1 mL of 50 mM Tris buffer (pH 7.5) and measuring the concentration of free FMN using an ε450 of 12.2 mM–1 cm–1.12 Both the wild type and mutants had a UV–vis peak at 456 nm with a shoulder characteristic of FMN-bound enzymes. The A450/A357 absorbance ratio was 1.1–1.2, consistent with previous results.13 The peak at 456 nm was readily bleached upon reduction with excess dithionite in the wild-type and mutant enzyme, indicating reduction of the FMN cofactor. Our A280/A456 ratio was never above 0.38 in contrast to a previously reported value of 0.67.11 Repeated attempts to introduce excess FMN failed to increase this ratio. However, our calculated ε456 of 10.8 mM–1 cm–1 was identical to that calculated previously11,13 and gave a range of 95–99% FMN incorporation. All calculations were performed using the yellow, fully oxidized form of the enzyme.
Cloning, Expression, Purification, and Characterization of P450cin
Although P450cin is expressed robustly in a PcWori vector used previously,2,14 we often had problems performing PCR-based site-directed mutagenesis. In many instances, the N-terminal portion of the P450cin gene was deleted while the rest of the vector remained intact. We believe this may be due to a calculated propensity to form a hairpin between the N-terminus of P450cin and the flanking PcWori vector sequence predicted using the Integrated DNA Technology oligo analyzer tool. We therefore performed PCR to amplify P450cin with NdeI and HindIII restrictions sites using the forward primer GA GGT CAT ATG ACA AGT CTG TTC ACC ACC GCC and the reverse primer GGA AAG CTT TCA TTC CGA CAG TCG CTTG. Restriction sites are shown in bold. The PCR product was gel purified and ligated into the Pet23a vector (Invitrogen) cut with NdeI and HindIII restriction enzymes. A stop codon at the end of the coding sequence ensured an untagged enzyme. P450cin was expressed and purified as described previously14 with the following modifications. Pet23a-P450cin was transformed into chemically competent BL21 DE3 cells (Invitrogen) and induced at 22 °C with 1 mM IPTG. The heme precursor, δ-aminolevulinic acid (200 μM), was also added, and the rate of shaking was set to 120 rpm for 36 h. Wild-type and mutant P450cin had an A280/A392 ratio between 1.2 and 1.3 in the presence of excess substrate. Protein concentrations were calculated using a substrate-bound ε392 of 132 mM–1 cm–1.2 The heme content was estimated to be >95% using a standard alkaline pyridine hemochromogen assay15 and an εred557 of 34.7 mM–1 cm–1 for a type B heme.
Crystal Growth, Data Collection, and Refinement of a Cdx Surface Entropy Reduction Mutant
The Cdx surface entropy reduction triple mutant (E134A/E135A/E138A), here termed SER-Cdx, containing a C-terminal six-His tag fusion was purified as described above. Protein was concentrated to 1.5 mM in 50 mM Tris (pH 7.5). Crystals were set up in EasyXtal 15-well hanging drop crystal trays (Qiagen). The wells contained 250 μL of 100 mM Hepes (pH 7) and 1.7–2.1 M ammonium sulfate. The drops consisted of 1 μL of protein mixed with 1 μL of well solution. Crystals were bright yellow and hexagonally shaped, growing from yellow droplets over a period of 3–5 days. Intensity data were collected at the Stanford Synchrotron Radiation Lightsource (Menlo Park, CA) on beamline 7-1 using a Q315R detector. Intensities were integrated and scaled using HKL2000, and the space group was determined to be P3221 (Table 1). Initial phases were not readily determined using the CPR FMN domain (FMN-CPR) as a search model, or upon conversion to polyalanines despite the high level of sequence identity (37%). Therefore, we used Sculptor16 to provide a better phasing model for molecular replacement. A Cdx-FMN-CPR sequence alignment file was fed into Sculptor implemented in the Phenix package17 along with the Protein Data Bank (PDB) file of the CPR-FMN domain without the FMN cofactor (PDB entry 1B1C). Sculptor performs a series of steps, including pruning and morphing of particular model side chains as well as B factor smearing to increase the level of agreement between the model and the target. Thereafter, phases were easily determined by molecular replacement using Phaser.18 Individual coordinates were refined with anisotropic B factors for protein and the FMN cofactor, while water B factors were refined isotropically. A summary of data collection and refinement statistics is presented in Table 1.
Table 1. Crystallographic Data Processing and Refinement Statistics for Cdx E134A/E135A/E138Aa.
| radiation source | SSRL 7-1 |
| space group | P3221 |
| unit cell dimensions | |
| a, b, c (Å) | 82.107, 82.107, 37.055 |
| α, β, γ (deg) | γ = 120 |
| resolution range (Å) | 99–1.21 (1.23–1.21) |
| wavelength (Å) | 1.0 |
| total no. of observations | 210090 |
| no. of unique reflections | 43778 (2151) |
| completeness (%) | 98 (98.9) |
| Rsym | 0.044 (0.195) |
| ⟨I/σ⟩ | 18.8 (6.32) |
| redundancy | 4.8 (3.8) |
| B factor, Wilson plot (Å2) | 11.3 |
| no. of reflections used in refinement | 43762 |
| resolution range (Å) used in refinement | 32.7–1.21 |
| no. of protein atoms fit | 1105 |
| no. of heteroatoms fit | 30 |
| no. of waters fit | 116 |
| Rwork (%) | 13.5 |
| Rfree (%) | 15.1 |
| rmsd for bond lengths (Å) | 0.015 |
| rmsd for bond angles (deg) | 1.592 |
| no. of observations per parameter | 4.1 |
Values for the highest-resolution shell are given in parentheses.
Catalytic Activity As Determined by NADPH Turnover
NADPH turnover assays were performed at room temperature on a Cary 3E UV–visible spectrophotometer. An established reconstituted assay was used to measure NADPH consumption,11 in which cindoxin reductase is replaced by Escherichia coli flavodoxin reductase (Fldr). In this reconstituted system, NADPH-derived electrons are transferred from Fldr to Cdx and then to P450cin for 1,8-cineole hydroxylation. For the Y96L mutant and the SER-Cdx mutant, 10 μM Fldr, 4 μM Cdx, and 0.25 μM P450cin were mixed in 50 mM Tris buffer (pH 7.8) containing 1 mM 1,8-cineole. NADPH turnover was initiated by adding NADPH from a 20 mM stock solution at a final concentration of 400 μM. In all cases, control reactions were performed in cineole-free buffer. NADPH consumption was followed by the loss of absorbance at 340 nm and the rate determined by dividing the slope of the curve by 6.22 mM–1 cm–1.19 Rates were subtracted from those of control reactions conducted in buffer lacking cineole, and the kcat was determined by dividing by the micromolar amount of P450cin used. In assays in which the concentrations of SER-Cdx and Cdx Y96L mutants were increased 10-fold, control reactions also had a 10-fold increase in these mutants to subtract background NADPH consumption. The remaining mutants used in the NADPH activity screening assays had varying redox partner ratios in addition to the appropriate wild-type controls. The percent activity was determined by dividing the kcat of the mutant by the kcat of the wild type. Assays that were preformed three or more times are presented with error bars, while those performed fewer times are presented without error bars.
Stopped-Flow OxyP450cin Formation
A stock solution of 2 mM P450cin was reduced by being titrated in a 100 mM stock of sodium dithionite to a final concentration of 20 mM in anaerobic buffer [50 mM KPi (pH 7.5) and 100 μM cineole] in an anaerobic glovebox. After 30 min, the sample was passed over a PD10 desalting column (Bio-Rad) pre-equilibrated in the same buffer, giving a final stock concentration of 160–200 μM. The exact concentration and confirmation of the reduced form of P450cin (determined by a Soret peak shift to 415 nm) were determined by UV–vis spectroscopy of the diluted protein in an anaerobic cuvette using the ferrous substrate-bound extinction coefficient (ε411 = 106 cm–1 mM–1).2 We have previously shown that binding of oxygen to P450cin results in a shift in the absorbance peak from 411 nm, characteristic of the Fe(II) state, to 418 nm, characteristic of the Fe(II)–O2 state.14 Oxygen binding and the subsequent decay were followed by tracking the absorbance at 418 nm on an SX.18MV stopped-flow apparatus (Applied Photophysics) upon rapid mixing of reduced P450cin and oxygenated buffer [50 mM KPi (pH 7.5) and 100 μM cineole] with and without Cdx. The syringes were kept at 4 °C using a circulating water bath that cooled the syringes and reaction cell. Final concentrations upon mixing were 10 μM Cdx, 5 μM P450cin, and 100 μM cineole. The initial increase at 418 nm occurs within 50 ms and was evident for shorter collection times of 2 s but not as obvious for log base collection times of 10 and 20 s used to follow the decay of the oxy complex. Curve fitting was performed using IgorPro (WaveMetrics, Inc.) over a time interval of 100 ms to 10 s.
Stopped-Flow P450cin–Cdx Electron Transfer
Single-electron transfer between P450cin and Cdx was conducted using an SX.18MV stopped-flow apparatus (Applied Photophysics), in a manner similar to that described previously13 with a few modifications. Briefly, protein samples were degassed with repeated cycles of purging with argon followed by degassing under vacuum. Samples and buffer purged of oxygen were transferred to an anaerobic glovebox. Cdx (16 μM) was reduced in anaerobic buffer (50 mM potassium phosphate, 20 unirs/mL catalase, 20 units/mL glucose oxidase, and 1 mM glucose) via titration in a solution of dithionite that was calibrated against cytochrome c. The solution was intermittently placed in an anaerobic cuvette and the UV–vis absorbance measured. Titration was halted when the intensity of the 456 nm UV–vis peak was reduced to approximately 0.02, leaving ∼1.85 μM oxidized Cdx (ε456ox = 10.8 mM–1 cm–1).11 This gave a total starting Cdx concentration of ∼14 μM and ensured that there was no excess dithionite present. The anaerobic cuvette was transferred back to the anaerobic chamber and drawn into a gastight Hamilton syringe. P450cin was diluted to 10 μM in CO-saturated buffer, giving final concentrations of 7 μM Cdx and 5 μM P450cin upon mixing. It was assumed that electron transfer was rate-limiting, and a single-exponential model was used to fit the data for the wild type (>0.5 s), the triple glutamate mutant (0.5 s), and the Y96L mutant (2 s). Graphs and calculations were generated using IgorPro.
Fldr–Cdx Electron Transfer
The transfer of an electron from Fldr to Cdx was assayed by following the decrease in absorbance at 456 nm characteristic of a transition from oxidized FMN to the reduced hydroquinone form. FAD-bound enzymes show a much lower absorbance than FMN-bound enzymes, allowing us to use a concentration of Fldr with no appreciable absorbance. This also had the effect of sufficiently slowing the electron transfer rate for quantitation. The absorbance of NADPH with a peak at 340 nm also does not overlap with the flavin peak at 456 nm at the concentrations used in this assay. Excess Cdx (20 μM) was mixed with a small amount of Fldr (0.25 μM) in 50 mM Tris buffer (pH 7.8). NADPH (200 μM) was added to initiate the reaction, and the absorbance was measured at 456 nm for 1.5 min using a Carry 3E UV–visible spectrophotometer. The initial rates were estimated from the slope of the curves for the first 20 s in which the curves were linear. kET was estimated by using an ε456ox of 10.8 mM–1 cm–1 11 and dividing the slope of the curve by the concentration of the rate-limiting enzyme (Fldr).
Brownian Dynamics–MD Docking
We generated an atomistic model of the P450cin–Cdx complex using a combination of Brownian dynamics (BD) and molecular dynamics (MD) simulations. The BD simulations were used to dock Cdx to P450cin, and a subsequent MD simulation was used to refine a particular complex selected from the BD trajectories. The approach we used is similar to that of Motiejunas et al.20 except our BD simulations did not include restraints derived from biochemical data. The BD method that we employed models proteins as atomically detailed rigid bodies and generates translational and rotational displacements using the Ermak–McCammon algorithm.21 The SDA program22 was used to conduct the BD simulations. The SDA energy function,22 from which the forces that drive the BD simulations are derived, contains four contributions: (1) interactions of the charges on one protein with the electrostatic potential due to another protein, (2) the electrostatic desolvation energy due to the charges on one protein entering the low-dielectric cavity of another protein, (3) the nonpolar desolvation energy due to the burial of the solvent-accessible surface of atoms on one protein in the cavity of another protein, and the (4) soft-core repulsion interactions between atoms on different proteins. For the sake of computational efficiency, these energy contributions are precomputed on cubic grids. The electrostatic potentials are obtained by finite-difference solution of the nonlinear Poisson–Boltzmann equation using the APBS program package,22 in which the solvent is represented by a dielectric continuum. The solvent dielectric constant was set to 78 with an ionic strength corresponding to 50 mM NaCl.
Crystal structures of both the substrate-free and substrate-bound forms of Cdx (PDB entries 4L77 and 4FMX, respectively) were used in the BD simulations. Initial configurations were prepared by placing the larger of the two proteins, P450cin, at the origin, where it was kept fixed in place during the BD simulations, and the smaller protein, Cdx, at a random position 300 Å away. The relative diffusion constants for Cdx diffusion around the fixed P450cin were calculated from diffusion constants for Cdx and P450cin, which were estimated on the basis of the crystal structures using the Hypropro 10 package.23 Each BD trajectory was prolonged for 500 μs, or until Cdx traveled 400 Å from the origin, at which point the simulation was stopped. The simulation time step was changed from 20 to 1 ps when the separation between the two proteins was <20 Å. For each system (substrate-free and substrate-bound Cdx), 20000 BD trajectories were generated.
The BD trajectories that produced encounters between Cdx and P450cin were clustered to identify a candidate complex to be refined with an atomistic MD simulation. The clustering revealed that both substrate-free and substrate-bound Cdx formed complexes with P450cin, although the substrate–P450cin complex gave slightly better clustering, so we chose the open form of P450cin for further analysis. We selected the P450cin–Cdx complex that had the shortest substrate-free Cdx flavin–P450cin heme distance, as well as the longest lifetime among the 20000 BD trajectories.
The complex was immersed in a rectangular box of water molecules with initial dimensions of 127 Å × 118 Å × 149 Å. Following energy minimization, a 300 ps MD trajectory was generated at a constant temperature (300 K) and pressure (1 bar) using NAMD version 2.8.24 The lengths of bonds to hydrogen atoms were held fixed using the SHAKE25 and SETTLE algorithms.26 The equations of motion were integrated with a reversible, mutiple-step algorithm27 with a time step of 2 fs for electrostatic forces and 1 fs for bonded and short-range nonbonded forces. The smooth particle mesh Ewald method28 with a fourth-order interpolation scheme was used to calculate the electrostatic interactions, and the real-space contributions were truncated at 12 Å. Nosé–Hoover–Langevin pistons29 were used for pressure control, and a Langevin dynamics scheme was used for temperature control. The CHARMM22 force field30 was used for the proteins, the TIP3P force field31 for water, and the CGenFF force field32 for the FMN cofactor.
Results
Analysis and Comparison of the SER-Cdx Crystal Structure
Although there are various P450cin structures available, including substrate-bound, substrate-free, and nitric oxide-bound structures,14 the crystal structure of its redox partner, Cdx, has remained elusive. After exhaustive crystallization trials, we were not able to improve upon one-dimensional plates that gave unusable highly anisotropic X-ray data. We therefore took advantage of an approach used previously to promote crystal packing by reducing the surface entropy.33−35 We mutated Glu134, Glu135, and Glu138 to alanines as directed by the Surface Entropy Reduction Prediction server (SERp) at the University of California at Los Angeles (Los Angeles, CA).35 The triple mutant readily yielded hexagonal crystals that diffracted beyond 1.2 Å. The resolution was limited by our particular choice of detector and not by the diffracting power of the crystals. Crystals were highly ordered and well-packed and belongs to space group P3221 (Table 1).
The sequence of Cdx is similar to those of human FMN-CPR, bacterial nitric oxide synthase, and sulfite reductase FMN domains, as well as to those of other flavodoxins with levels of sequence identity of >39%. There are several highly conserved regions (Figure 1A), including the phosphate binding loop (P-loop), the tyrosine-containing loops (Y1 and Y2 loops), and the β-sheets proceeding these loops (β3 and β4). The SER-Cdx (SER stands for surface entropy reduction) crystal structure is shown in Figure 1B. The crystal structure reflects the sequence conservation with an α–β–α fold topology characteristic of all flavodoxins and FMN-CPR. Alternating parallel β-strands (β2−β1−β3−β4−β5 arrangement) form a five-layer central core with standard interstrand H-bonds between amide nitrogen and carbonyl carbons. The β-strands sweep toward the FMN binding domains, while the α-helices run in the opposite direction around the periphery of the protein. Helices 3 and 5 are completely α-helical, while helices 1, 2, and 4 are capped by shorter 310-helical segments.
Figure 1.

Crystal structure of cindoxin (Cdx). (A) Sequence alignment, including major elements of secondary structure. (B) Overall structure with the main elements of secondary structure labeled. (C) Close-up of the FMN binding pocket. (D) Stereo model showing key interactions discussed in the text.
As expected, the overall structure of SER-Cdx is similar to that of FMN-CPR with a few notable differences. In agreement with other bacterial flavodoxins, SER-Cdx does not have the extra N-terminal helix that is present in FMN-CPR. Also, the partitioning of β5 into segments “a” and “b” does not occur in Cdx like it does in FMN-CPR and most other bacterial flavodoxins. Instead, a long loop travels from helix 5 to a short β5 strand near the flavin. Some degree of conformational rigidity may be maintained in this loop as the Gln118 ε-nitrogen H-bonds with the Thr122 γ-oxygen. In some “long chain” flavodoxins, this loop is the site of an ∼20-amino acid insertion36 that is absent in Cdx.
FMN Binding Site
A high degree of sequence conservation is evident in the loops surrounding the FMN cofactor (Y1, Y2, and P) as well as many of the adjoining β-sheets and α-helices (Figure 1). This high degree of sequence conservation is undoubtedly a result of specific protein–flavin interactions in the FMN binding site depicted in panels B–D of Figure 1. The FMN cofactor is situated at the apex of five pairs of β-strands and α-helices, making contacts with the Y1 and Y2 loops. Three conserved aromatic residues, Tyr59, Tyr97, and Phe100, play a prominent role in FMN binding (Figure 1). The isoalloxazine ring is sandwiched between Y1 loop Tyr59 (bridging β3 and H3) and Y2 loop Tyr97 (bridging β4 and H4). Try59 sits on the re side with a 40° tilt with respect to the FMN plane, while Tyr97 resides on the si side, making coplanar π–π stacking interactions with the isoalloxazine ring. The analogous residues are important for FMN binding in rat CPR.37 Tyr97 is universally conserved as a Tyr/Phe and has been shown to influence the FMNsq/hq redox potential in Desulfovibrio vulgaris.38 The sequence conservation of the β4 sheet prior to the Y2 loop in Cdx is probably needed to properly position Tyr97 over the isoalloxazine ring. In particular, the highly conserved Thr58 H-bonds to the Ala103 amide nitrogen in H4 via its hydroxyl group (Figure 1C). The Thr58 carbonyl also H-bonds with FMN ribityl O2 while O3 points into the solvent.
Y2 loop Phe100 is also conserved as an aromatic Phe/Tyr and is close to the isoalloxazine ring but is too far for significant stacking or van der Waals contacts. Despite this, Paine et al. showed that mutations to Leu or Gln at this position result in a 50% decrease in FMN binding affinity.39 In the same study, these mutants were shown to have significant NMR backbone 1H and 15N chemical shifts for helix and loop residues near the FMN binding site, even after correcting for changes expected due to the loss of phenylalanine. These data suggest that the conserved Phe/Tyr residue plays an indirect role in FMN binding by positioning the Y2 loop for favorable interaction with the FMN ring system.
H-Bonds between the protein and the pyridine heteroatoms as well as ribityl oxygen atoms are consistent with what is observed in other flavodoxins as well as CPR-FMN.3 In particular, the pyridine N1 and O2 atoms H-bond with Y2 loop backbone atoms (Thr99 and Asn101) analogous to that of human CPR-FMN. However, the Thr99 side chain hydroxyl makes an additional H-bond with the pyridine O4 atom that cannot be accomplished in FMN-CPR because of the His at this position. The ribityl O4 atom H-bonds with side chains Asn13 (δ2 N atom) and Thr127 (hydroxyl group), which is similar to what has been seen in CPR-FMN.
The FMN phosphate binding loop (P-loop) connecting β1 and H1 shows a high degree of sequence conservation and features H-bonding between Thr9/Thr11 hydroxyls and phosphate oxygen atoms (Figure 1). FMN phosphate–protein interactions may also be stabilized by the amino end of the H1 dipole that terminates adjacent to the negatively charged phosphate groups as previously suggested for CPR-FMN.3 The high degree of conservation of the Y1 loop, Thr58, and the β4 sheet creates a loop-FMN structural relationship that is remarkably consistent when overlaid with other FMN-binding proteins. Much of this conservation is probably also important for establishing unique flavoprotein-FMN redox potentials as previously discussed.38,40,41
A superposition of FMN-CPR and Cdx shows an expected strong degree of structural homology between Cdx and FMN-CPR (Figure 2A). However, there are substantial deviations in the terminal helices (H1 and H5) as well as the β2–H2 loop and H2 helix of Cdx. Note that the helix numbering used for Cdx is different from that of FMN-CPR because of an extra N-terminal helix for the latter, as well as defining a short three-residue α-helix as a loop in Cdx. These are also regions with the lowest degree of sequence homology. The triple-glutamate mutant crystallizes with a buried hydrophobic patch between two molecules related by a 2-fold rotation (Figure 2B). This interaction is likely to be key in obtaining high-quality diffracting crystals as the E134A and E135A single mutants failed to crystallize. The E134A/E135A/E138A triple-mutant methyl groups point into a hydrophobic core created by crystal packing at the interface of the two molecules. E134A forms van der Waals contacts with the same residue on its symmetry-related partner. E135A is buried in the Ala16, Thr19, and Thr20 γ-carbons, while the E138A methyl sits close to the Thr20 γ-carbon. The wild-type enzyme would have exhibited abundant ionic repulsion under the same scenerio, making crystallization in this packing configuration highly unlikely.
Figure 2.

(A) Comparison between Cdx (green) and the FMN domain of mammalian cytochrome P450 reductase (blue). (B) Cdx showing the relationship between symmetry-related molecules in the crystal lattice.
Redox partner interactions are governed by electrostatic complementarity where the electronegative surface of the flavoprotein pairs with the electropositive portion of the P450. Cdx carries a high negative charge with a theoretical pI of 4.06 as a consequence of having 23 acidic residues and only seven basic amino acids. Furthermore, an APBS calculation predicts a charge of −13e after in silico mutation of the alanines back to glutamates.42 As with other P450 systems, the electronegative surface surrounding the exposed edge of the FMN complements the electropositive region on the proximal side of P450cin that is the closest approach of the heme to the surface. To develop a model of the Cdx–P450cin complex, we turned to a combined BD–MD simulation-based approach. This approach provided a structural model giving us insight into specific protein interactions and ultimately guided our structure–function mutagenesis studies.
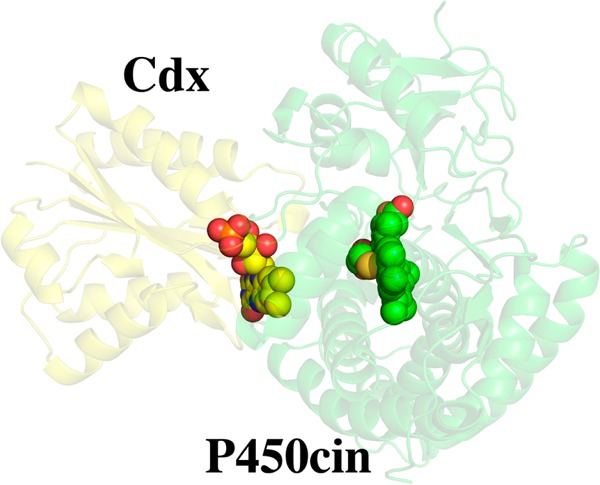
Cdx–P450cin Model
We used a docking approach combining BD and MD to generate an atomistic model of the Cdx–P450cin complex. As described in Experimental Procedures, initial docking was conducted using rigid-body BD, and the complex was refined by MD simulation, which samples the local side chain flexibility not permitted in the BD simulations. The model generated by BD and MD depicted in Figure 3 shows several hydrophobic and complementary electrostatic contacts that are likely important for complex formation. P450cin Arg346 and Cdx Asp94 form an interconnected network of charge–polar interactions with each other and with the FMN ligand (Figure 3B). Specifically, Arg346 forms a salt bridge with Asp94 and a charge–polar interaction with the FMN ribityl hydroxyl group. Asp94, in turn, completes the network by forming an amide nitrogen H-bond with the FMN ring O2 atom. Asp94 may also play a general role in P450 redox recognition as it is conserved across bacterial flavodoxins, although it is an Asn in human CPR. Cdx Tyr96 protrudes into a hydrophobic cleft created by P450cin Val105, Ala106, and Phe109 of helix C and loop CD, as well as Leu348. Tyr96 also forms a π–π stacking interaction with P450cin Arg102. In summary, the major interactions in our model include a combination of electrostatic interactions between Arg346 and the FMN hydroxyl group/Cdx Asp94, and hydrophobic interactions between P450cin and Cdx Tyr96.
Figure 3.

Brownian dynamics- and molecular dynamics-generated model of the P450cin (green)–Cdx (yellow) complex. The structure depicted is the final configuration from the 300 ps MD simulation. Panels A and B show the interface in two different orientations, while panel C shows the overall structure. (D) Enzyme activity of various mutants.
Testing the Hypothetical Model
We next generated a series of mutants to test our hypothetical model. A summary of the results is presented in Figure 3D and Table 2. In all, 15 single mutations near the FMN binding site failed to show a marked reduction in catalytic activity. This is reminiscent of a number of mutations made in FMN-CPR where mutating only Asp148 gave a significant reduction (70%) in activity.3 Despite these modest effects, the Cdx triple mutant used for crystallography reduced NADPH turnover activity by 92% of that of the wild type (the EEEA mutant in Figure 3D). This observation coupled with the fact that the single mutants, E134A, E135A, and E138A, do not significantly effect activity suggests that this region may provide a large electronegative patch for initial nonspecific binding rather than being responsible for particular interprotein electrostatic interactions.
Table 2. Enzyme Activity of Wild-Type Cdx and Various Mutants.
| cindoxin | NADPH turnover, kcat (min–1) |
|---|---|
| 10 μM Fldr, 1 μM Cdx, 0.5 μM P450cin | |
| wild type | 291.5 ± 7.5 |
| D131A | 131.3 ± 7.9 |
| D132A | 210.2 ± 40.8 |
| E134A | 173.2 ± 32.9 |
| E135A | 288.9 ± 3.9 |
| E15A | 103.9 ± 11.1 |
| 10 μM Fldr, 4 μM Cdx, 0.25 μM P450cin | |
| wild type | 847.4 ± 10.8 |
| E138A | 756.1 ± 39.8 |
| Y96L | 96.1 ± 2.8 |
| Y96L (40 μM) | 353 |
| 10 μM Fldr, 1 μM Cdx, 0.25 μM P450cin | |
| wild type | 153.6 ± 5.7 |
| E134A/E135A/E138A | 12.2 ± 7.9 |
| C17A | 72.5 ± 5.0 |
| 4 μM Fldr, 0.5 μM Cdx, 1 μM P450cin | |
| wild type | 65.0 |
| E10A | 65.3 |
| T11A | 90.4 |
| E61A | 42.1 |
| E63A | 95.8 |
| T98A | 74.4 |
| Y96F | 46.6 |
| S128A | 62.4 |
The involvement of Cdx Tyr96 and P450cin Arg102 and Arg346 in the modeled complex interface prompted us to examine each mutant individually. The Y96F mutant had near-wild-type NADPH turnover activity, suggesting that the Tyr hydroxyl group is not important for binding. We next tested the Y96L mutant. This mutant gives the characteristic flavoprotein spectrum with an A280/A456 ratio almost identical to that of the wild type, indicating that FMN binding is not compromised. The Y96L mutant, however, exhibits a marked decrease in kcat by >8-fold from 847 to 96 min–1. However, increasing the Y96L Cdx concentration to 40 μM (10-fold increase) increases activity to ∼40% of the wild-type activity with a kcat of 353 min–1. This concentration dependence on activity suggests that Y96L is involved in protein–protein binding but is probably not critical to electron transfer.
We also conducted stopped-flow kinetics to isolate the effect of the Y96L mutant on the first electron transfer step between Cdx and P450cin (Table 3). The hydroquinone species of Cdx transfers a single electron to P450cin to produce a ferrous iron center that in the presence of CO gives the characteristic 450 nm Soret band. This characteristic allows us to easily use stopped-flow spectroscopy to follow the first electron transfer to P450cin.13 Under these conditions, wild-type Cdx and the Y96L mutant give rates (k) of ∼6.6 s–1 (χ2 = 4.4 × 10–5) and ∼2.1 s–1 (χ2 = 1.0 × 10–4), respectively. The difference in the two plots in Figure 4A clearly shows that the first electron transfer is hampered in the Y96L mutant. However, this difference still cannot account for the 8-fold reduction seen in NADPH turnover assays. We therefore assayed the transfer of an electron from Fldr to Cdx to discern whether part of the reduction in activity was due to impaired recognition between these two proteins (Table 3). The nature of this assay dictates that Fldr is limiting, so we cannot directly compare rates between assays. However, we can discuss relative rates as shown in Figure 4B. Wild-type Cdx is reduced by Fldr at rate that is more than twice as fast (kET = 48.1 ± 5.7 min–1) as that of the Y96L mutant (kET = 18.8 2.9 ± min–1). The NADPH turnover assay utilizes a large excess of Fldr, so it is unlikely that moderate differences seen in the Fldr–Cdx assay (where Fldr is limiting) could explain the large decrease in the rate of NADPH consumption for this mutant. Because the effects of the Y96L mutant on the first electron transfer are moderate, it is likely that the second electron transfer step in which oxy-P450cin is reduced to give product is severely hampered. Even so, it is possible that even with excess Fldr the rate of reduction of Cdx remains close to 18.8 min–1, so that the large decrease in the rate of NADPH turnover in the Y96L mutant is due to the combined effect of both slow reduction and oxidation of the Cdx Y96L mutant.
Table 3. Kinetic Data Illustrating the Importance of Key Interface Residues Predicted from the Computer-Generated Model.
| Cdx–P450cin ET [k (s–1)] | Fldr–Cdx ET [kET (min–1)] | percent wild-type NADPH turnover | |
|---|---|---|---|
| wild type | 6.6 | 48.1 ± 5.7 | 100 |
| Cdx Y96L | 2.1 | 18.8 ± 2.9 | 11 |
| SER-Cdx | 9.3 | 19.9 ± 3.3 | 8 |
| P450cin R346A | – | – | 5 |
| P450cin R102A | – | – | 10 |
Figure 4.

(A) Rate of reduction of P450cin by wild-type and mutant Cdx. The P450cin and Cdx concentrations were 7 and 5 μM, respectively. (B) Rate of transfer of an electron from 0.125 μM flavodoxin reductase (Fldr) and 20 μM Cdx. (C) Rate of decay of oxy-P450cin.
The P450cin R102A and R346A mutants both showed substantial decreases in NADPH turnover activity with kcat values of 82.4 ± 13.7 and 44.0 ± 13.6 min–1, respectively. Decreases in wild-type activity of 90% (R102A) and nearly 95% (R346A) support our model in which Arg102 forms π–π base stacking interactions with Cdx Tyr96 and P450cin Arg346 ion pairs with Cdx Asp94. Increasing the concentration of R102A P450cin 10-fold restores activity to only ∼18% of that of the wild type, with a kcat value of 154.4 ± 12.8 min–1. Increasing the R346A mutant concentration 20-fold (5 μM) failed to give a measurable increase in the turnover activity for the Arg346 mutant.
The glutamates in the SER-Cdx mutant (E134, E136, and E138) used for crystallization are not near the proposed interaction interface. Given that the single-alanine mutants do not effect the activity, it was plausible that the total net charge is important for an initial nonspecific recognition but that a specific orientation is required for electron transfer. However, despite a large decrease in NADPH activity, the SER-Cdx mutant showed a rate constant similar to that of the wild type (k ∼ 9.3 s–1; χ2 = 4.0 × 10–5) for the first electron transfer (Figure 4A and Table 3). In addition, the rate of transfer of electrons from Fldr to Cdx is reduced by only 50% (Figure 4B and Table 3). This observation leaves open the possibility that the decreased kcat for NADPH turnover is due to a weakened ability to transfer the second electron to oxy-P450cin, but it also is possible that on–off rates important under steady state conditions have been altered.
Decay Kinetics of Oxy-P450cin
It has been known for some time that the binding of oxidized Pdx to oxy-P450cam dramatically decreases the stability of oxy-P450cam.43,44 This is very likely due to Pdx favoring the more open conformation of P450cam in which the O2 binding site is solvent-exposed.7,8 Given our docking results, we were interested in understanding if a similar phenomenon occurs when Cdx binds to P450cin. Oxy-P450cin is not nearly as stable as oxy-P450cam, but the decay of oxy-P450cin can readily be followed using stopped-flow spectroscopy (Figure 4C). The spontaneous decay of oxy-P450cin is biphasic with a kfast of 0.77 s–1 (t1/2 ∼ 0.9 s) and a kslow of 0.08 s–1 (t1/2 ∼ 8.5 s), with the slower rate accounting for ∼75% of the reaction. In the presence of oxidized Cdx, the rate of oxy-P450cin decay is monophasic with a k of 0.69 s–1 (t1/2 ∼ 1 s). Therefore, Cdx decreases the stability of oxy-P450cin ∼10-fold, while Pdx decreases the stability of oxy-P450cam 150-fold.44 We also found that the addition of Cdx to substrate-bound P450cin, which is fully high-spin with a Soret band at 392 nm, does not shift P450cin to the low-spin form with a Soret band at 418 nm. In sharp contrast, the addition of Pdx to substrate-bound P450cam shifts P450cam to the low-spin form.45 We now know that this change is due to an active site opening that allows water to enter and coordinate the heme iron. These results show that Cdx must induce some changes that result in destabilization of the oxy complex, but these changes are not large enough to allow water to enter the active site and coordinate the heme iron. Therefore, whatever structural changes Cdx may induce are modest compared to those resulting from the binding of Pdx to P450cam.
Discussion
As expected, the crystal structure of Cdx is very similar to those of related FMN proteins involved in P450 electron transfer. However, one unique feature of Cdx is Tyr96, which extends out into solution. This residue is a Thr in the FMN domain of mammalian CPR. The computer docking studies predict that Tyr96 will be positioned between the FMN of Cdx and heme of P450cin, and indeed, the mutagenesis studies show that Tyr96 is quite important. Although the Y96L mutant has a substantially reduced activity in steady state assays, the activity does increase with an increasing mutant Cdx concentration, indicating that the role of Tyr96 is primarily in binding and not electron transfer. The docking studies also indicate that P450cin Arg346 and Arg102 are important, and the mutagenesis supports these predictions. The effect of the Arg102 mutant is complicated by the fact that this Arg also interacts with a P450cin heme propionate. Arg346, however, extends out into solution, so the functional effects of mutating Arg346 are likely confined to Cdx binding and electron transfer. R346A exhibits only 5% of the wild-type activity, and the activity remains low even when the Cdx concentration is increased, indicating that Arg346 is involved in more than just binding of Cdx. Precisely what additional role Arg346 might play is open to speculation, but our computer-generated model has Arg346 closely approaching the Cdx FMN and Asp94. Therefore, it is possible that Arg346 plays an important role in electron transfer.
Interestingly, only the triple-Ala mutant affects the activity; each individual mutation (E134A, E135A, or E138A) has little effect. Our model also shows that these residues are not involved in direct binding. The role of this large electronegative patch could be to promote rapid formation of an electrostatically bound complex. This could be followed by Cdx sampling the surface of P450cin until it settles into a stable complex that is optimal for electron transfer. However, the triple mutant delivers the first electron at approximately the same rate as the wild type, so the decrease in steady state activity is likely due to changes in the second electron transfer step, reduction of oxy-P450cin to give the product.
Our results also indicate that Cdx does not play the same major effector role as Pdx does in the P450cam system. Cdx does not shift P450cin from high-spin to low-spin, so unlike binding of Pdx to P450cam, binding of Cdx is unlikely to cause a major closed-to-open conformational change. Nevertheless, Cdx does increase the rate of decay of the oxy-P450cin complex. Our docking model shows that Cdx very closely approaches the heme thiolate ligand, and such a close approach should affect the electronic properties of the thiolate ligand and coordinated ligands, as has been observed when Pdx binds to P450cam. In addition, the optimal computer docking model uses the substrate-free open conformation of P450cin. The open-to-closed transition involves not only opening of the substrate access channel but also structural changes at the Cdx docking site. Thus, Cdx binding may favor binding to a partially open state that destabilizes oxy-P450cin but is not sufficiently altered to give the large changes in the active site observed in the fully open P450cin structure.14 Subtle changes in the local hydrogen bonds to the thiolate ligand44 could also contribute to stability changes.
Finally, subtle differences between the P450cam and P450cin active sites may explain why the former requires a greater effector role with respect to its redox partner. A key amino acid in the proposed effector function of P450cam is Asp251 (Figure 5). This highly conserved Asp is critical to activity in P450cam, and the corresponding Asp in P450cin, Asp241, is also essential.46 It is believed that the essential role of Asp251 is the delivery of protons from the solvent to the iron-linked dioxygen.47 Proton shuttling is important because it is required for protonation of the distal O2 O atom to promote O–O bond heterolysis and formation of Compound I. However, Asp251 ion pairs with Lys178 and Arg189 in the closed conformation. This strong ion pair would likely prevent Asp251 from shuttling protons into the active site (Figure 5). The ion pairs are ruptured when Pdx binds, thus freeing Asp251 to serve its proton shuttling function. The structure of P450cin in this region is substantially different. Asp241 is not ion paired with any basic side chain but instead is connected to bulk solvent via a continuous H-bonded network involving His176 (Figure 5). As a result, the active site would not need to open to the extent observed in P450cam to shuttle solvent protons into the active site. P450cin redox partner binding may not play as significant a role in triggering the catalytic machinery required for proton-coupled electron transfer.
Figure 5.

Comparison between the conserved Asp region in P450cin and P450cam.
Acknowledgments
Simulations were conducted on the Greenplanet cluster at the University of California (Irvine, CA), which is supported by National Science Foundation Grant CHE-0840513. We thank Dr. Novelle Kimmich for the initial Cdx cloning work.
Glossary
Abbreviations
- CYP
cytochrome P450
- Cdx
cindoxin
- Pdr
putidaredoxin reductase
- Pdx
putidaredoxin
- Fldr
flavodoxin reductase
- NADPH
nicotinamide adenine dinucleotide phosphate
- BD
Brownian dynamics
- MD
molecular dynamics
- APBS
Adapative Possion-Boltzmann Solver
- rmsd
root-mean-square deviation.
Author Present Address
∥ Y.M.: Department of Pharmaceutical Chemistry, University of California, San Francisco, Box 280, 600 16th St., Room N572, San Francisco, CA 94158.
The authors declare no competing financial interest.
This work was supported by National Institutes of Health Grants GM33688 (T.L.P.) and GM86685 (D.J.T.) and an Institutional Cancer Biology Training Grant to S.A.H. (NIH/NCI T32CA009054).
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Base as entry 4OXX.
Funding Statement
National Institutes of Health, United States
References
- Hannemann F.; Bichet A.; Ewen K. M.; Bernhardt R. (2007) Cytochrome P450 systems: Biological variations of electron transport chains. Biochim. Biophys. Acta 1770, 330–344. [DOI] [PubMed] [Google Scholar]
- Hawkes D. B.; Adams G. W.; Burlingame A. L.; Ortiz de Montellano P. R.; De Voss J. J. (2002) Cytochrome P450cin (CYP176A): Isolation, expression, and characterization. J. Biol. Chem. 277, 27725–27732. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Modi S.; Smith G.; Paine M.; McDonagh P. D.; Wolf C. R.; Tew D.; Lian L. Y.; Roberts G. C.; Driessen H. P. (1999) Crystal structure of the FMN-binding domain of human cytochrome P450 reductase at 1.93 Å resolution. Protein Sci. 8, 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rylott E. L.; Jackson R. G.; Edwards J.; Womack G. L.; Seth-Smith H. M.; Rathbone D. A.; Strand S. E.; Bruce N. C. (2006) An explosive-degrading cytochrome P450 activity and its targeted application for the phytoremediation of RDX. Nat. Biotechnol. 24, 216–219. [DOI] [PubMed] [Google Scholar]
- Sevrioukova I. F.; Li H.; Zhang H.; Peterson J. A.; Poulos T. L. (1999) Structure of a cytochrome P450-redox partner electron-transfer complex. Proc. Natl. Acad. Sci. U.S.A. 96, 1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strushkevich N.; MacKenzie F.; Cherkesova T.; Grabovec I.; Usanov S.; Park H.-W. (2011) Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc. Natl. Acad. Sci. U.S.A. 108, 10139–10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S.; Li H.; Poulos T. L. (2013) Structural Basis for Effector Control and Redox Partner Recognition in Cytochrome P450. Science 340, 1227–1230. [DOI] [PubMed] [Google Scholar]
- Hiruma Y.; Hass M. A.; Kikui Y.; Liu W. M.; Olmez B.; Skinner S. P.; Blok A.; Kloosterman A.; Koteishi H.; Lohr F.; Schwalbe H.; Nojiri M.; Ubbink M. (2013) The Structure of the Cytochrome P450cam-Putidaredoxin Complex Determined by Paramagnetic NMR Spectroscopy and Crystallography. J. Mol. Biol. 425, 4353–4365. [DOI] [PubMed] [Google Scholar]
- Shimada H.; Nagano S.; Hori H.; Ishimura Y. (2001) Putidaredoxin-cytochrome P450cam interaction. J. Inorg. Biochem. 83, 255–260. [DOI] [PubMed] [Google Scholar]
- Klock H. E.; Lesley S. A. (2009) The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol. Biol. 498, 91–103. [DOI] [PubMed] [Google Scholar]
- Hawkes D. B.; Slessor K. E.; Bernhardt P. V.; De Voss J. J. (2010) Cloning, Expression and Purification of Cindoxin, an Unusual FMN-Containing Cytochrome P450 Redox Partner. ChemBioChem 11, 1107–1114. [DOI] [PubMed] [Google Scholar]
- Alessandro Aliverti B. C., and Maria A. (1999) Vanoni Flavoprotein Protocols, Springer Protocols, Vol. 131, Springer, Berlin. [Google Scholar]
- Kimmich N.; Das A.; Sevrioukova I.; Meharenna Y.; Sligar S. G.; Poulos T. L. (2007) Electron transfer between cytochrome P450cin and its FMN-containing redox partner, cindoxin. J. Biol. Chem. 282, 27006–27011. [DOI] [PubMed] [Google Scholar]
- Madrona Y.; Tripathi S.; Li H.; Poulos T. L. (2012) Crystal structures of substrate-free and nitrosyl cytochrome P450cin: Implications for O2 activation. Biochemistry 51, 6623–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry E. A.; Trumpower B. L. (1987) Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal. Biochem. 161, 1–15. [DOI] [PubMed] [Google Scholar]
- Bunkoczi G.; Read R. J. (2011) Improvement of molecular-replacement models with Sculptor. Acta Crystallogr. D67, 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmeyer H. U. (1975) [New values for the molar extinction coefficients of NADH and NADPH for the use in routine laboratories (author’s translation)]. Z. Klin. Chem. Klin. Biochem. 13, 507–508. [PubMed] [Google Scholar]
- Motiejunas D.; Gabdoulline R.; Wang T.; Feldman-Salit A.; Johann T.; Winn P. J.; Wade R. C. (2008) Protein-protein docking by simulating the process of association subject to biochemical constraints. Proteins 71, 1955–1969. [DOI] [PubMed] [Google Scholar]
- Ermak D. L.; McCammon J. A. (1978) Brownian dynamics with hydrodynamic interactions. J. Chem. Phys. 69, 1352–1360. [Google Scholar]
- Gabdoulline R. R.; Wade R. C. (1998) Brownian dynamics simulation of protein-protein diffusional encounter. Methods 14, 329–341. [DOI] [PubMed] [Google Scholar]
- Ortega A.; Amoros D.; Garcia de la Torre J. (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. (1977) Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. [Google Scholar]
- Miyamoto S.; Kollman P. A. (1992) SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water molecules. J. Comput. Chem. 13, 952–962. [Google Scholar]
- Grubmüller H.; Heller H.; Windemuth H.; Schulten K. (1991) Generalized Verlet Algorithm for Efficient Molecular Dynamics Simulations with Long-range Interactions. Mol. Simul. 1991, 121–142. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 98, 8577–8593. [Google Scholar]
- Martyna G. J.; Tobias D. J.; Klein M. L.; Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. (1995) Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 103, 4613–4621. [Google Scholar]
- MacKerell A. D. Jr. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; MacKerell A. D. Jr. (2010) CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force field. J. Comput. Chem. 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derewenda Z. S. (2004) Rational protein crystallization by mutational surface engineering. Structure 12, 529–535. [DOI] [PubMed] [Google Scholar]
- Derewenda Z. S.; Vekilov P. G. (2005) Entropy and surface engineering in protein crystallization. Acta Crystallogr. D62, 116–124. [DOI] [PubMed] [Google Scholar]
- Goldschmidt L.; Cooper D. R.; Derewenda Z. S.; Eisenberg D. (2007) Toward rational protein crystallization: A Web server for the design of crystallizable protein variants. Protein Sci. 16, 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho J. (2006) Flavodoxins: Sequence, folding, binding, function and beyond. Cell. Mol. Life Sci. 63, 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen A. L.; Porter T. D.; Wilson T. E.; Kasper C. B. (1989) Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J. Biol. Chem. 264, 7584–7589. [PubMed] [Google Scholar]
- Zhou Z.; Swenson R. P. (1996) The cumulative electrostatic effect of aromatic stacking interactions and the negative electrostatic environment of the flavin mononucleotide binding site is a major determinant of the reduction potential for the flavodoxin from Desulfovibrio vulgaris [Hildenborough]. Biochemistry 35, 15980–15988. [DOI] [PubMed] [Google Scholar]
- Paine M. J.; Ayivor S.; Munro A.; Tsan P.; Lian L. Y.; Roberts G. C.; Wolf C. R. (2001) Role of the conserved phenylalanine 181 of NADPH-cytochrome P450 oxidoreductase in FMN binding and catalytic activity. Biochemistry 40, 13439–13447. [DOI] [PubMed] [Google Scholar]
- Chang F. C.; Swenson R. P. (1997) Regulation of oxidation-reduction potentials through redox-linked ionization in the Y98H mutant of the Desulfovibrio vulgaris [Hildenborough] flavodoxin: Direct proton nuclear magnetic resonance spectroscopic evidence for the redox-dependent shift in the pKa of histidine-98. Biochemistry 36, 9013–9021. [DOI] [PubMed] [Google Scholar]
- Ludwig M. L.; Pattridge K. A.; Metzger A. L.; Dixon M. M.; Eren M.; Feng Y.; Swenson R. P. (1997) Control of oxidation-reduction potentials in flavodoxin from Clostridium beijerinckii: The role of conformation changes. Biochemistry 36, 1259–1280. [DOI] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. (2001) Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscomb J. D.; Sligar S. G.; Namtvedt M.; Gunsalus I. C. (1976) Autooxidation and hydroxylation reactions of oxygenated cytochrome P-450cam. J. Biol. Chem. 251, 1116–1124. [PubMed] [Google Scholar]
- Glascock M. C.; Ballou D. P.; Dawson J. H. (2005) Direct observation of a novel perturbed oxyferrous catalytic intermediate during reduced putidaredoxin-initiated turnover of cytochrome P-450-CAM: Probing the effector role of putidaredoxin in catalysis. J. Biol. Chem. 280, 42134–42141. [DOI] [PubMed] [Google Scholar]
- Unno M.; Christian J. F.; Benson D. E.; Gerber N. C.; Sligar S. G.; Champion P. M. (1997) Resonance Raman investigations of cytochrome P450cam complexed with putidaredoxin. J. Am. Chem. Soc. 119, 6614–6620. [Google Scholar]
- Stok J. E.; Yamada S.; Farlow A. J.; Slessor K. E.; De Voss J. J. (2013) Cytochrome P450(cin) (CYP176A1) D241N: Investigating the role of the conserved acid in the active site of cytochrome P450s. Biochim. Biophys. Acta 1834, 688–696. [DOI] [PubMed] [Google Scholar]
- Gerber N. C.; Sligar S. G. (1994) A role for Asp-251 in cytochrome P-450cam oxygen activation. J. Biol. Chem. 269, 4260–4266. [PubMed] [Google Scholar]