

Abstract
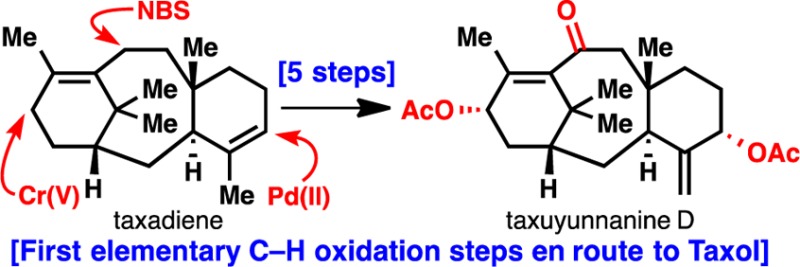
The first successful effort to replicate the beginning of the Taxol oxidase phase in the laboratory is reported, culminating in the total synthesis of taxuyunnanine D, itself a natural product. Through a combination of computational modeling, reagent screening, and oxidation sequence analysis, the first three of eight C–H oxidations (at the allylic sites corresponding to C-5, C-10, and C-13) required to reach Taxol from taxadiene were accomplished. This work lays a foundation for an eventual total synthesis of Taxol capable of delivering not only the natural product but also analogs inaccessible via bioengineering.
Only eight C–H oxygenation events separate the minimally oxidized hydrocarbon taxadiene (1) from Taxol (2).1 Known by enzymologists as the oxidase phase, the approximate order of oxidation is proposed to occur at C-5, C-10/C-13, C-9, C-7/C-2, and then C-1/C-4/C-20 (Figure 1A).2 Divergent oxidation pathways and acylation patterns lead to hundreds of members in the taxane family.3 Intriguingly, the first three of eight oxygenation events involve the formal activation of allylic C–H bonds. In an ongoing effort to replicate the two phases of Taxol biosynthesis in the laboratory, we focused on uncovering the innate reactivity of 1 so as to achieve a controlled oxidation of these positions (C-5, C-10, and C-13). In order to obtain material to launch these investigations, an artificial cyclase phase for taxanes was devised, culminating in the first enantioselective, scalable total synthesis of (+)-taxadiene (1).4 In fact, decagram-scale reproduction of that route was independently accomplished by Albany Molecular Research Inc. to provide “taxadieneone,”4 the precursor for 1.5 We present a systematic approach to mimicking the early stages of the Taxol oxidase phase and may form the basis of an eventual scalable total synthesis of 2. Extensive empirical studies into the fundamental reactivity of taxadiene (1), computational modeling, and reagent development were all enlisted to achieve the first total synthesis of (−)-taxuyunnanine D (3)6 in only five steps from 1.
Figure 1.
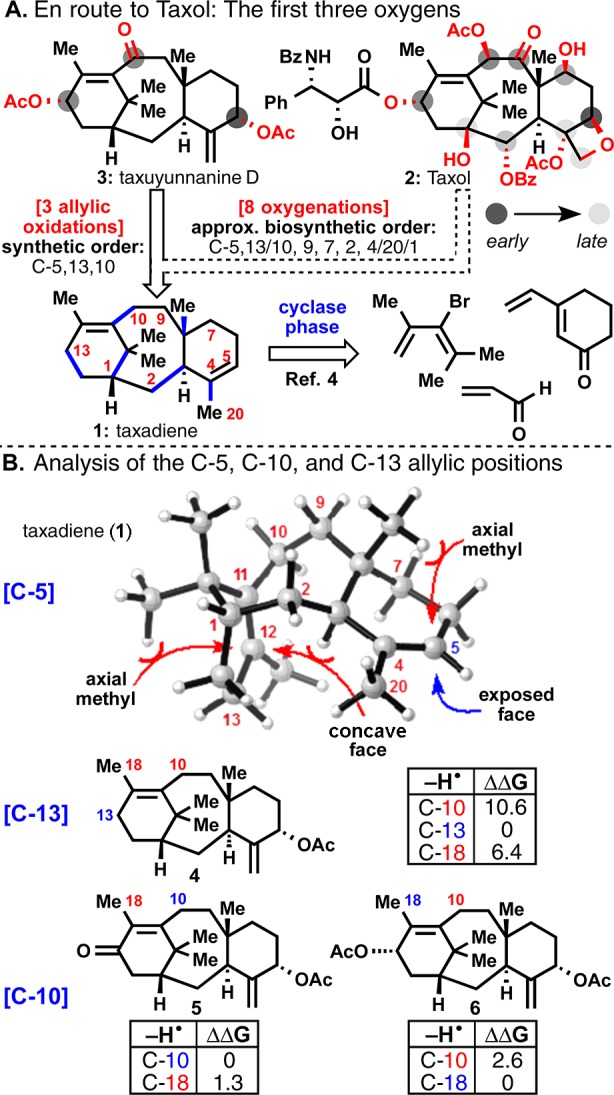
Planning stages for taxane oxidase phase. (A) Retrosynthetic rationale for the oxidase phase toward (−)-taxuyunnanine D (3). (B) Reasoning for the synthetic order of allylic oxidation: C-5 (steric hindrance, based on MM2-minimized model of 1),7,14 C-13 (most stable radical), and then C-10 (C-13 ketone stabilizes C-10 radical). ΔΔG values are relative energies of indicated allylic radical species in kcal/mol (see Supporting Information, SI).
As a doubly unsaturated and strained hydrocarbon, taxadiene (1) is “spring loaded” for oxidation. Thus, numerous possible oxidative pathways are conceivable. Presumably due to the relative ease by which allylic C–H bonds can be broken, C-5, C-10, and C-13 appear to be oxidized first in the biosynthetic pathway.2 The Croteau/Williams team’s enzymatic studies7 demonstrated that C-5 of 1 would likely be oxidized first in both the biosynthesis and in the laboratory, and this might be rationalized by relative steric hindrance around the C-11(12) and C-4(5) olefins (Figure 1B). After C-5 oxidation, the question of whether C-10 or C-13 would be more easily oxidized is complicated by potentially competitive C-18 oxidation. The relative C–H bond strength for radical oxidation reactions can be estimated by comparing the stability of the radical species resulting from homolytic C–H bond cleavage. DFT calculations (using Gaussian098 and UB3LYP/6-31+G(d,p)9 level of theory) modeling such radical species indicate a radical centered on C-13 of 4 (ΔG for C-13 hydrogen atom abstraction, relative to that on C-10 and C-18, is 0 kcal/mol) would be more stable than that centered on C-10 or C-18 (ΔΔG = 10.6 and 6.4 kcal/mol, respectively). This might be rationalized by the additional strain of converting the sp3-hybridized C-10 methylene to an sp2-hybridized methine, in the already strained eight-membered ring. Such strain might even be enough to outweigh any preference for a secondary allylic radical over a primary one. After C-13 oxidation, there are only two probable carbon atoms subject to allylic oxidation, and DFT calculations suggest that a C-13 ketone might help drive selectivity to C-10 oxidation if this substrate were to undergo a radical-based oxidation (compare 5 and 6). With this basic reasoning in mind, extensive empirical investigations began.
Over the course of hundreds of experiments, five different mechanism-based categories of allylic oxidation10 were evaluated for each of the consecutive targeted oxidations (Table 1, see also SI for an extended summary): (1) π-allyl metal complex oxidation, (2) stoichiometric transition-metal-mediated single-electron oxidation, (3) radical halogenation, (4) transition-metal-catalyzed hydrogen abstraction, and (5) pericyclic oxidation. The screening strategy thus required that each category of oxidant be screened for each allylic C–H bond.11 In accord with initial predictions, the C-5 position of 1 (prepared as described in ref (4)) was exclusively oxidized first using Pd-catalyzed allylic acetoxylation under conditions pioneered by Åkermark and Bäckvall.12 All other attempts at C-5 oxidation were unsuccessful, leading to either decomposition or indecipherable product mixtures. With a solution to the C-5 oxidation in hand, this product was then evaluated under the five oxidant categories mentioned above. With halogenation conditions, the electron-rich C-11(12) olefin reacted preferentially to allylic positions. Pericyclic oxidations such as SeO2 and 1O2 allylic oxidation reactions were selective for C-18 oxidation, and catalytic metal-mediated oxidation only gave degradation. A chromium-based oxidant was identified to selectively oxidize C-13 (vide infra). Although transition-metal oxidants such as Cr(VI) oxides are known to effect C-13 oxidation,13 this has never been reported on such minimally oxidized taxanes. Finally, the product containing both C-5 and C-13 oxidation was extensively screened across the same panel in order to functionalize C-10. These efforts were complicated by the additional option of having C-13 at either the ketone or alcohol oxidation state, so the above oxidation panel was performed using both substrates 5 and 6 (see Figure 1B). A selective C-10 oxidation could only be identified when C-13 was a ketone, and the solution was to use radical halogenation conditions. As with the other allylic positions, only one category gave useful products. These extensive trials demonstrated the singularity of each of these allylic positions and laid the groundwork to achieve the total synthesis of a taxane expressing oxygenation at only the C-5, C-10, and C-13 positions: taxuyunnanine D (3).
Table 1. Summary of Allylic C–H Oxidation Panel Used to Develop Selective Oxidations of 1.

The synthesis, outlined in Scheme 1, commences with the C-5 oxidation discussed above (Step 1). A chemical counterpart for the enzymatic C-5 oxidation of Δ4(5)-taxadiene is without precedent.14 Thus, palladium-catalyzed acetoxylation with Pd(OAc)2, benzoquinone, and acetic acid afforded the C-5α acetate 4 in 35% yield with no other isomers observed. After extensive screening of solvents, additives, and co-oxidants, it was discovered that addition of four equivalents of anisole increased the yield from ∼35% to 49%. The origin of this improvement is currently under investigation, but it was qualitatively observed that less palladium black was produced when anisole is employed, implying that anisole may facilitate reoxidation of Pd(0) to Pd(II) in this case.
Scheme 1. Synthesis of (−)-Taxuyunnanine D (3) from (+)-Taxadiene (1).

(a) Pd(OAc)2, p-benzoquinone (BQ), anisole, acetic acid, 50°C; (b) Cr(V) reagent 9, MnO2, 15-crown-5, trifluorotoluene, 80°C; (c) NBS, benzoyl peroxide, CCl4, reflux; then AgOTf, Et3SiOH, toluene, 0°C; (d) DIBAL, toluene, −78°C; then MeOH, 0°C; then Ac2O, DMAP, Et3N; (e) IBX, DMSO, 80°C.
Step 2, the oxygenation of C-13, proved to be quite challenging. C-13 allylic oxidation is known using chromium(VI) oxide 3,5-dimethylpyrazole complex13a (CrO3·DMP) or pyridinium chlorochromate13b (PCC), but both transformations used substrates containing oxidation at C-10. Subjecting acetate 4 to both of these conditions (Scheme 2A) afforded enone 5 in about 30% yield along with oxidative cleavage (7) and epoxidation (8) of the C-11(12) olefin as byproducts. An intriguing report15 showed that hydroxyacid-bound Cr(V) complexes are much less reactive toward 1,2-diol oxidative cleavage than Cr(VI) reagents, so 9 was employed in an effort to reduce alternative oxidation pathways. Reagent 9, which has only rarely been employed in synthesis,15,16 can be obtained simply by mixing hydroxyacid 16 with sodium dichromate (Scheme 2B).17d Cr(V) complexes such as 9 have been observed and isolated since the 1970s,17 but nothing has been reported concerning their reactivity with olefins to effect allylic oxidation or epoxidation. Treating acetate 4 with 30 equiv of Cr(V) reagent 9 afforded enone 5 in 53% yield, with byproduct enone 10 produced in 35% yield (Scheme 1). It was later determined that using excess MnO2 as a co-oxidant reduced the number of equivalents of Cr(V) reagent 9 to 5 without affecting the yield. Interestingly, enone 10 expresses an oxidation pattern resembling other known taxanes.3
Scheme 2. Cr(V) Complex 9 Continued Studies.
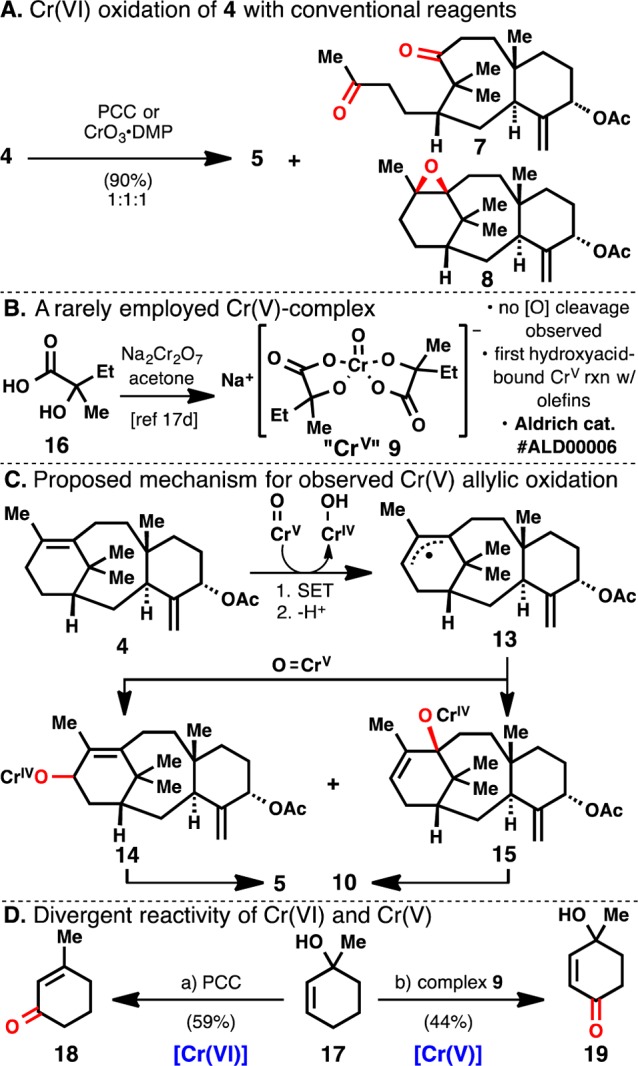
(A) Byproducts 7 and 8 from Cr(VI) oxidation. (B) Synthesis of 9. (C) Proposed mechanism for the formation of 10 with Cr(V) reagent 9. (D) Orthogonal reactivity of Cr(VI) and Cr(V) reagents. (a) PCC, NaOAc, DCM, 0°C; (b) Cr(V) reagent 9, MnO2, 15-crown-5, trifluorotoluene, 80°C.
The final oxidation step (Step 3) involved allylic oxidation of C-10, another unknown chemical oxidation of taxanes. As discussed above, only radical-based oxidation was capable of functionalizing this position. Thus, allylic bromination of enone 5 with NBS and benzoyl peroxide cleanly gave bromination at C-10 and, if more than 1 equiv of NBS was used, C-18. Submitting 5α,13α-diacetate 6 to identical conditions first brominates C-18 and then C-10, indicating the electron-withdrawing C-13 ketone stabilizes a C-10 radical (in accord with our predictions, see Figure 1B). After bromination, silver-mediated substitution using a silanol nucleophile18 could be accomplished in an overall one-pot procedure using AgOTf and triethylsilanol to afford silyl ether 11 in 80% yield. Rigorously dry conditions are required; otherwise, a mixture of 11 and the C-10β alcohol was observed.
Following the selective allylic oxidations at C-5, C-13, and C-10, completion of the synthesis of taxuyunnanine D (3) required only two steps. A one-pot reduction/acylation of the C-13 ketone (DIBAL, then MeOH quench, then Ac2O) delivered diacetate 12 in 80% yield. Finally, treatment of 12 with IBX in DMSO to remove the silyl group and oxidize the resulting allylic alcohol in one pot19 produced (−)-taxuyunnanine D (3) in 88% yield.
The uniqueness and exceptional reactivity of Cr(V) reagent 9 toward allylic oxidation (Step 2) warrants further discussion. The difference in product distributions in the Cr(VI) and Cr(V) allylic oxidations is clearly suggestive of different mechanistic pathways. Proposals for Cr(V) oxidation of dialkyl sulfides suggest that chromium initiates oxidation through a single-electron transfer (SET).16 Along these lines, Cr(V) reagent 9 might oxidize 4 to an allylic radical 13 through SET and deprotonation (Scheme 2C). Recombination of 13 with another chromium-oxo species in either of the two carbon atoms bearing radical character gives two possible regioisomeric intermediates 14 and 15. The former would furnish desired enone 5, whereas the latter is presumably unable to undergo oxidative rearrangement. Hydrolysis of the O–Cr bond in 15 followed by another allylic oxidation would yield enone 10. The hypothesis that Cr(V) reagent 9 cannot mediate the oxidative rearrangement of a tertiary allylic alcohol was central to this mechanistic proposal, so it was tested on a simpler system. Thus, tertiary allylic alcohol 17 was subjected to both PCC and Cr(V) conditions (Scheme 2D). PCC effected the well-known Babler–Dauben oxidative rearrangement20 to give enone 18, but Cr(V) gave direct allylic oxidation product 19. Neither reaction showed any trace of the other’s product in their respective crude NMR spectrum (see SI). This implies that SET occurs faster than binding to the alcohol to effect oxidative rearrangement or binding to an alcohol is simply unproductive toward the Babler–Dauben reaction. This might be explained by a different oxidation potential required for SET, unavailable binding sites on complex 9, or incorrect geometry for allylic transposition if an alkoxide complex is formed. This reactivity with tertiary allylic alcohols appears to be unknown in the literature and should prove useful for such transformations.
The study of Taxol’s biosynthesis is still an area of immense interest from a bioengineering standpoint.21 The focus of our work is to understand and learn how the conversion of a minimally oxidized taxane to Taxol might be achieved entirely in a laboratory setting. This paper has outlined the first fundamental steps toward achieving this goal in a systematic fashion using a two-phase approach.22 It appears that the wisdom of biosynthesis correlates to laboratory findings23 in that the allylic positions of 1 (C-5, C-13, and C-10) are innately primed, matching the biosynthetic order, for early oxidation en route to Taxol. This realization, however, required extensive exploration using a panel of complementary allylic oxidation methods (see SI) culminating in the first total synthesis of 3. The tools, lessons, and insights reported herein lay the necessary groundwork to access higher oxidized taxane natural products and analogs currently inaccessible through biosynthesis.
Acknowledgments
The authors thank S. J. McKerrall for computational analyses, D.-H. Huang and L. Pasternack for NMR spectroscopic assistance, J. A. Golen and A. L. Rheingold for X-ray crystallographic analysis, I. Prochnow and M. Giambrone for valuable technical assistance, and Y. Ishihara for insightful discussions. Financial support for this work was provided by NIH/NIGMS(GM-097444) and Ministerio de Educación y Ciencia-Fulbright programme (postdoctoral support for A.M.).
Supporting Information Available
Experimental procedures, analytical data, and full ref (8). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Arrhenius Laboratory, Department of Organic Chemistry, Stockholm University, 106 91 Stockholm, Sweden
Author Contributions
‡ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ishihara Y.; Baran P. S. Synlett 2010, 1733. [Google Scholar]
- Croteau R.; Ketchum R. E. B.; Long R. M.; Kaspera R.; Wildung M. R. Phytochem. Rev. 2006, 5, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-F.; Shi Q.-W.; Dong M.; Kiyota H.; Gu Y.-C.; Cong B. Chem. Rev. 2011, 111, 7652. [DOI] [PubMed] [Google Scholar]
- Mendoza A.; Ishihara Y.; Baran P. S. Nat. Chem. 2012, 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasutsky S. G.; Jacobo S. H.; Tweedie S. R.; Krishnamoorthy R. Personal communication from AMRI, a manuscript describing this work is currently being prepared.
- Zhang H.; Takeda Y.; Minami Y.; Yoshida K.; Matsumoto T.; Mu O.; Sun H. Chem. Lett. 1994, 957. [Google Scholar]
- Hefner J.; Rubenstein S. M.; Ketchum R. E. B.; Gibson D. M.; Williams R. M.; Croteau R. Chem. Biol. 1996, 3, 479. [DOI] [PubMed] [Google Scholar]
- Frisch M. J., et al. Gaussian09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2009.
- Becke A. D. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]
- Nakamura A.; Nakada M. Synthesis 2013, 45, 1421. [Google Scholar]
- For a recent example of kilogram-scale allylic oxidation in the synthesis of a terpene, see:Zhong Y. L.; Gauthier D. R.; Shi Y. J.; McLaughlin M.; Chung J. Y. L.; Dagneau P.; Marcune B.; Krska S. W.; Ball R. G.; Reamer R. A.; Yasuda N. J. Org. Chem. 2012, 77, 3297. [DOI] [PubMed] [Google Scholar]
- a Heumann A.; Åkermark B. Angew. Chem., Int. Ed. 1984, 23, 453. [Google Scholar]; b Bäckvall J. E.; Nordberg R. E. J. Am. Chem. Soc. 1981, 103, 4959. [Google Scholar]; c Heumann A.; Reglier M.; Waegell B. Angew. Chem., Int. Ed. 1982, 21, 366. [Google Scholar]; d Grennberg H.; Bäckvall J. E. Chem.—Eur. J. 1998, 4, 1083. [Google Scholar]
- a Kende A. S.; Johnson S.; Sanfilippo P.; Hodges J. C.; Jungheim L. N. J. Am. Chem. Soc. 1986, 108, 3513. [Google Scholar]; b Nicolaou K. C.; Nantermet P. G.; Ueno H.; Guy R. K.; Couladouros E. A.; Sorensen E. J. J. Am. Chem. Soc. 1995, 117, 624. [Google Scholar]
- SeO2 oxidation of Δ4(5)-taxadiene gives a mixture of products:; Huang Q. L.; Pennington J. D.; Williams H. J.; Scott A. I. Synth. Commun. 2006, 36, 2577. [Google Scholar]; For clean SeO2 oxidation of Δ4(20)-taxadiene, see:; Rubenstein S. M.; Vazquez A.; Sanz-Cervera J. F.; Williams R. M. J. Labelled Compd. Rad. 2000, 43, 481. [Google Scholar]
- Krumpolc M.; Roček J. Inorg. Chem. 1985, 24, 617. [Google Scholar]
- Ganesan T. K.; Rajagopal S.; Bharathy J. B. Tetrahedron 2000, 56, 5885. [Google Scholar]
- a Srinivas. V.; Roček J. J. Am. Chem. Soc. 1974, 96, 127. [Google Scholar]; b Krumpolc M.; Roček J. J. Am. Chem. Soc. 1976, 98, 872. [Google Scholar]; c Krumpolc M.; Deboer B. G.; Roček J. J. Am. Chem. Soc. 1978, 100, 145. [Google Scholar]; d Krumpolc M.; Roček J. J. Am. Chem. Soc. 1979, 101, 3206. [Google Scholar]
- Keck G. E.; Poudel Y. B.; Cummins T. J.; Rudra A.; Covel J. A. J. Am. Chem. Soc. 2011, 133, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. K.; Huang J. H.; Shen X.; Hu Q.; Tang C. J.; Li L. Org. Lett. 2002, 4, 2141. [DOI] [PubMed] [Google Scholar]
- a Babler J. H.; Coghlan M. J. Synth. Commun. 1976, 6, 469. [Google Scholar]; b Dauben W. G.; Michno D. M. J. Org. Chem. 1977, 42, 682. [Google Scholar]
- Ajikumar P. K.; Xiao W. H.; Tyo K. E. J.; Wang Y.; Simeon F.; Leonard E.; Mucha O.; Phon T. H.; Pfeifer B.; Stephanopoulos G. Science 2010, 330, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For other two-phase approaches to terpene synthesis, see:; a Renata H.; Zhou Q.; Baran P. S. Science 2013, 339, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cherney E. C.; Green J. C.; Baran P. S. Angew. Chem., Int. Ed. 2013, 52, 9019. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jørgensen L.; McKerrall S. J.; Kuttruff C. A.; Ungeheuer F.; Felding J.; Baran P. S. Science 2013, 341, 878. [DOI] [PubMed] [Google Scholar]; d Chen K.; Baran P. S. Nature 2009, 459, 824. [DOI] [PubMed] [Google Scholar]; g Foo K.; Usui I.; Götz D. C. G.; Werner E. W.; Holte D.; Baran P. S. Angew. Chem., Int. Ed. 2012, 51, 11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschenmoser has described a similar phenomenon in the corrin space, see:Eschenmoser A. Angew. Chem., Int. Ed. 1988, 27, 5. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.