

Abstract
Synthetically useful radical thiol–ene reactions can be initiated by visible light irradiation in the presence of transition metal polypyridyl photocatalysts. The success of this method relies upon the use of p-toluidine as an essential additive. Using these conditions, high-yielding thiol–ene reactions of cysteine-containing biomolecules can be accomplished using biocompatibile wavelengths of visible light, under aqueous conditions, and with the thiol component as the limiting reagent. We present evidence that p-toluidine serves as a redox mediator that is capable of catalyzing the otherwise inefficient photooxidation of thiols to the key thiyl radical intermediate. Thus, we show that co-catalytic oxidants can be important in the design of synthetic reactions involving visible light photoredox catalysis.
Introduction
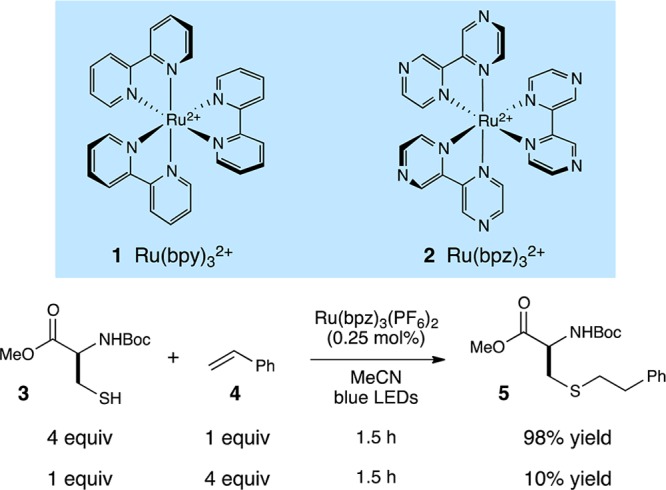
The ability of transition metal polypyridyl complexes (e.g., Scheme 1, 1 and 2) to efficiently convert visible light energy to electrochemical potential has long been appreciated in applications ranging from solar energy conversion to the study of biological electron transport phenomena.1 The design of synthetically useful chemical transformations involving these photocatalysts, however, has only recently begun to attract the attention of organic chemists.2 Interest in this approach arises from a number of advantages of visible light transition metal photocatalysis, including the accessibility of a diverse range of reactive intermediates under mild conditions and the ability to use convenient household visible light sources instead of specialized UV photochemical equipment. One of the most important features of this strategy has been the ability to tune the relative electrochemical potentials of the substrate and photocatalyst. This has been achieved by introducing Lewis acidic co-catalysts that can increase the propensity of an electrophilic substrate toward one-electron reduction3 and by utilizing ligand-modified polypyridyl complexes with varying redox potentials.4 Thus, the factors governing the thermodynamics of organic substrate activation in visible light photoredox catalysis have become well understood and are commonly manipulated in the design of photocatalytic reactions. Strategies to control the kinetics of the key electron-transfer activation events, on the other hand, have not yet received extensive investigation. Herein, we report a new strategy for the design of co-catalytic photochemcial reactions in which a soluble redox mediator is used to accelerate a thermodynamically feasible but kinetically unfavorable photoredox activation step.
Scheme 1. Photocatalytic Radical Thiol–Ene Reaction9.
In the past several years, our laboratory has investigated the ability of Ru(bpy)32+ and its derivatives to catalyze organic transformations initiated by one-electron photooxidations of electron-rich alkenes.4,5 In an effort to expand this concept to reactions of heteroatomic functional groups, we became interested in developing a thiol–ene ″click″ reaction6 initiated by visible light irradiation. The diversity of suitable coupling partners, the high degree of compatibility with common organic functional groups, and the facility with which this reaction produces covalent linkages between complex coupling partners has resulted in numerous applications in materials chemistry and in bioconjugation chemistry.7
Generally, these reactions employ radical initiators that are activated thermally or by irradiating with UV light. A radical thiol–ene reaction photoinitiated by Ru(bpy)32+ would provide a useful complement to the most common initiation methods, particularly for applications involving functionalization of cysteine-containing peptides, for a number of reasons.8 First, while natural biopolymers can undergo a variety of photoinduced reactions upon exposure to UV light, they are transparent to the long wavelengths of visible light (ca. 450 nm) involved in photoexcitation of Ru polypyridyl complexes and will thus be resistant to visible light-induced photochemical decomposition. Second, while traditional radical photoinitiators for radical thiol–ene reactions are expended after one initiation, we have been successful in designing radical chain processes that involve photocatalyst-regenerating turnover steps, which allows for extremely low catalyst loadings. Finally, the biscationic Ru(II) photocatalysts are freely water-soluble and should enable the solution-phase bioconjugation of native cysteine-containing polypeptides and proteins without the use of organic cosolvents.
We recently reported a preliminary investigation of photocatalytic radical thiol–ene reactions,9 in which we found that Ru(bpz)32+ (2) can serve as an effective photocatalyst for visible light initiated couplings of a wide range of thiols and alkenes (Scheme 1).9 On the one hand, the results of these experiments were promising; N-Boc cysteine methyl ester (3) participates in a high-yielding thiol–ene coupling with styrene (4) at quite low photocatalyst loadings (0.25 mol %). The conditions that proved to be optimal in these initial studies, however, are not suitable for functionalization of higher-order cysteine-containing biomolecules. In particular, we found that the reaction required rather high concentrations (0.2 M in olefin) and a large excess (0.8 M) of the thiol component. Experiments using the thiol as the limiting reagent resulted in dramatically lower, synthetically impractical yields.
The need for very high concentrations of thiol was initially quite surprising, as the oxidation potential of the photoexcited Ru*(bpz)32+ complex (+1.4 V vs SCE)10 is significantly more positive than that of the thiol substrates utilized in our study (ca. +0.50 V for benzyl thiol);11 the one-electron photooxidation of alkyl thiols by Ru*(bpz)32+ should therefore be thermodynamically quite favorable. On the other hand, prior studies of the photooxidation of thiols by ruthenium polypyridyl complexes have demonstrated that this process is inefficient; Matsuda found that the fluorescence of Ru*(bpy)32+ is not significantly quenched in the presence of aliphatic thiols.12 Thus, while the direct photooxidation of thiols by ruthenium polylpyridyl complexes is thermodynamically very favorable, it appears that a significant kinetic barrier to this process renders the overall photocatalytic thiol–ene coupling quite inefficient.
With the eventual goal of developing a useful method for the visible light initiated photocatalytic modification of biologically relevant cysteine-containing molecules, we wondered if we might learn how to overcome this kinetic barrier and develop an efficient protocol for photocatalytic radical thiol–ene couplings that can be conducted in water with the thiol as the limiting reagent.
Results and Discussion
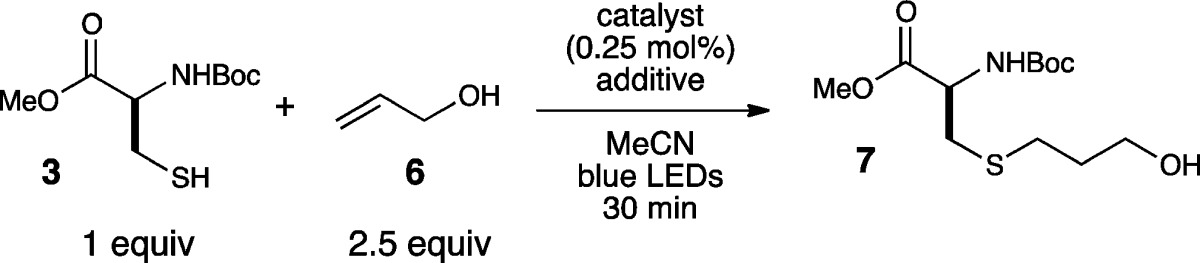
We began our investigation by examining the radical thiol–ene coupling of N-Boc cysteine methyl ester (3) with allyl alcohol (6) under a variety of conditions (Table 1). Consistent with our previously reported results, we found that reactions using the thiol as the limiting reagent were impractically sluggish (entry 1). Our efforts to increase the rate of the reaction focused initially on the use of basic additives; we hoped that deprotonation of the thiol would afford a more electron-rich thiolate anion that might undergo photooxidation to produce the requisite thiol radical more readily.12 However, we observed instead that strong inorganic bases had a deleterious effect on the reaction (entries 2–4). Similarly, most of the standard organic bases we assessed in this reaction failed to provide any beneficial effect in the thiol–ene coupling (entries 5–7). In the course of a more exhaustive survey of basic additives, however, we made the surprising observation that a variety of aniline additives produced a dramatic increase in the rate of the reaction (entries 8–10). The optimal additive, p-toluidine, afforded complete conversion after just 30 min of irradiation.
Table 1. Additive Effects in Photocatalytic Thiol–Ene Reactions.
| entry | catalyst | additive | yielda (%) |
|---|---|---|---|
| 1 | Ru(bpz)3(PF6)2 | none | 10 |
| 2 | Ru(bpz)3(PF6)2 | Na2CO3 | 0 |
| 3 | Ru(bpz)3(PF6)2 | K2CO3 | 0 |
| 4 | Ru(bpz)3(PF6)2 | NaOAc | 0 |
| 5 | Ru(bpz)3(PF6)2 | i-Pr2NEt | <5 |
| 6 | Ru(bpz)3(PF6)2 | pyridine | <5 |
| 7 | Ru(bpz)3(PF6)2 | DMAP | <5 |
| 8 | Ru(bpz)3(PF6)2 | N,N-dimethylaniline | 24 |
| 9 | Ru(bpz)3(PF6)2 | aniline | 82 |
| 10 | Ru(bpz)3(PF6)2 | p-toluidine | 99 |
| 11 | none | p-toluidine | 0 |
| 12b | Ru(bpz)3(PF6)2 | p-toluidine | 0 |
| 13c | Ru(bpz)3(PF6)2 | p-toluidine | 80 |
| 14 | Ru(bpy)3(PF6)2 | p-toluidine | 96 |
| 15c | Ru(bpy)3(PF6)2 | p-toluidine | 52 |
| 16d | Ru(bpz)3(PF6)2 | p-toluidine | 75 |
Yields determined by 1H NMR using a calibrated internal standard.
Reaction conducted in the dark.
Reaction conducted in rigorously degassed solvent.
Reaction irradiated with a 23 W compact fluorescent light bulb instead of blue LEDs.
We were intrigued by this unexpected result and became curious about the origin of this dramatic rate acceleration observed in the presence of p-toluidine. First, we performed a series of control experiments verifying the necessity of each component of the reaction mixture. Reactions conducted in the absence of the ruthenium photocatalyst or in the dark produced no observable thiol–ene adduct (entries 11 and 12). We found that the reaction can be conducted either open to the air or under rigorously degassed conditions, although the latter resulted in somewhat slower conversion (entry 13). Reactions conducted using the less oxidizing parent Ru(bpy)32+ photocatalyst instead of Ru(bpz)32+ also gave thiol–ene product, but at slower rates consistent with the poorer oxidizing power of Ru(bpy)32+ compared to Ru(bpz)32+ (entries 14 and 15). Finally, while we observed optimal conversion using blue LED strips as the illumination source, convenient broad-spectrum household light bulbs could also be used to promoted the thiol–ene coupling (entry 16).
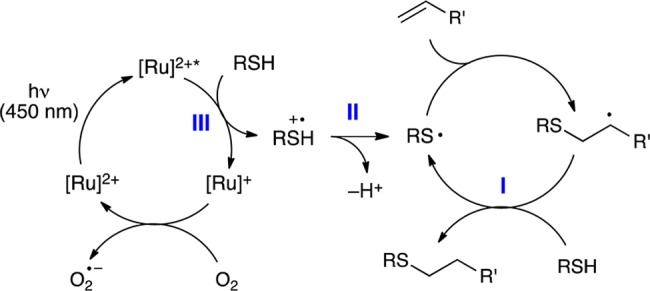
The results of these control studies suggest that the mechanism of this reaction is roughly similar to the proposal we offered for the photocatalytic radical thiol–ene reaction in the absence of aniline additives (Scheme 2). Namely, visible light photoexcitation of the Ru(bpz)32+ chromophore affords a long-lived redox-active MLCT state that must convert a thiol into its corresponding thiyl radical via one-electron oxidation and deprotonation. This thiyl radical is the key intermediate in all radical thiol–ene reactions, which propagates via a radical addition chain mechanism to produce the observed addition product. Finally, to account for the beneficial effect of an aerobic atmosphere, we propose that the photochemically inactive reduced Ru(bpz)3+ complex can be turned over by oxygen to regenerate the photocatalyst13 and initiate further radical chains.
Scheme 2. Proposed Mechanism of Direct Photocatalytic Thiol–Ene Coupling.
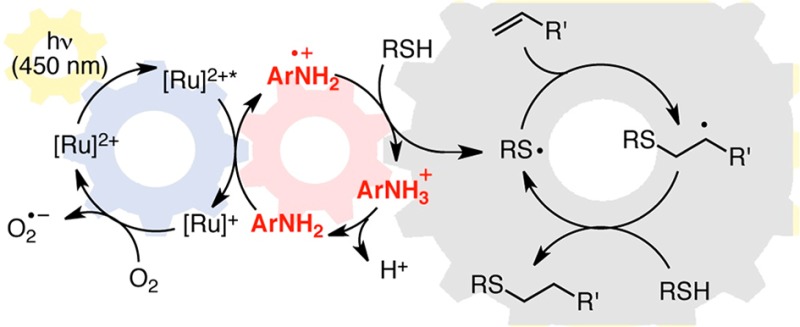
We considered several possible roles for the p-toluidene additive that could account for the dramatic increase in the rate of product formation. First, we speculated that the aniline could be serving as an auxiliary H atom donor to facilitate turnover of the radical chain reaction (I). We also wondered if the aniline might be a general base capable of either accelerating the deprotonation of the thiol or of the intermediate thiol radical cation (II). Finally, we considered whether the aniline might be involved in mediating the photocatalytic electron-transfer step itself in some way (III). Each of these three hypotheses invokes a different role for the additive, either as a hydrogen atom donor (I), as a base (II), or as a redox mediator (III); as such, one would expect different structure–activity relationships depending on each of these possible roles. We therefore examined radical thiol–ene additions in the presence of a number of structurally varied aromatic additives. These data are summarized in Table 2.
Table 2. Effect of Additive Variation in the Radical Thiol–Ene Reaction of 3 and 6a.
| entry | additive | yieldb (%) | N–H BDE (kcal/mol)c | conjugate acid pKa (H2O)d | E0 (V vs SCE)e |
|---|---|---|---|---|---|
| 1 | 1,4-phenylenediamine | 0 | 82.5 | 6.08 | 0.17 |
| 2 | p-aminophenol | 5 | 84.7 | 5.50 | 0.33 |
| 3 | none | 10 | 0.45 | ||
| 4 | p-anisidine | 34 | 84.6 | 5.29 | 0.56 |
| 5 | 1,2,3,4,-tetrahydroquinoline | 53 | 83.4 | 4.90 | 0.60 |
| 6 | N,N-dimethylaniline | 22 | 5.06 | 0.67 | |
| 7 | p-toluidine | 99 | 86.9 | 5.07 | 0.72 |
| 8 | N-methylaniline | 82 | 85.7 | 4.85 | 0.74 |
| 9 | aniline | 82 | 88.4 | 4.58 | 0.86 |
| 10 | diphenylamine | 88 | 81.9 | 0.79 | 0.86 |
| 11 | p-bromoaniline | 27 | 88.3 | 3.91 | 0.92 |
| 12 | p-nitroaniline | 62 | 92.5 | 0.98 | 1.03 |
| 13 | 1,2,3,4-tetrahydroisoquinoline | 0 | 90.0 | 9.55 | 1.06 |
| 14 | 4-aminopyridine | 0 | 93.6 | 8.96 | 1.20 |
| 15 | anisole | 0 | 1.66 |
Reactions conducted by irradating 0.10 M 3, 0.25 M 6, and 0.05 M additive in MeCN with a blue LED for 30 min.
Yields determined by NMR analysis against a calibrated internal standard.
Calculated at the B3LYP/6-311++G(3df,3pd) level of theory. See Supporting Information for details.
Values taken from ref (16).
E0 values in reference to SCE in MeCN solution.
We first considered the possibility that the additive could be involved in radical chain transfer. Many prior studies of the mechanism of the radical thiol–ene addition have proposed that the rate-limiting step involves slow hydrogen atom transfer from a thiol to the intermediate carbon-centered radical in a chain transfer step.14 We speculated that an aniline might serve as an efficient H-atom shuttle, rapidly reacting with the carbon-centered radical to generate a resonance-stabilized anilino radical that could subsequently react with thiol to regenerate the propagating thiyl radical (Scheme 3). However, we were able to rule this possibility out for several reasons. Experiments conducted using aniline additives lacking N–H bonds (e.g., N,N-dimethylaniline, Table 2, entry 6) nevertheless provided a substantial increase in the rate of the reaction. Moreover, a plot of N–H bond dissociation energy vs the yield of the radical thiol–ene after 30 min of irradiation showed no dependence on the H-atom donating ability of the aniline (Figure 1). Thus, we concluded that the primary effect of the p-toluidine additive was not merely to increase the rate of chain transfer by serving as an H-atom shuttle.
Scheme 3. H-Atom Shuttle Hypothesis.

Figure 1.

Relationship of N–H BDE to thiol–ene yield.
We next considered the possibility that aniline might be behaving as a base catalyst, in line with our original rationale for screening basic additives. We imagined that this might be occurring in one of two ways. First, while Matsuda did not observe photooxidation of aliphatic thiols by Ru*(bpy)32+, later work from the same group indicated that deprotonated thiolates are efficiently oxidized and produce disulfides.15 We hypothesized that the aniline might deprotonate the thiol to generate a more easily oxidized thiolate (Scheme 4, top). Alternatively, aniline could be involved in deprotonation of an intermediate thiol radical cation produced upon one-electron photooxidation of the thiol (Scheme 4, bottom).11 These hypotheses, however, are inconsistent with the observation that inorganic bases had an inhibitory effect on the reaction (Table 1, entries 2–4). Indeed, we observe no relationship between the efficiency of this reaction and the pKa of the conjugate ammonium salt of the additives screened (Figure 2).16 Thus, we do not believe that the basicity of the aniline is responsible for the observed rate acceleration.
Scheme 4. Brønsted Base Hypothesis.

Figure 2.

Relationship of pKa to thiol–ene yield.
Finally, we considered the possibility that the aniline might be involved in mediating electron transfer between the excited photocatalyst and the thiol. Studies of the reductive quenching of Ru*(bpy)32+ by aromatic amines have established that these photooxidation reactions generally occur at rapid rates with a wide range of aromatic amines (ca. 108 M–1 s–1).17 Similarly, while the rate of hydrogen atom abstraction by arylamine radical cations has not, to the best of our knowledge, been measured, we expect this process to be reasonably rapid as well, as the second-order rate constant for reactions of aliphatic amines with thiols has been measured by laser flash photolysis to be 3 × 106 M–1 s–1.18 We speculated that the slow intrinsic rate of direct photooxidation of thiol by Ru*(bpz)32+ might be circumvented by diffusion-controlled oxidation of the aniline followed by fast reaction with the thiol substrate to afford the propagating thiyl radical species (Scheme 5). If this mechanism were operative, one would expect that the most effective aniline additives would possess redox potentials that are intermediate between that of Ru*(bpz)32+ and that of the thiol, such that both oxidation of the aniline by the photocatalyst and the oxidation of the thiol by the aminium radical cation were thermodynamically favorable steps.
Scheme 5. Redox Mediator Hypothesis.

Indeed, when the yields of thiol–ene adduct are plotted against the redox potential of the additive (Figure 3), a clear trend emerges in which the additives that result in an increase in the rate of thiol–ene addition cluster within a window of potentials bounded by +1.4 V (the redox potential of the photocatalyst excited state) and +0.50 V (the redox potential of aliphatic thiols). The structure–activity relationship therefore strongly suggests that the role of the aniline additive is to serve as a redox mediator that is involved in the key thiol oxidation step.
Figure 3.

Relationship of Eox to thiol–ene yield.
Scheme 6 outlines our working model for the mechanism of the photocatalytic radical thiol–ene reaction in the presence of p-toluidine, which is a modest perturbation of the hypothesis proposed in Scheme 2 for the reaction without this additive. In particular, we propose that the addition of an oxidizable aniline additive provides a kinetically facile method for the generation of the thiol radical via photocatalytic generation of an aniline radical cation that is capable of activating a thiol, either via direct hydrogen atom abstraction or sequential electron- and proton-transfer steps, to afford the chain-propagating thiyl radical species.
Scheme 6. Redox Mediated Radical Thiol–Ene Reaction.

The p-toluidine additive in this reaction, therefore, might be considered an oxidative redox mediator. The use of soluble organic redox mediators in electrochemical oxidation reactions is well precedented.19 The use of aromatic amines, in particular, to provide homogeneous oxidizing species that can diffuse away from the surface of the cathode has been important in the development of electrochemical Wacker,20 thioacetal hydrolysis,21 and fluorination reactions.22 In the context of photoredox catalysis, pyridinium cations have been widely utilized to mediate electron transfer reactions from a photocatalyst to a hydrogen-evolving catalyst.23 They have also been shown to play a similar role in the photocatalytic reductive debromination of vicinal dibromides.24 The use of oxidative redox mediators in photoredox catalysis, however, has been less extensively explored25 and to the best of our knowledge has not been previously utilized in transition metal catalyzed photoredox reactions.
This mechanistic model is a further example of a surprising feature of photoredox catalysis that has been emerging from several research groups, in which various co-catalytic additives can have profound effects on the mechanism of a photocatalytic process. We have previously documented the importance of Lewis acid3 and Brønsted acid4 activation in a variety of photocatalytic reactions developed in our laboratories. Similarly, the addition of organocatalysts and transition metal catalysts have been demonstrated by groups such as MacMillan,26 Sanford,27 and Rovis28 to profoundly alter the course of photoredox reactions. These results raise the intriguing prospect of electrocatalytic coactivation of organic substrates using the same broad family of visible light photocatalysts investigated to date.
In order to demonstrate that the beneficial effect of p-toluidine on the photocatalytic thiol–ene reaction was not limited to cysteine derivatives, we next studied the effect of this additive on the thiol–ene reaction of a range of coupling partners. The results of this study, summarized in Table 3, demonstrate that the effect is indeed quite general. The conditions we previously reported for photocatalytic thiol–ene couplings of organic-soluble substrates required a 4-fold excess of the thiol in order for the reaction to proceed at a reasonable rate. Consistent with this observation, we found that the rate of thiol–ene reactions with thiol as the limiting reagent were generally quite slow.
Table 3. Use of Thiol as Limiting Reagenta.
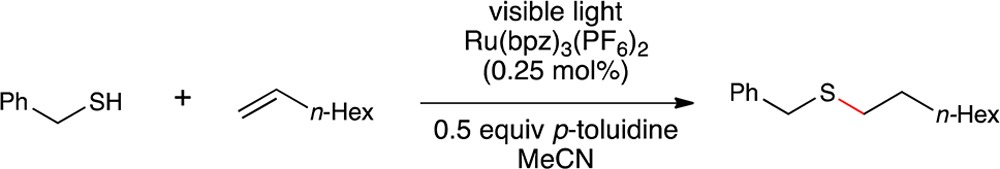
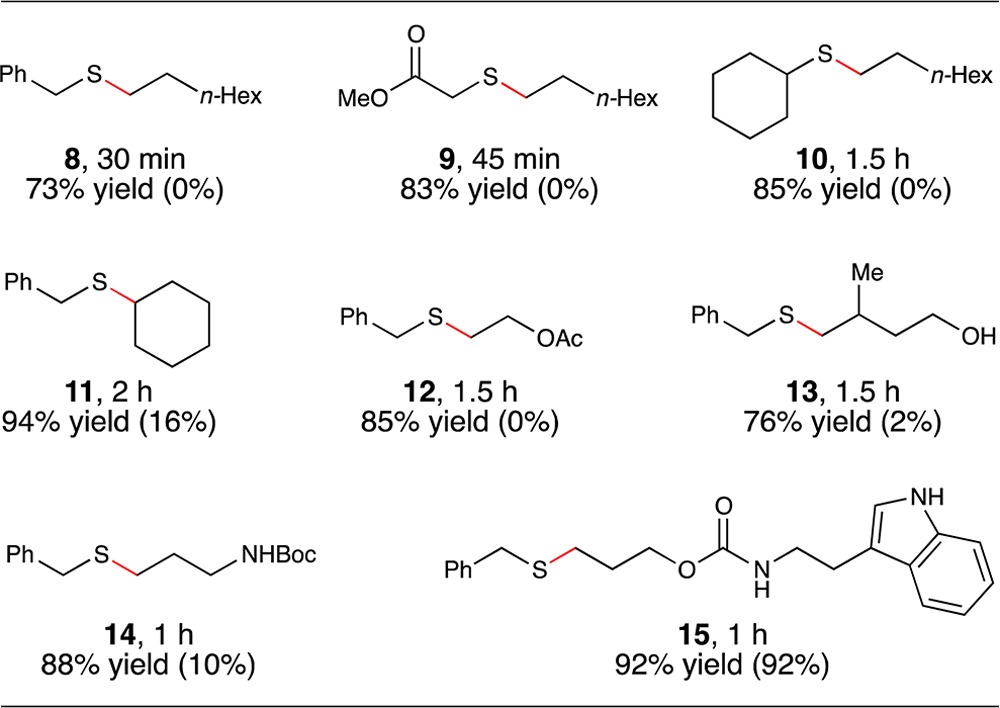
Reactions irradiated with a blue LED. Yields determined by 1H NMR using a calibrated internal standard. Values in parentheses are yields of reactions conducted without p-toluidine.
The addition of 0.5 equiv of p-toluidine, however, produced a significant rate increase for most coupling reactions examined in this study. The reaction between primary aliphatic thiols and monosubstituted aliphatic olefins went to excellent conversions in the presence of the additive (Table 3, products 8 and 9). Secondary thiols also coupled efficiently (product 10), as well as 1,2-disubstiuted olefins (product 11). The functional group compatibility of this process is excellent; the thiol–ene products arising from coupling with vinyl acetate (product 12), 1,1-disubstituted homoallylic alcohol (product 13), and N-Boc-allylamine (product 14) could be isolated in excellent yield. We also tested electron-rich heterocyclic scaffolds that might potentially be sensitive to oxidative destruction (product 15). However, N-Alloc-tryptamine reacts with benzyl thiol in excellent yields, both in the presence and absence of p-toluidine. The oxidation potential of indole falls into the range we found to be suitable for redox mediation of this reaction (+1.0 V); we interpret the significant thiol–ene coupling in the absence of p-toluidine as evidence for the ability of tryptamine to serve as its own redox mediator.
Finally, we performed an initial assessment of the suitability of this reaction for thiol–ene modification of cysteine-containing biomolecules under aqueous conditions by examining the conjugation of glutathione (16) with a variety of coupling partners of potential biological relevance (Table 4). These included alkene-modified azides, PEG oligomers, protected sugars, and biotin (entries 1–4). These aqueous reactions were not noticeably different in any way from the organic reactions we had investigated to this point, and in each case, the thiol–ene reaction proceeded smoothly under conditions of limiting glutathione. We also examined the compatibility of this reaction with representative proteinogenic functional groups by examining the reaction of glutathione with several N-Alloc-protected amino acids (entries 5–10). Notably, the reaction was insensitive to the presence of unprotected alcohols (entry 6), basic groups (entry 7), acidic groups (entry 8), and oxidizable heteroatomic and aromatic functionalities (entries 9 and 10). These preliminary results suggest that the conditions described in this manuscript may indeed enable thiol–ene modifications of even more complex biomolecules using visible light, aqueous conditions, and limiting thiol, which may make this method ideal for biological applications.
Table 4. Substrate Scope of Bioconjugates with Glutathione.
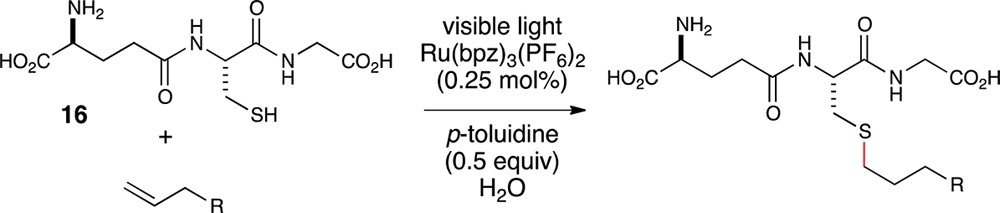
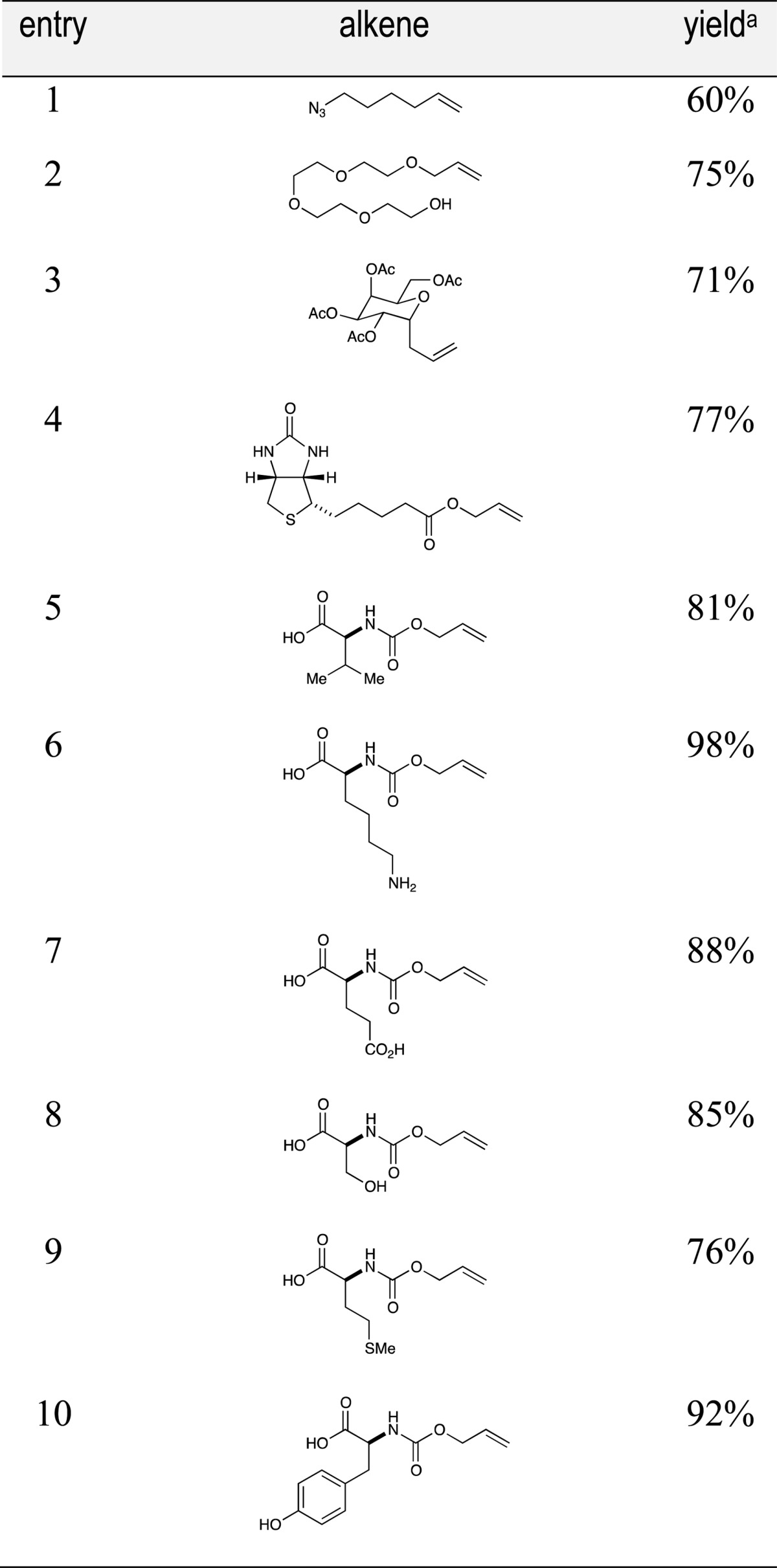
Isolated yields represent the averaged results of two reproducible experiments.
Conclusion
In summary, we have developed a protocol for highly efficient radical thiol–ene conjugation of structurally varied thiols and alkenes using the thiol component as the limiting reagent. This reaction is enabled by the use of p-toluidine as an essential redox mediator, which circumvents the slow rate of direct photooxidation of thiol with Ru*(bpz)32+ by facilitating two more rapid electron-transfer events that generate the key thiyl radical intermediate. This reaction has excellent substrate scope and translates readily to aqueous conditions that are ideal for bioconjugation reactions of thiol-functionalized biomolecules.
These results have several important implications. First, radical thiol–ene reactions are becoming increasingly recognized as an important class of ″click″ reactions for applications in chemical biology. The ability to perform these coupling reactions using completely biologically compatible wavelengths of visible light, under native aqueous conditions, and without the need for high concentrations of thiol suggest that this reaction could be a useful complement to existing thermal and UV-promoted radical thiol–ene methodology. More broadly, the results of this study demonstrate that unfavorable electron-transfer kinetics in a photoredox reaction can be mitigated by the use of redox mediators that can catalyze the key photoelectrochemical activation step. We are interested in studying the kinetics of this co-catalytic process in greater detail and applying our fundamental insights to the development of ever more efficient photocatalytic processes in multiple synthetic contexts.
Experimental Section
General Information
Photochemical reactions were irradiated with a 6″ strip of blue LED lights purchased from Creative Lightings. Ru(bpz)3(PF6)2 was synthesized using known methods.29 The alkenes used for the thiol–ene couplings reported in Table 4, entries 1–4, were synthesized using previously reported procedures.31−33,35 All other reagents were purchased from commercial sources and purified immediately prior to use. Chromatography was performed with 60 Å silica gel (230–400 mesh). 1H and 13C NMR data for all previously uncharacterized compounds were obtained using 500 MHz spectrometers and are referenced to TMS (0.00 ppm) and CDCl3 (77 ppm), respectively. Mass spectra were obtained by electrospray ionization and a time-of-flight analyzer.
Allyl (2-(1H-Indol-3-yl)ethyl)carbamate
To a flame-dried 100 mL round-bottomed flask were added 600 mg (3.74 mmol) of tryptamine and 18 mL CH3CN under an atmosphere of N2. The mixture was cooled to 0 °C, and 524 mg (4.34 mmol, 1.2 equiv) allyl chloroformate was added followed by 344 mg (4.34 mmol, 1.2 equiv) pyridine. The resulting dark orange reaction was stirred at 0 °C for 2 h, diluted with 10 mL EtOAc, and quenched with 5% (w/v) aqueous citric acid. The organic layer was separated, and the aqueous layer was extracted with EtOAc (10 mL). The organic layers were combined, washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to afford a thick orange residue. Purification via column chromatography on SiO2 gel (hexanes/EtOAc = 3:1) afforded 781 mg (3.20 mmol, 85% yield) of a pale orange oil. IR (thin film) 3411, 3333, 3081, 3057, 2940, 2859, 1698, 1547, 1526, 1460, cm–1; 1H NMR (500 MHz, CDCl3) δ 8.04 (s, 1H), 7.61 (d, J = 7.9 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.21 (td, J = 7.9, 7.5, 1.1 Hz, 1H), 7.16–7.08 (m, 1H), 7.04 (s, 1H), 5.91 (td, J = 10.8, 5.2 Hz, 1H), 5.34–5.25 (m, 1H), 5.20 (d, J = 10.4 Hz, 1H), 4.79 (s, 1H), 4.56 (d, J = 5.7 Hz, 2H), 3.54 (q, J = 6.6 Hz, 2H), 2.98 (t, J = 6.8 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 156.2, 136.4, 133.0, 127.3, 122.2, 122.0, 119.5, 118.8, 117.6, 112.9, 111.2, 65.4, 41.2, 25.8; HRMS (ESI) calcd for [C14H17N2O2]+ requires m/z 245.1285, found m/z 245.1292.
(S)-2-(((Allyloxy)carbonyl)amino)-6-aminohexanoic Acid, TFA Salt
In a 50 mL round-bottom flask, Nε-Boc-l-Lys (1.00 g, 4.06 mmol) was dissolved in 4 M NaOH (8 mL) with vigorous stirring. After 10 min, allyl chloroformate (440 μL, 4.14 mmol) was added to the solution dropwise, resulting in a modest exotherm. The solution was stirred for an additional 10 min and was then diluted with water and 4 M NaOH (8 mL each). After 2.5 h of stirring, the solution was washed with 25 mL Et2O, and the remaining aqueous phase was acidified to pH 2 with 1 M HCl (100 mL), resulting in a cloudy white suspension. The product was extracted into EtOAc (3 × 50 mL), dried over Na2SO4, filtered, and concentrated to a white semisolid. Without any additional purification, this material was dissolved in CH2Cl2 (20 mL), and trifluoroacetic acid (20 mL) was added over the course of 5 min. After 30 min, TLC analysis (9:1 CH2Cl2/MeOH, ninhydrin stain) indicated complete conversion. The reaction was concentrated directly and subjected to high vacuum, affording 793 mg (57%) of the Alloc-protected amino acid as a viscous, colorless oil. IR (thin film) 3073, 2938, 1665, 1530 cm–1; 1H NMR (500 MHz, D2O) δ 5.94 (ddt, J = 16.3, 10.5, 5.2 Hz, 1H), 5.31 (app. d, J = 17.3 Hz, 1H), 5.24 (dt, J = 10.6, 1.3 Hz, 1H), 4.57 (dt, J = 5.1, 1.0 Hz, 2H), 4.16 (dd, J = 9.1, 4.9 Hz, 1H), 2.98 (t, J = 7.6 Hz, 2H), 1.95–1.83 (m, 1H), 1.80–1.62 (m, 3H), 1.54–1.39 (m, 2H). 13C NMR (126 MHz, D2O) δ 176.3, 162.9 (q, 2JCF = 35.6 Hz, TFA), 158.1, 132.5, 117.2, 116.3 (q, 1JCF = 291.7 Hz, TFA), 66.0, 53.7, 39.1, 30.0, 26.1, 22.0. HRMS (ESI) calcd for [C10H19N2O4]+ requires m/z 231.1340, found m/z 231.1347.
Methyl 2-((tert-Butoxycarbonyl)amino)-3-((3-hydroxypropyl)thio) Propanoate (7)
In a 1.5 dram vial were placed 43 mL (0.630 mmol) allyl alcohol, 58.8 mg Boc-Cys-OMe (0.250 mmol), 12.5 mg (0.119 mmol) p-toluidine, and 0.5 mg (0.578 μmol) Ru(bpz)3(PF6)2 in 0.25 mL MeCN. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 30 min. Measured 99% yield by NMR analysis using TMSPh as an internal standard. Analytically pure material may be obtained by flash column chromatography (SiO2, 2:1 EtOAC/hexanes). IR (thin film) 3397, 2981, 2955, 1699, 1575 cm–1; 1H NMR (500 MHz, CDCl3) δ 5.38 (d, J = 8.3 Hz, 1H), 4.59–4.52 (m, 1H), 3.77 (s, 5H), 3.00 (dd, J = 14.0, 4.9 Hz, 1H), 2.90 (dd, J = 13.9, 6.0 Hz, 1H), 2.68 (dq, J = 12.6, 6.4, 5.9 Hz, 2H), 2.04 (s, 1H), 1.82 (qt, J = 14.0, 7.0 Hz, 2H), 1.45 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 171.6, 155.3, 80.3, 60.9, 53.0, 52.6, 34.8, 31.7, 29.0, 28.3; HRMS (ESI) calcd for [C12H23NO5S + Na]+ requires m/z 316.1190, found m/z 316.1180.
General Procedure for Radical Thiol–Ene Reactions (Table 3)
Condition A: To an oven-dried 1.5 dram vial were added 0.833 mmol olefin, 0.33 mmol thiol, 1.7 μmol Ru(bpz)3(PF6)2, 0.167 mmol p-toluidine, and 0.67 mL MeCN. The vial was sealed with a Teflon cap and irradiated with blue LEDs. Condition B: Reactions set up identically as in the procedure described for A, except without p-toluidine. Yields were determined by 1H NMR analysis using TMSPh as an internal standard.
Benzyl(octyl)sulfane (Table 3, 8)
Condition A: To an oven-dried 1.5 dram vial were added 130 μL (0.833 mmol) octene, 39 mL (0.333 mmol) benzyl mercaptan, 17.1 g (0.159 mmol) p-toluidine, 1.4 mg (1.7 μmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 45 min. Measured 74% yield by NMR analysis. Condition B: 130 μL (0.833 mmol) octene, 39 μL (0.333 mmol) benzyl mercaptan, 1.4 mg (1.7 μmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. Measured 0% yield by NMR analysis. Spectral data were identical in all respects to previously reported values.30
Methyl 2-(Octylthio)acetate (Table 3, 9)
Condition A: To an oven-dried 1.5 dram vial were added 130 μL (0.833 mmol) octene, 30 mL (0.333 mmol) methyl thiolglycolate, 18.9 mg (0.176 mmol) p-toluidine, 1.6 mg (1.9 mmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 45 min. Measured 83% yield by NMR analysis. Condition B: 130 μL (0.833 mmol) octene, 30 μL (0.333 mmol) methyl thiolglycolate, 1.4 mg (1.6 μmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. Measured 0% yield by NMR analysis. Spectral data were identical in all respects to previously reported values.4
Cyclohexyl(octyl)sulfane (Table 3, 10)
Condition A: To an oven-dried 1.5 dram vial were added 130 μL (0.833 mmol) octene, 41 μL (0.333 mmol) cyclohexyl mercaptan, 17.6 mg (0.164 mmol) p-toluidine, 1.1 mg (1.2 μmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 2 h. Measured 85% yield by NMR analysis. Condition B: 130 μL (0.833 mmol) octene, 41 μL cyclohexylthiol (0.333 mmol), 1.1 mg (1.2 μmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. Measured 0% yield by NMR analysis. IR (thin film) 3422, 2927, 2853 cm–1; 1H NMR (500 MHz, CDCl3) δ 2.67–2.57 (m, 1H), 2.55–2.48 (m, 2H), 2.01–1.91 (m, 2H), 1.82–1.71 (m, 2H), 1.66–1.52 (m, 3H), 1.41–1.15 (m, 16H), 0.87 (t, J = 7.0 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 43.5, 33.8, 31.8, 30.2, 30.1, 29.3, 29.2, 29.1, 26.2, 25.9, 22.7, 14.1; HRMS (ESI) calcd for [C14H28S + H]+ requires m/z 229.1985, found m/z 229.1983.
Benzyl(cyclohexyl)sulfane (Table 3, 11)
Condition A: To an oven-dried 1.5 dram vial were added 85 μL (0.833 mmol) cyclohexene, 43 μL (0.333 mmol) benzyl mercaptan, 18.9 mg (0.176 mmol) p-toluidine, 2.0 mg (2.3 μmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 2 h. Measured 94% yield by NMR analysis. Condition B: 85 μL (0.833 mmol) cyclohexene, 43 μL (0.333 mmol) benzyl mercaptan, 1.3 mg (1.5 μmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. Measured 16% yield by NMR analysis. Spectral data were identical in all respects to previously reported values.4
2-(Benzylthio)ethyl Acetate (Table 3, 12)
Condition A: To an oven-dried 1.5 dram vial were added 90 μL (0.833 mmol) allylacetate, 39 μL (0.333 mmol) benzyl mercaptan, 19.5 mg (0.181 mmol) p-toluidine, 1.1 mg (1.2 μmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 1.5 h. Measured 85% yield by NMR analysis. Condition B: 90 μL (0.833 mmol) allylacetate, 39 μL (0.333 mmol) benzyl mercaptan, 1.3 mg (1.5 μmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. Measured 0% yield by NMR analysis. Spectral data were identical in all respects to previously reported values.4
4-(Benzylthio)-3-methylbutan-1-ol (Table 3, 13)
Condition A: To an oven-dried 1.5 dram vial were added 85 μL (0.833 mmol) 3-methyl-3-buten-1-ol, 39 μL (0.333 mmol) benzyl mercaptan, 17.2 mg (0.160 mmol) p-toluidine, 1.1 mg (1.2 μmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 1 h. Measured 76% yield by NMR analysis. Condition B: 85 μL (0.833 mmol) 3-methyl-3-buten-1-ol, 39 μL (0.333 mmol) benzyl mercaptan, 1.5 mg (1.7 μmol) Ru(bpz)3(PF6)2, and 0.67 mL acetonitrile. Measured 2% yield by NMR analysis. Spectral data were identical in all respects to previously reported values.4
tert-Butyl (3-(Benzylthio)propyl)carbamate (Table 3, 14)
Condition A: To an oven-dried 1.5 dram vial were added 130 mg (0.833 mmol) tert-butyl allylcarbamate, 39 μL (0.333 mmol) benzyl mercaptan, 18.3 mg (0.17 mmol) p-toluidine, 1.4 mg Ru(bpz)3(PF6)2 (1.6 μmol), and 0.67 mL acetonitrile. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 1 h. Measured 88% yield by NMR analysis. Condition B: 130 mg (0.833 mmol) tert-butyl allylcarbamate, 39 μL (0.333 mmol) benzyl mercaptan, 1.7 mg Ru(bpz)3(PF6)2 (2.0 μmol), and 0.67 mL acetonitrile. Measured 10% yield by NMR analysis. Spectral data were identical in all respects to previously reported values.4
3-(Benzylthio)propyl (2-(1H-Indol-3-yl)ethyl)carbamate (Table 3, 15)
Condition A: To an oven-dried 1.5 dram vial equipped with a stir bar were added 203 mg (0.833 mmol) Alloc-tryptamine, 39 μL (0.333 mmol) benzyl mercaptan, 17.8 mg (0.17 mmol) p-toluidine, 1.3 mg Ru(bpz)3(PF6)2 (1.5 μmol) and 0.67 mL acetonitrile. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 1 h. Measured 91% yield by NMR analysis. In order to isolate analytically pure material, the solution was first extracted with 10% NaOH (aq) (1 × 2 mL) to remove unreacted thiol. The aqueous layer was extracted with EtOAc (2 × 3 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated prior to column chromatography on SiO2 gel (gradient of hexanes/acetone = 4:1–3:1) to afford 226 mg of an inseparable 1:1.56 mixture of desired thioether product to residual Alloc tryptamine. This mixture was transferred to a flame-dried, nitrogen-purged 250 mL round bottomed flask equipped with a stir bar. To the flask was added 43 mL CH2Cl2, and the mixture was stirred until homogeneous. Thereafter, 49.8 mg (0.0431 mmol) Pd(PPh3)4 and 320 μL (2.59 mmol) PhSiH3 were added, and the reaction was stirred under nitrogen for 30 min until 1H NMR analysis indicated complete disappearance of Alloc-tryptamine. Subsequently, saturated NaHCO3 (aq) (4 mL) was added, the mixture was shaken, and the organic layer was separated and dried over Na2SO4. The mixture was filtered and concentrated prior to column chromatography on SiO2 gel (hexanes/acetone = 4:1) to afford 91 mg (0.25 mmol, 74% yield over two steps) of desired thioether product as a pale yellow oil. Condition B: 202 mg (0.833 mmol) Alloc-tryptamine, 43 μL benzyl mercaptan (0.333 mmol), 1.3 mg Ru(bpz)3(PF6)2 (1.5 μmol), and 0.67 mL acetonitrile. Measured 92% yield by NMR analysis. IR (thin film) 3417, 3333, 2949, 2919, 1700, 1524, 1454, 1251 cm–1; 1H NMR (500 MHz, CDCl3) δ 7.99 (s, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.37 (dt, J = 8.1, 0.9 Hz, 1H), 7.35–7.27 (m, 4H), 7.25–7.16 (m, 2H), 7.13 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 7.03 (s, 1H), 4.68 (s, 1H), 4.10 (t, J = 6.3 Hz, 2H), 3.70 (s, 2H), 3.51 (q, J = 6.6 Hz, 2H), 2.97 (s, 2H), 2.44 (t, J = 7.3 Hz, 2H), 1.83 (p, J = 6.7 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 156.4, 138.3, 136.4, 128.8, 128.5, 127.3, 127.0, 122.2, 122.0, 119.49, 118.7, 112.9, 111.2, 63.3, 41.1, 36.2, 28.7, 27.6, 25.7; HRMS (ESI) calcd for [C21H28N3O2S]+ requires m/z 386.1897, found m/z 386.1892.
General Procedure for Bioconjugation Reactions with Glutathione (Table 4)
To a 1.5 dram vial were added 0.625 mmol olefin, 0.25 mmol thiol, 0.63 μmol Ru(bpz)3(PF6)2, 0.125 mmol p-toluidine and 1.25 mL Millipore water. The vial was sealed with a Teflon cap and irradiated with blue LEDs. Unless otherwise indicated, the crude reaction mixture was loaded directly onto a methanol-charged Sephadex column and eluted with methanol at a flow rate of 1 mL/min. Fractions expected to contain the desired product were concentrated in vacuo and analyzed by 1H NMR in D2O. Fractions containing >90% pure product were combined and used for calculating the yield.
(S)-2-Amino-5-(((R)-3-((6-azidohexyl)thio)-1-((carboxymethyl)amino)-1-oxopropan-2-yl)amino)-5-oxopentanoic Acid (Table 4, Entry 1)
To a 1.5 dram vial were added 75.4 mg (0.602 mmol) azide,31 76.6 mg glutathione (0.250 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 13.4 mg (0.125 mmol) p-toluidine, and 1.25 mL Millipore water. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 2 h. Over the course of the reaction a white solid crashed out of solution. This solid was isolated via filtration; the solid was washed with water and dried in vacuo to give analytically pure product. Obtained 63.8 mg (0.148 mmol, 59% yield) of white solid. Experiment 2: 78.8 mg (0.257 mmol) glutathione, 77.1 mg (0.615 mmol) azide, 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 14.4 mg p-toluidine (0.134 mmol) and 1.25 mL water. Isolated 64.9 mg (0.150 mmol, 60% yield). IR (ATR) 3348, 3042, 2856, 2093, 1673, 1645, 1516 cm–1; 1H NMR (500 MHz, DMSO-d6) δ 8.64 (t, J = 5.2 Hz, 1H), 8.42 (d, J = 8.5 Hz, 1H), 4.42 (dt, J = 9.0, 4.5 Hz, 1H), 3.76–3.61 (m, 2H), 3.33 (dt, J = 13.8, 6.5 Hz, 1H and H2O), 2.88 (dd, J = 13.6, 4.3 Hz, 1H), 2.62 (dd, J = 13.5, 9.7 Hz, 1H), 2.50 (m, 4H, and DMSO) 2.33 (dddd, J = 15.5, 7.5 Hz, 2H), 1.90 (ddp, J = 20.8, 13.8, 7.3 Hz, 2H), 1.57–1.42 (m, 4H), 1.42–1.24 (m, 4H); 13C NMR (126 MHz, DMSO) δ 172.2, 171.4, 171.2, 170.9, 53.6, 53.0, 51.0, 41.7, 33.9, 31.9, 31.6, 29.3, 28.6, 28.1, 27.3, 26.2; HRMS (ESI) calcd for [C16H28N6O6S]+ requires m/z 433.1864, found m/z 433.1861.
(18R,23S)-23-Amino-18-((carboxymethyl)carbamoyl)-1-hydroxy-20-oxo-3,6,9,12-tetraoxa-16-thia-19-azatetracosan-24-oic Acid (Table 4, Entry 2)
To a 1.5 dram vial were added 137.4 mg (0.587 mmol) allyl(tetra)ethylene glycol,32 77.9 mg glutathione (0.253 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 13.7 mg (0.128 mmol) p-toluidine, and 1.25 mL Millipore water. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 2 h. Upon completion of the reaction, the mixture was directly loaded onto a column of Sephadex LH-20 for purification (100% methanol). Obtained 96.6 mg (0.178 mmol, 71% yield) of white solid. Experiment 2: 143.1 mg (0.611 mmol) allyl(tetra)ethylene glycol, 76.4 mg glutathione (0.249 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 13.3 mg (0.124 mmol) p-toluidine, and 1.25 mL Millipore water. Isolated 98.1 mg (0.181 mmol, 71% yield). IR (ATR) 2667, 1639, 1539, 1449, 1402 cm–1;1H NMR (500 MHz, D2O) δ 4.46 (dd, J = 8.7, 5.1 Hz, 1H), 3.86 (s, 2H), 3.71 (t, J = 6.3 Hz, 1H), 3.65–3.49 (m, 18H), 2.96 (dd, J = 14.1, 5.1 Hz, 1H), 2.78 (dd, J = 14.1, 8.8 Hz, 1H), 2.55 (tdd, J = 7.2, 2.4, 1.2 Hz, 2H), 2.48–2.38 (m, 2H), 2.06 (q, J = 7.1 Hz, 2H), 1.76 (p, J = 6.7 Hz, 2H); 13C NMR (126 MHz, D2O) δ 174.7, 173.5, 173.5, 172.7, 71.7, 69.6, 69.5, 69.5, 69.5, 69.40, 69.2, 60.3, 53.7, 53.1, 41.5, 32.7, 31.1, 28.4, 28.0, 26.0 (one aliphatic carbon signal is absent due to accidental equivalence); HRMS (ESI) calcd for [C21H39N3O11S]+ requires m/z 542.2379, found m/z 542.2386.
(S)-2-Amino-5-(((R)-1-((carboxymethyl)amino)-1-oxo-3-((3-((2R,3S,4R,5S,6R)-3,4,5-triacetoxy-6-(acetoxymethyl)tetrahydro-2H-pyran-2-yl)propyl)thio)propan-2-yl)amino)-5-oxopentanoic Acid (Table 4, Entry 3)
To a 1.5 dram vial were added 232.5 mg (0.624 mmol) allylated tetraacetate galactose,33 76.6 mg glutathione (0.250 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 12.8 mg (0.120 mmol) p-toluidine, and 1.25 mL Millipore water. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 18 h. Upon completion of the reaction, the mixture was directly loaded onto a column of Sephadex LH-20 for purification (100% methanol). Obtained 119.0 mg (0.175 mmol, 70% yield) of white solid. Experiment 2: 78.8 mg (0.257 mmol) glutathione, 231.1 mg (0.624 mmol) allylated galactose, 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 14.4 mg p-toluidine (0.134 mmol), and 1.25 mL water. Isolated 120.2 mg (0.177 mmol, 71% yield). Spectral data were identical in all respects to previously reported values.34
(2S)-2-Amino-5-(((2R)-1-((carboxymethyl)amino)-1-oxo-3-((3-((4-((4S)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)butanoyl)oxy)propyl)thio)propan-2-yl)amino)-5-oxopentanoic Acid (Table 4, Entry 4)
To a 1.5 dram vial were added 171.2 mg (0.602 mmol) allyl biotin,35 76.6 mg glutathione (0.250 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 13.4 mg (0.125 mmol) p-toluidine, and 1.25 mL Millipore water. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 2 h. Over the course of the reaction a white solid crashed out of solution. This solid was isolated via filtration; the solid was washed with water and dried in vacuo to give analytically pure product. Obtained 63.8 mg (0.148 mmol, 59% yield) of white solid. Experiment 2: 78.8 mg (0.257 mmol) glutathione, 174.9 mg (0.615 mmol) allyl biotin, 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 14.4 mg p-toluidine (0.134 mmol) and 1.25 mL water. Isolated 64.9 mg (0.150 mmol, 60% yield). IR (solution in DMF) 2325, 2089 cm–1; 1H NMR (500 MHz, DMSO-d6) δ 8.64 (t, J = 5.2 Hz, 2H), 8.42 (d, J = 8.5 Hz, 1H), 4.42 (dt, J = 9.0, 4.5 Hz, 2H), 3.76–3.61 (m, 4H), 3.33 (dt, J = 13.8, 6.5 Hz, 4H), 2.88 (dd, J = 13.6, 4.3 Hz, 1H), 2.62 (dd, J = 13.5, 9.7 Hz, 1H), 2.55–2.44 (m, 8H), 2.33 (dddd, J = 15.5, 7.5 Hz, 3H), 1.90 (ddp, J = 20.8, 13.8, 7.3 Hz, 4H), 1.57–1.42 (m, 7H), 1.42–1.24 (m, 6H); 13C NMR (126 MHz, DMSO) δ 172.2, 171.4, 171.2, 170.9, 53.6, 53.0, 51.0, 41.7, 33.9, 31.9, 31.6, 29.3, 28.6, 28.1, 27.3, 26.2; HRMS (ESI) calcd for [C16H28N6O6S]+ requires m/z 433.1864, found m/z 433.1861.
(2S,11R,16S)-16-Amino-11-((carboxymethyl)carbamoyl)-2-isopropyl-4,13-dioxo-5-oxa-9-thia-3,12-diazaheptadecane-1,17-dioic Acid (Table 4, Entry 5)
To a 1.5 dram vial were added 124.4 mg (0.618 mmol) Alloc-Val-OH, 77.0 mg glutathione (0.251 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 12.7 mg (0.118 mmol) p-toluidine, and 1.25 mL Millipore water. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 2 h. Upon completion of the reaction, the mixture was directly loaded onto a column of Sephadex LH-20 for purification (100% methanol). Obtained 105.8 mg (0.208 mmol, 83% yield) of white solid. Experiment 2: 123.6 mg (0.614 mmol) Alloc-Val-OH, 77.0 mg glutathione (0.251 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 13.0 mg (0.121 mmol) p-toluidine and 1.25 mL Millipore water. Isolated 99.2 mg (0.195 mmol, 78% yield). IR (ATR) 3301, 3076, 2925, 1701, 1654.cm–1; 1H NMR (500 MHz, D2O) δ 4.47 (dd, J = 8.5, 5.2 Hz, 1H), 4.05 (t, J = 5.8 Hz, 2H), 3.94 (d, J = 5.3 Hz, 1H), 3.88 (s, 2H), 3.74 (t, J = 6.3 Hz, 1H), 2.96 (dd, J = 14.1, 5.0 Hz, 1H), 2.78 (dd, J = 14.0, 8.8 Hz, 1H), 2.60–2.51 (m, 1H), 2.43 (m, 2H), 2.07 (q, J = 7.3 Hz, 3H), 1.82 (m, 2H), 0.85 (dd, J = 22.8, 6.8 Hz, 6H); 13C NMR (126 MHz, D2O) δ 176.4, 174.7, 173.3, 173.3, 172.7, 158.5, 64.0, 59.9, 53.6, 53.0, 41.3, 32.7, 31.1, 29.8, 28.1, 27.9, 25.9, 18.4, 16.9; HRMS (ESI) calcd for [C19H32N4O10S]+ requires m/z 509.1912, found m/z 509.1936.
(2S,11R,16S)-16-Amino-2-(4-aminobutyl)-11-((carboxymethyl)carbamoyl)-4,13-dioxo-5-oxa-9-thia-3,12-diazaheptadecane-1,17-dioic Acid, TFA Salt (Table 4, Entry 6)
To a 1.5 dram vial were added 77.1 mg (0.251 mmol) glutathione, 215.0 mg (0.625 mmol) Alloc-Lys-OH.TFA salt, 0.5 mg (0.63 μmol) Ru(bpz)3(PF6)2, 13.4 mg (0.125 mmol) p-toluidine,and 1.25 mL Millipore water. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 2 h. The reaction mixture was then loaded directly onto a Sephadex column and eluted with MeOH to afford the product as a yellow solid (155.4 mg, 0.239 mmol, 96%). Experiment 2: 77.6 mg (0.253 mmol) glutathione, 215 mg (0.625 mmol) Alloc-Lys-OH.TFA salt, 0.5 mg (0.63 μmol) Ru(bpz)3(PF6)2, 13.4 mg (0.125 mmol) p-toluidine, and 1.25 mL Millipore water. Obtained 162.6 mg product (0.250 mmol, 99%). IR (ATR) 3088, 2952, 1697, 1529 cm–1; 1H NMR (500 MHz, D2O) δ 4.55 (dd, J = 8.5, 5.2 Hz, 1H), 4.18–4.06 (m, 3H), 3.98 (s, 2H), 3.87 (t, J = 6.4 Hz, 1H), 3.04 (dd, J = 14.1, 5.1 Hz, 1H), 2.98 (t, J = 7.6 Hz, 2H), 2.86 (dd, J = 14.0, 8.7 Hz, 1H), 2.64 (t, J = 5.9 Hz, 2H), 2.61–2.45 (m, 2H), 2.17 (q, J = 7.4 Hz, 2H), 1.95–1.82 (m, 3H), 1.79–1.59 (m, 3H), 1.53–1.38 (m, 2H); 13C NMR (126 MHz, D2O) δ 176.7, 174.7, 173.2, 173.1, 172.9, 163.0 (q, 2JCF = 35.4 Hz, TFA), 158.4, 116.4 (q, 1JCF = 291.7 Hz, TFA), 64.1, 54.0, 53.4, 53.1, 41.3, 39.2, 32.8, 31.1, 30.2, 28.2, 27.9, 26.2, 25.9, 22.1; HRMS (ESI) calcd for [C20H35N5O10S + H]+ requires m/z 538.2178, found m/z 538.2155.
(3S,12R,17S)-17-Amino-12-((carboxymethyl)carbamoyl)-5,14-dioxo-6-oxa-10-thia-4,13-diazaheptadecane-1,3,17-tricarboxylic Acid (Table 4, Entry 7)
To a 1.5 dram vial were added 144 mg (0.625 mmol) Alloc-Glu-OH, 76.8 mg glutathione (0.250 mmol), 0.5 mg (0.625 μmol) Ru(bpz)3(PF6)2, 13.4 mg (0.125 mmol) p-toluidine, and 1.25 mL Millipore water. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 2 h. Upon completion of the reaction, mixture was directly loaded onto a column of Sephadex LH-20 for purification (100% methanol). Obtained 116 mg (0.215 mmol, 86% yield). Experiment 2: 144 mg (0.625 mmol) Alloc-Glu-OH, 76.8 mg glutathione (0.250 mmol), 0.5 mg (0.625 μmol) Ru(bpz)3(PF6)2, 13.4 mg (0.125 mmol) p-toluidine, and 1.25 mL Millipore water. Isolated 126 mg (0.233 mmol, 90% yield). IR (ATR) 2957, 1703, 1646, 1535 cm–1; 1H NMR (500 MHz, D2O) δ 4.56 (dd, J = 8.7, 5.1 Hz, 1H), 4.19 (dd, J = 9.3, 4.9 Hz, 1H), 4.16–4.10 (m, 2H), 3.97 (s, 2H), 3.83 (t, J = 6.3 Hz, 1H), 3.04 (dd, J = 14.1, 5.1 Hz, 1H), 2.86 (dd, J = 14.2, 8.7 Hz, 1H), 2.66–2.63 (m, 2H), 2.59–2.47 (m, 4H), 2.21–2.08 (m, 3H), 1.99–1.93 (m, 1H), 1.94–1.82 (m, 2H); 13C NMR (126 MHz, D2O) δ 177.2, 176.0, 174.7, 173.3, 173.2, 172.7, 158.2, 64.0, 53.5, 53.0, 48.8, 41.3, 32.7, 31.1, 30.1, 28.1, 27.9, 26.0, 25.9. HRMS (ESI) calcd for [C19H30N4O12S]+ requires m/z 539.1654, found m/z 539.1675.
(2S,11R,16S)-16-Amino-11-((carboxymethyl)carbamoyl)-2-(hydroxymethyl)-4,13-dioxo-5-oxa-9-thia-3,12-diazaheptadecane-1,17-dioic Acid (Table 4, Entry 8)
To a 1.5 dram vial were added 119.8 mg (0.633 mmol) Alloc-Ser-OH, 77.2 mg glutathione (0.251 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 13.3 mg (0.124 mmol) p-toluidine, and 1.25 mL Millipore water. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 2 h. Upon completion of the reaction, mixture was directly loaded onto a column of Sephadex LH-20 for purification (100% methanol). Obtained 96.7 mg (0.195 mmol, 78% yield) of white solid. Experiment 2: 117.3 mg (0.620 mmol) Alloc-Ser-OH, 77.0 mg glutathione (0.251 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 12.2 mg (0.114 mmol) p-toluidine, and 1.25 mL Millipore water. Isolated 113.4 mg (0.228 mmol, 91% yield). IR (ATR) 3299, 3077, 2940, 1697, 1647 cm–1; 1H NMR (500 MHz, D2O) δ 4.56 (dd, J = 8.4, 5.2 Hz, 1H), 4.32 (t, J = 4.1 Hz, 1H), 4.15 (t, J = 5.4 Hz, 2H), 4.00 (s, 2H), 3.90 (qd, J = 11.7, 4.5 Hz, 2H), 3.04 (dd, J = 14.1, 5.1 Hz, 1H), 2.86 (dd, J = 14.0, 8.6 Hz, 1H), 2.68–2.49 (m, 5H), 2.28–2.13 (m, J = 7.4 Hz, 2H), 1.91 (p, J = 6.6 Hz, 2H); 13C NMR (126 MHz, D2O) δ 174.5, 174.1, 173.0, 172.9, 172.1, 163.0 (q, J = 35.5 Hz, TFA) 158.3, 116.3 (q J = 291.6 Hz, TFA), 64.2, 61.3, 56.1, 53.1, 52.7, 41.2, 32.8, 31.0, 28.2, 28.0, 25.6; HRMS (ESI) calcd for [C17H28N4O11S]+ requires m/z 497.1549, found m/z 497.1551.
(2S,11R,16S)-16-Amino-11-((carboxymethyl)carbamoyl)-2-(2-(methylthio)ethyl)-4,13-dioxo-5-oxa-9-thia-3,12-diazaheptadecane-1,17-dioic Acid (Table 4, Entry 9)
To a 1.5 dram vial were added 145.8 mg (0.625 mmol) Alloc-Met-OH, 77.2 mg glutathione (0.251 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 13.7 mg (0.128 mmol) p-toluidine, and 1.25 mL Millipore water. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 2 h. Upon completion of the reaction, mixture was directly loaded onto a column of Sephadex LH-20 for purification (100% methanol). Obtained 105.6 mg (0.195 mmol, 78% yield) of white solid. Experiment 2: 146.6 mg (0.628 mmol) Alloc-Met-OH, 75.2 mg glutathione (0.245 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 14.1 mg (0.131 mmol) p-toluidine and 1.25 mL Millipore water. Isolated 98.2 mg (0.182 mmol, 73% yield). IR (ATR) 3300, 3076, 2965, 2921, 1701, 1654 cm–1; 1H NMR (400 MHz, D2O) δ 4.58 (dd, J = 8.7, 5.1 Hz, 1H), 4.32 (dd, J = 9.4, 4.5 Hz, 1H), 4.16 (d, J = 35.3 Hz, 2H), 3.99 (s, 2H), 3.85 (t, J = 6.4 Hz, 1H), 3.06 (dd, J = 14.1, 5.2 Hz, 1H), 2.88 (dd, J = 14.1, 8.7 Hz, 1H), 2.76–2.46 (m, 6H), 2.18 (q, J = 7.4 Hz, 3H), 2.11 (s, 3H), 1.92 (p, J = 6.7 Hz, 3H); 13C NMR (101 MHz, D2O) δ 176.5, 174.7, 173.3, 173.3, 172.7, 158.2, 64.1, 53.6, 53.2, 53.1, 41.4, 32.8, 31.2, 30.2, 29.5, 28.2, 28.0, 26.0, 14.1. HRMS (ESI) calcd for [C19H32N4O10S2]+ requires m/z 541.1633, found m/z 541.1634.
(2S,11R,16S)-16-Amino-11-((carboxymethyl)carbamoyl)-2-(4-hydroxybenzyl)-4,13-dioxo-5-oxa-9-thia-3,12-diazaheptadecane-1,17-dioic Acid (Table 4, Entry 10)
To a 1.5 dram vial were added 164.3 mg (0.619 mmol) Alloc-Tyr-OH, 75.7 mg glutathione (0.246 mmol), 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 13.2 mg (0.123 mmol) p-toluidine and 1.25 mL Millipore water. The vial was sealed with a Teflon cap and irradiated with blue LEDs for 2 h. Obtained 116.7 mg (0.204 mmol, 89% yield) of a colorless oil. Experiment 2: 78.0 mg (0.254 mmol) glutathione, 162.2 mg (0.611 mmol) Alloc-Tyr-OH, 0.4 mg (0.462 μmol) Ru(bpz)3(PF6)2, 13.5 mg p-toluidine (0.126 mmol), and 1.25 mL water. Isolated 137.5 mg (0.240 mmol, 95% yield). IR (ATR) 3216, 2961, 1649, 1515 cm–1; 1H NMR (500 MHz, D2O) δ 7.06 (d, J = 7.8 Hz, 2H), 6.75 (d, J = 8.0 Hz, 2H), 4.45 (dd, J = 8.3, 5.4 Hz, 1H), 4.31 (dd, J = 8.8, 5.0 Hz, 1H), 3.96 (m, 2H), 3.89 (s, 2H), 3.78 (t, J = 6.4 Hz, 1H), 3.06 (dd, J = 14.2, 4.5 Hz, 1H), 2.92 (dd, J = 14.2, 5.2 Hz, 1H), 2.76 (m, 2H), 2.45 (m, 4H), 2.08 (q, J = 6.9 Hz, 2H), 1.71 (m, 2H); 13C NMR (126 MHz, D2O) δ 175.8, 174.6, 173.1, 172.8, 157.9, 154.3, 128.6, 115.3, 63.8, 55.6, 53.4, 53.0, 48.8, 41.2, 36.2, 32.7, 31.0, 28.0, 27.8, 25.8; HRMS (ESI) calcd for [C23H32N4O11S]+ requires m/z 573.1862, found m/z 573.1855.
Acknowledgments
E.L.T. wishes to thank the members of the Yoon group for enabling the completion of this work. We thank Prof. Eric Strieter and Ellen Valkovich for their valuable perspective on thiol–ene chemistry. Financial support was provided by the National Institutes of Health (GM095666). The NMR spectroscopy facility at UW-Madison is funded by the NSF (CHE-1048642).
Supporting Information Available
Experimental details for BDE calculations and CV measurements, experiments veryfing the necessity for constant irradiation, and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Center for Molecular Electrocatalysis, Pacific Northwest National Laboratory, P.O. Box 999, Richland, Washington 99352, United States.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Dempsey J. L.; Winkler J. R.; Gray H. B. Chem. Rev. 2010, 110, 7024–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hartings M. R.; Kurnikov I. V.; Dunn A. R.; Winkler J. R.; Gray H. B.; Ratner M. A. Coord. Chem. Rev. 2010, 254, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Barton J. K.; Olmon E. D.; Sontz P. A. Coord. Chem. Rev. 2011, 255, 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gill M. R.; Thomas J. A. Chem. Soc. Rev. 2012, 41, 3179–3192. [DOI] [PubMed] [Google Scholar]; e Concepcion J. J.; Jurss J. W.; Brennaman A. O. T.; Iha N. Y. M.; Templeton J. L.; Meyer T. J. Acc. Chem. Res. 2009, 42, 1954–1965. [DOI] [PubMed] [Google Scholar]; f Alstrum-Acevedo J. H.; Brennaman M. K.; Meyer T. J. Inorg. Chem. 2005, 44, 6802–6827. [DOI] [PubMed] [Google Scholar]
- For recent reviews, see:; a Zeitler K. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. [DOI] [PubMed] [Google Scholar]; b Yoon T. P; Ischay M. A.; Du J. Nat. Chem. 2010, 2, 527–532. [DOI] [PubMed] [Google Scholar]; c Narayanam J. M. R.; Stephenson C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; d Teply F. Collect. Czech. Chem. Commun. 2011, 76, 859–917. [Google Scholar]; e Tucker J. W.; Stephenson C. R. J. J. Org. Chem. 2012, 77, 1617–1622. [DOI] [PubMed] [Google Scholar]; f Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 133, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ischay M. A.; Anzovino M. E.; Du J.; Yoon T. P. J. Am. Chem. Soc. 2008, 130, 12886–12887. [DOI] [PubMed] [Google Scholar]; b Du J.; Yoon T. P. J. Am. Chem. Soc. 2009, 131, 14604–14605. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lu Z.; Shen M.; Yoon T. P. J. Am. Chem. Soc. 2011, 133, 1162–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hurtley A. E.; Cismesia M. A.; Ischay M. A.; Yoon T. P. Tetrahedron 2011, 67, 4442–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Tyson E. L.; Farney E. P.; Yoon T. P. Org. Lett. 2012, 14, 1110–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lin S.; Ischay M. A.; Fry C. G.; Yoon T. P. J. Am. Chem. Soc. 2011, 133, 19350–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Parrish J. D.; Ischay M. A.; Lu Z.; Guo S.; Peters N. R.; Yoon T. P. Org. Lett. 2012, 14, 1640–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ischay M. A.; Ament M. S.; Yoon T. P. Chem. Sci. 2012, 3, 2807–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischay M. A.; Lu Z.; Yoon T. P. J. Am. Chem. Soc. 2010, 132, 8572–8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]; b Finn M. G.; Fokin V. V. Chem. Soc. Rev. 2010, 39, 1231–1232. [DOI] [PubMed] [Google Scholar]
- For reviews, see:; a Hoyle C. E.; Lee T. Y.; Roper T. J. Polym. Sci. Part A: Polym. Chem. 2004, 42, 5301–5338. [Google Scholar]; b Sletten E.; Bertozzi C. R. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hoyle C. E.; Bowman C. N. Angew. Chem., Int. Ed. 2010, 49, 1540–1573. [DOI] [PubMed] [Google Scholar]
- a Dondoni A.; Massi A.; Nanni P.; Roda A. Chem.—Eur. J. 2009, 15, 11444–11449. [DOI] [PubMed] [Google Scholar]; b Trang V. H.; Valkevich E. M.; Minami S.; Chen Y.-C.; Ge Y.; Strieter E. R. Angew. Chem., Int. Ed. 2012, 51, 13085–13088. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Valkevich E. M.; Guenette R. M.; Sanchez N. A.; Chen Y.-C.; Ge Y.; Strieter E. R. J. Am. Chem. Soc. 2012, 134, 6916–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Deforest C. A.; Anseth K. S. Nat. Chem. 2011, 3, 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson E. L.; Ament M. S.; Yoon T. P. J. Org. Chem. 2013, 78, 2046–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutchley R. J.; Lever A. B. P. J. Am. Chem. Soc. 1980, 102, 7128–7129. [Google Scholar]
- Bordwell F. G.; Zhang X.-M.; Satish A. V.; Cheng J.-P. J. Am. Chem. Soc. 1994, 116, 6605. [Google Scholar]
- Miyashita T.; Matsuda M. Bull. Chem. Soc. Jpn. 1985, 58, 3031–3032. [Google Scholar]
- a Winterle J. S.; Kliger D. S.; Hammond G. S. J. Am. Chem. Soc. 1976, 98, 3719–3721. [Google Scholar]; b Demas J. N.; Harris E. W.; McBride R. P. J. Am. Chem. Soc. 1977, 99, 3547. [Google Scholar]; c Lin S.; Ischay M. A.; Fry C. G.; Yoon T. P. J. Am. Chem. Soc. 2011, 133, 19350–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cramer N. B.; Davies T.; O’Brien A. K.; Bowman C. N. Macromolecules 2003, 36, 4631–4636. [Google Scholar]; b Cramer N. B.; Reddy S. K.; O’Brien A. K.; Bowman C. N. Macromolecules 2003, 36, 7964–7969. [Google Scholar]
- Miyashita T.; Matsuda M. Bull. Chem. Soc. Jpn. 1981, 54, 1740–1742. [Google Scholar]
- a Brown H. C.; McDaniel D. H.; Hafliger O. In Determination of Organic Structures by Physical Methods; Braude E. A., Nachod F. C., Eds.; Academic Press: New York, 1955; Vol. 1, pp 567–662. [Google Scholar]; b Okazaki H.; Onishi K.; Soeda M.; Ikefuji Y.; Tamura R.; Mochida I. Bull. Chem. Soc. Jpn. 1990, 63, 3167–3174. [Google Scholar]
- a Anderson C. P.; Salmon D. J.; Meyer T. J.; Young R. C. J. Am. Chem. Soc. 1977, 99, 1980–1982. [Google Scholar]; b Bock C. R.; Connor J. A.; Cutierrez A. R.; Meyer T. J.; Whitten D. G.; Sullivan B. P.; Nagle J. K. J. Am. Chem. Soc. 1979, 101, 4815–4824. [Google Scholar]
- Horner J. H.; Martinez F. N.; Musa O. M.; Newcomb M.; Shahin H. E. J. Am. Chem. Soc. 1995, 117, 11124–11133. [Google Scholar]
- For reviews, see:; a Steckhan E. Angew. Chem., Int. Ed. 1986, 25, 683–701. [Google Scholar]; b Steckhan E. Top. Curr. Chem. 1987, 142, 1–69. [Google Scholar]; c Efimov O. N.; Strelets V. V. Coord. Chem. Rev. 1990, 99, 15–53. [Google Scholar]
- Inokuchi T.; Ping L.; Hamaue F.; Izawa M.; Torii S. Chem. Lett. 1994, 23, 121–124. [Google Scholar]
- Platen M.; Steckham E. Tetrahedron Lett. 1980, 21, 511–514. [Google Scholar]
- a Shen Y.; Suzuki K.; Atobe M.; Fuchigami T. J. Electroanal. Chem. 2003, 540, 189–194. [Google Scholar]; b Fuchigami T.; Tetsu M.; Tajima T.; Ishii H. Synlett 2001, 8, 1269–1271. [Google Scholar]; c Fuchigami T.; Mitomo K.; Ishii H.; Konno A. J. Electroanal. Chem. 2001, 507, 30–33. [Google Scholar]
- a Happ B.; Winter A.; Hager M. D.; Schubert U. S. Coord. Chem. Rev. 2012, 41, 2222–2255. [DOI] [PubMed] [Google Scholar]; b Andreiadis E. S.; Chavarot-Kerlidou M.; Fontecave M.; Artero V. Photochem. Photobiol. 2011, 87, 946–964. [DOI] [PubMed] [Google Scholar]; c Teets T. S.; Nocera D. G. Chem. Commun. 2011, 47, 9268–9274. [DOI] [PubMed] [Google Scholar]; d Sakai K.; Ozawa H. Coord. Chem. Rev. 2007, 251, 2753–2766. [Google Scholar]
- a Goren Z.; Willner I. J. Am. Chem. Soc. 1983, 105, 7764–7765. [Google Scholar]; b Maidan R.; Goren Z.; Becker J. Y.; Willner I. J. Am. Chem. Soc. 1984, 106, 6217–6222. [Google Scholar]
- For example of the use of aromatic cosensitizers in photoinduced electron transfer reactions, see:; a Pac C.; Nakasone A.; Sakurai H. J. Am. Chem. Soc. 1977, 99, 5806–5808. [Google Scholar]; b Arnold D. R.; Snow M. S. Can. J. Chem. 1988, 66, 3012–3026. [Google Scholar]; c Yoshimi Y.; Itou T.; Hatanaka M. Chem. Commun. 2007, 5244–5246. [DOI] [PubMed] [Google Scholar]
- a Nicewicz D. A.; MacMillan D. W. C. Science 2008, 322, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nagib D. A.; Scott M. E.; MacMillan D. W. C. J. Am. Chem. Soc. 2009, 131, 10875–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shih H.-W.; Vander Wal M. N.; Grange R. L.; MacMillan D. W. C. J. Am. Chem. Soc. 2010, 132, 13600–13603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kalyani D.; McMurtrey K. B.; Neufeldt S. R.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 18566–18569. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ye Y.; Sanford M. S. J. Am. Chem. Soc. 2012, 134, 9034–9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRocco D. A.; Rovis T. J. Am. Chem. Soc. 2012, 134, 8094–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rillema D. P.; Allen G.; Meyer T. J.; Conrad D. Inorg. Chem. 1983, 22, 1617–1622. [Google Scholar]
- Tyson E. L.; Ament M. S.; Yoon T. P. J. Org. Chem. 2013, 78, 2046–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henessey E. T.; Betley T. A. Science 2013, 340, 590–591. [DOI] [PubMed] [Google Scholar]
- Xu W. Z.; Zhang X.; Kadla J. F. Biomacromolecules 2012, 13, 350–357. [DOI] [PubMed] [Google Scholar]
- Wittrock S.; Becker T.; Kunz H. Angew. Chem., Int. Ed. 2007, 46, 5226–5230. [DOI] [PubMed] [Google Scholar]
- Dondoni A.; Massi A.; Nanni P.; Roda A. Chem.—Eur. J. 2009, 15, 11444–11449. [DOI] [PubMed] [Google Scholar]
- Merbouh N.; Wallner F.; Cociorva O. M.; Seeberger P. H. Org. Lett. 2007, 9, 651–653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.