

Abstract
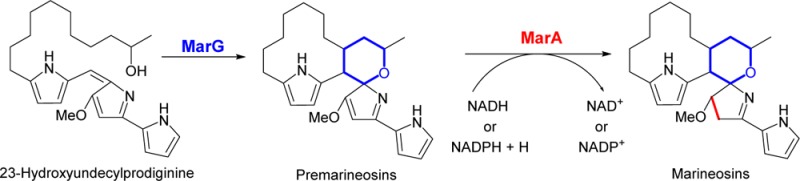
The marine Streptomyces sp. CNQ-617 produces two diastereomers, marineosins A and B. These are structurally related to alkyl prodiginines, but with a more complex cyclization and an unusual spiroaminal skeleton. We report the identification of the mar biosynthetic gene cluster and demonstrate production of marineosins through heterologous expression in a S. venezuelae host named JND2. The mar cluster shares the same gene organization and has high homology to the genes of the red cluster (which directs the biosynthesis of undecylprodiginine) but contains an additional gene, named marA. Replacement of marA in the JND2 strain leads to the accumulation of premarineosin, which is identical to marineosin with the exception that the middle pyrrole (Ring B) has not been reduced. The final step of the marineosin pathway is thus a MarA catalyzed reduction of this ring. Replacement of marG (a homologue of redG that directs undecylprodiginine cyclization to give streptorubin B) in the JND2 strain leads to the loss of all spiroaminal products and the accumulation of 23-hydroxyundecylprodiginine and a shunt product, 23-ketoundecylprodiginine. MarG thus catalyzes the penultimate step of the marineosin pathway catalyzing conversion of 23-hydroxyundecylprodiginine to premarineosin. The preceding steps of the biosynthetic marineosin pathway likely mirror that in the red-directed biosynthetic process, with the exception of the introduction of the hydroxyl functionality required for spiroaminal formation. This work presents the first experimentally supported scheme for biosynthesis of marineosin and provides a new biologically active molecule, premarineosin.
Introduction
Prodiginines, such as prodigiosin (1), undecylprodiginine (2), streptorubin B (3), and metacycloprodiginine (4) (Figure 1), are a family of linear and cyclic oligopyrrole red-pigmented compounds with anticancer, antimalarial, and immunosuppressant activities.1−5 The biosynthesis of members of this class of natural products has been extensively studied.6−13 The biosynthesis of 2 and 3 in Streptomyces coelicolor A(3)2 is directed by the red gene cluster and proceeds via a bifurcated pathway that culminates with the formation of two subunits, 2-undecylpyrrole (2-UP) and 4-methoxy-[2,2′-bipyrrole]-5-carboxaldehyde (MBC) (Scheme 1). RedH then catalyzes the condensation of these two subunits to generate 2, and in the final step of the pathway a non-heme iron Rieske-oxygense, RedG, catalyzes the oxidative cyclization of the alkyl chain of 2 to generate the prodiginine 3 (Scheme 1).10,14 A RedG homologue in S. longispororuber, named mcpG, catalyzes the cyclization of 2 to 4 (Figure 1).14
Figure 1.

Structures of naturally occurring prodiginines (1–4) and marineosins (5 and 6).
Scheme 1. Bifurcated Pathway Leading to the Biosynthesis of Undecylprodiginine (2) and Streptorubin B (3) in S. coelicolor A(3)2.

Red color is for parts of the structure derived from acetate; green, from glycine and l-serine; blue, from l-proline. A = adenylation domain, KS = ketide synthase, AT = acyltransferase, GT = glycine transferase, ST = serine transferase, HBP = 4-hydroxy-5-hydroxymethyl-2,2′-bipyrrole, 2-UP = 2-undecylpyrrole subunit, MBC = 4-methoxy-[2,2′-bipyrrole]-5-carboxaldehyde subunit.
Marineosins A (5) and B (6) (Figure 1) are two prodiginine analogues isolated from the marine Streptomyces sp. CNQ-617 and were shown to have strong and selective anticancer activity.15 Marineosins are novel prodiginines in that they have a different regioselectivity for the alkyl chain cyclization, a partially reduced tripyrrole nucleus, and an oxygen. The oxygen is incorporated into a spiroaminal ring. Spiroaminal moieties are present in a number of marine-derived natural products such as crambescidin 800 and crambescidin 816 (compounds with strong antiviral and antimalarial activities) but are generally rare in natural products.16,17 The intriguing structure and biological activity of marineosins has spurred efforts toward total synthesis attempts. Although synthesis of spiroaminal containing compounds has been reported by several research groups,16,17 the total synthesis of marineosins is still incomplete.18,19 The ability to access marineosin analogues and evaluate their biological activity is thus currently elusive.
The unique structure of marineosins cannot be directly explained on the basis of the current knowledge of prodiginines biosynthesis and has spurred two disparate biosynthetic hypotheses.15,18 The first hypothesis was formation of an enone-undecylprodiginine intermediate that undergoes a hetero Diels–Alder cyclization to yield marineosins A (5) and B (6) (Supplementary Figure S1).15 The proposed hetero-Diels–Alder cyclization was subsequently found to be energetically unfavorable.19 A second hypothesis proposed that a homologue of the non-heme iron Rieske oxygenase, RedG must be encoded by the marineosin gene cluster (Supplementary Figure S2). This RedG-homologue consecutively catalyzes the oxidative cyclization of the alkyl chain of 2 to form the macrocyclic ring and hydroxylation of the alkyl chain to initiate the formation of the spiroaminal ring of marineosin. Finally, the spiroaminal ring formation is induced by a series of transformations, such as a 1,5-sigmatropic hydride shift, cyclization, and tautomerization (Supplementary Figure S2).18 To date, no experimentally supported biosynthetic pathway for the marineosins has been reported.
In this study we sought to identify the marineosin (mar) gene cluster, in order to both provide the tools to study the biosynthetic process and access novel marineosin analogues for biological testing. We report cloning, sequencing, and analysis of this cluster and successful production of marineosin by expression of this cluster in a heterologous host. Through construction of gene mutants, the final steps in the marineosin biosynthetic process have been delineated and shown to differ from those previously proposed. In addition, a number of new marineosin intermediates and shunt metabolites have been purified and characterized, and their antimalarial activity was determined.
Experimental Section
General
NMR spectra were measured in acetone-d6 using a Bruker AMX-600 MHz spectrometer calibrated using residual acetone as an internal standard. High resolution mass spectra analyses and Tandem MS analyses were performed using ThermoElectron LTQ-Orbitrap high resolution mass spectrometer. LC–MS/UV analyses were performed using a MicroTOF-Q mass spectrometer equipped with an Agilent 1100 series HPLC with MWD detector. HPLC purification of marineosin and analogues was performed using either a Waters HPLC equipped with a DWD detector or an Agilent HPLC equipped with a MWD detector.
Bacterial Strains, Plasmids, and Reagents
Topo-pcr8 vector and chemically competent Oneshot Top10 E. coli were purchased from Life Technologies and were used in routine cloning and subcloning of plasmid and cosmid DNA. E. coli strains used in Red/ET-mediated recombination were provided by the John Innes Institute. A list of major strains and cosmids generated in this study can be found in Supplementary Tables S1 and S2. All antibiotics, solvents, and HPLC grade solvents were purchased from either Sigma-Aldrich or Fisher-Scientific. Marineosin standard was provided by Dr. William Fenical (Scripps Institute of Oceanography, San Diego, CA).
DNA Manipulation
PCR amplifications were carried out using either Platinum high-fidelity (Life Technologies) or Phusion high-fidelity polymerase (New England Biolabs). Genomic and plasmid DNA purification, restriction analysis, transformation, and conjugation of E. coli and S. venezuelae strains were performed as described.21,24,25 A list of primers used throughout this study can be found in Supplementary Table S3. All manipulations were done according to manufacturer’s protocol unless otherwise stated.
Construction of Genome-Integrative pMAR Cosmid and the Heterologous Expression of mar Gene Cluster
The screening of the genomic library of Streptomyces sp. CNQ-617 led to the identification of 8A7 cosmid carrying the entire mar gene cluster. The mar cluster was sequenced and annotated and the complete sequence was deposited into Genbank (accession KF711829). The ampicillin-resistance gene bla of 8A7 cosmid was replaced with oriT, ΦC31 int, and aac(3)IV to allow for site-specific integration of 8A7 cosmid into the attB site of S. venezuelae genome using Red/ET-mediated recombination (Supplementary Figure S3).23,26 Gene replacement was confirmed via PCR, and the engineered cosmid was named pMAR. The donor E. coli strain harboring the pMAR cosmid E. coli ET12537/pUZ8002/pMAR was conjugated with wild type S. venezuelae to generate S. venezuelae JND2 expressing the marineosin gene cluster. For more information about construction of the genomic library, please refer to Supporting Information.
Isolation and Characterization of Marineosin A Isolated from S. venezuelae JND2
Seed cultures were used to inoculate 1-L fermentations (in 4-L baffled flasks) of S. venezuelae JND2 at 1% ratio. Cultures were then incubated at 30 °C and 220 rpm for 7 days, and cells were harvested by centrifugation at 7000 rpm for 10 min. The harvested cells were disrupted by the addition of acetone followed by sonication. The acetone extract was filtered through cotton wool and evaporated till dryness. Marineosin A (5) was purified using HPLC as specified in Supplementary Table S4 and S5. The 1H NMR, MS, tandem-MS, and LC–MS analyses of 5 were compared to that of a standard marineosin sample.
Construction of marA and marG Gene Replacement Mutants
The marA and marG genes in the pMAR cosmid were replaced with the spectinomycin resistance marker aadA gene using Red/ET mediated recombination23 to generate pMARΔAand pMARΔG cosmids, respectively. pIJ788 plasmid was used as a template for the amplification of the aadA gene using recombination primers specified in Supplementary Table S3. Gene replacements were confirmed via PCR amplification using outer primers (Supplementary Table S3). The pMARΔA cosmid was transformed into dam-/dcm-E. coli ET12537/pUZ8002 to generate an E. coli ET12537/pUZ8002/pMARΔA donor strain. This was subsequently conjugated with wild type S. venezuelae to generate S. venezuelae JND2ΔA strain. Fresh protoplasts of wild type S. venezuelae were transformed with pMARΔG cosmid to generate S. venezuelae JND2ΔG strain.
Isolation and Characterization of Premarineosin A (9) and 16-Ketopremarineosin A (10) from S. venezuelae JND2ΔA
Seed cultures of S. venezuelae JND2ΔA were used to inoculate 60 and 100 mL of SCM media in 300- and 500-mL baffled flasks, respectively, supplied with the appropriate antibiotics. Seed cultures were used at 1% ratio. It should be noted that no production was observed when larger fermentations were used. The cultures were incubated at 30 °C, 220 rpm for 6 days. The cells were harvested as before, and the cells were extracted with acetone followed by sonication. The filtered, dried crude acetone extract was dissolved in methanol and was partially purified by HPLC using a gradient specified in Supplementary Table S6. Peaks with retention times of 15 and 21 min, corresponding to 16-ketopremarineosin A (10) and premarineosin A (9), respectively, were collected and evaporated under vacuum, and residual water was eliminated via lyophilization. The semipure compounds were subjected to another round of HPLC purification using the same specified conditions to yield 1.0 and 0.5 mg of 10 and 9, respectively.
Isolation and Characterization of 23-Hydroxyundecylprodiginine (7) and 23-Ketoundecylprodiginine (8) from S. venezuelae JND2ΔG
Seed cultures were used to inoculate 1-L fermentations (in 4-L baffled flasks) of S. venezuelae JND2ΔG at 1% ratio, and then the fermentations were incubated at 30 °C, 220 rpm for 7 days. Cells were harvested by centrifugation at 7000 rpm for 10 min. Harvested cells were disrupted by the addition of acetone and sonication. The acetone extract was dried under vacuum, and the residual water was extracted with ethyl acetate 3 times. The combined ethyl acetate fractions were evaporated under vacuum. The dried extract was dissolved in methanol and purified according to the gradient specified in Supplementary Table S7a. The peak with retention time of 25 min was collected and evaporated under vacuum, and the residual water was removed by lyophilization. The partially purified sample was dissolved in chloroform and loaded onto 20 cm × 20 cm preparative silica gel 60A TLC plates (Whatman) that were developed using 50% ethyl acetate/hexane 3 times. The top and bottom layers were excised from the TLC plate, extracted with acetone, and then dried under vacuum to yield semipure 23-ketoundecylprodiginine (8) and 23-hydroxyundecylprodiginine (7), respectively. The individual compounds 7 and 8 were obtained in pure form after final HPLC purification using the gradient specified in Supplementary Table S7b.
LC–MS/UV Analysis of S. venezuelae JND2, JND2ΔG, and JND2ΔA Strains
Spores of S. venezuelae JND2 were propagated onto SPA agar containing 50 μg/mL apramycin, and spores of S. venezuelae JND2ΔG and JND2ΔA were propagated onto SPA agar plates containing 50 μg/mL apramycin and spectinomycin antibiotics. All plates were incubated at 30 °C for 5–7 days. Fresh spores were used to inoculate seed cultures of 10 mL of SCM media21 supplied with the appropriate antibiotics, and the cultures were incubated at 30 °C and 220 rpm for 72 h. The cells were harvested by centrifugation at 4000 rpm for 5 min and then extracted with acetone and sonication. The acetone extract was dried under vacuum, and then the crude extract was dissolved in methanol (0.5 mg/mL) to be used in MS and LC–MS/UV analysis according to protocols in Supplementary Tables S8–S10. Analyses were done in comparison to extracts of wild type S. venezuelae prepared under the same conditions.
Results and Discussion
A red Gene Cluster Homologue Directs the Biosynthesis of Marineosin in Streptomyces CNQ-617
We hypothesized that the streptorubin B biosynthetic pathway (Scheme 1) and marineosin pathway would have strong similarities and that the differences between the two structures would likely be in the formation of the 2-UP subunit (Scheme 1) and in the latter stages of the biosynthetic process. The similarities in the structures suggested they likely follow an identical route to the MBC subunit. The biosynthesis of the MBC subunit of undecylprodiginine (2) in S. coelicolor A(3)2 starts with the oxidation of the amino acid l-proline into pyrroline in a step catalyzed by the acyl-CoA dehydrogenase RedW (Scheme 1). Thus a 32P-labeled redW probe was used to screen a SuperCos1 genomic library of Streptomyces sp. CNQ-617 (as described in the Experimental Section). A secondary screen to determine if these cosmids contained other red homologues was then carried out. Degenerate PCR primers for redG, which encodes a non-heme iron Rieske oxygenase (RedG) that catalyzes oxidative cyclization of undecylprodiginine (2) into streptorubin B (3), were used on the basis that there must be a redG homologue in the marineosin pathway. The 15 positive cosmids from redW screening of the genomic cosmid library of Streptomyces CNQ-617 were thus screened using marG_FWD and marG_REV primers (Supplementary Table S3), leading to the identification of the 8A7 cosmid. Shotgun sequencing of the 8A7 insert revealed 21 genes with strong homology to the red biosynthetic genes. Not only was every gene involved in the streptorubin biosynthetic process present in the putative mar gene cluster, but the gene organization was identical (Figure 2). The homology between the red and mar gene products varied between 53 and 79% (Table 1). The lowest homology (less than 60%) was noticed among regulatory and hypothetical proteins such as RedD RedY, RedZ, RedV, and RedU and their respective Mar homologues. The highest homology was noticed between RedR and RedP and their respective Mar homologues, MarR and MarP (79% and 75%, respectively). It should be noted that RedP and RedR initiate the biosynthesis of the 2-UP subunit of 2 (Scheme 1), suggesting that the mar and red pathways are probably identical at these steps. Other proteins involved in the biosynthesis of the 2-UP subunit such as RedQ, RedJ, and RedL share a homology of 61–69% relative to their Mar counterparts (Table 1). Most of the genes involved in the biosynthesis of the MBC subunit of 2 (Scheme 1) such as RedW, RedO, RedN, RedM, and RedI share a homology of 69–79% with the Mar proteins (Table 1). The PEP-utilizing RedH protein (responsible for the condensation of the 2-UP and MBC subunits) has a homology of 70% to MarH, while the Rieske oxygenase RedG (responsible for the oxidative cyclization of 2 to 3) has a homology of 64% to the MarG homologue (Table 1). Sequencing of the 8A7 insert also revealed the presence of 6 kbp of primary metabolic genes at the 3′ end of the mar gene cluster. At the far end of the cluster and before these primary metabolic genes was a gene that we designated marA. A conserved domain analysis of the primary amino acid sequence of MarA using Conserved Domain Database (CDD) suggests that MarA belongs to the Hot-Dog superfamily of proteins that is functionally divided mainly into dehydratases/hydratases and thioesterases.20 No other apparent homologies to known proteins or conserved domains were noticed from the primary amino acid sequence of MarA (Supplementary Figure S4).
Figure 2.
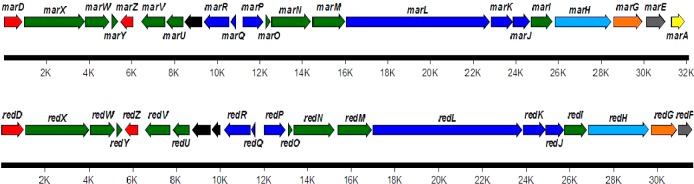
Comparison between the mar gene cluster (top panel) and the red cluster (bottom). Both gene clusters show almost identical gene organization. Red color is for regulatory proteins; green, involved in the biosynthesis of the MBC subunit; dark blue, involved in the biosynthesis of the 2-UP subunit; light blue, condensation of the MBC and 2-UP subunits; orange, oxidative cyclization; yellow, pyrrole reduction; gray, membrane protein; black, hypothetical proteins.
Table 1. Marineosin Gene Cluster and Proposed Functions.
| gene | length (aa) | proposed function | nearest NCBI BLAST hit (identity, similarity) |
|---|---|---|---|
| marD | 300 | RedD, putative transcriptional regulator | Streptomyces coelicolor A3(2) NP_629999 (56%, 67%) |
| marX | 963 | RedX, polyketide synthase | Streptomyces coelicolor A3(2) NP_630000 (59%, 69%) |
| marW | 387 | RedW, acyl-CoA dehydrogenase | Streptomyces coelicolor A3(2) NP_630001 (78%, 86%) |
| marY | 105 | RedY, proposed in binding MBC and HBP bipyrrole intermediates | Streptomyces coelicolor A3(2) NP_630002 (58%, 70%) |
| marZ | 216 | RedZ, response regulator | Streptomyces coelicolor A3(2) NP_733677 (54%, 71%) |
| marV | 388 | RedV, hypothetical protein | Streptomyces coelicolor A3(2) NP_630003 (53%, 64%) |
| marU | 279 | RedU, putative 4′-phosphopantetheinyl transferase | Streptomyces coelicolor A3(2) NP_630004 (59%, 68%) |
| marT | 295 | Hypothetical protein | Streptomyces coelicolor A3(2) NP_630005 (65%, 76%) |
| marR | 409 | RedR, 3-oxoacyl-ACP-synthase II | Streptomyces coelicolor A3(2) NP_630007 (79%, 88%) |
| marQ | 110 | RedQ, acyl carrier protein | Streptomyces coelicolor A3(2) NP_630008 (67%, 76%) |
| marP | 333 | RedP, 3-oxoacyl-ACP synthase | Streptomyces coelicolor A3(2) NP_630009 (75%, 84%) |
| marO | 87 | RedO, putative peptidyl carrier protein | Streptomyces coelicolor A3(2) NP_630010 (72%, 81%) |
| marN | 632 | RedN, 8-amino-7-oxononanoate synthase | Streptomyces coelicolor A3(2 NP_630011 (72%, 79%) |
| marM | 531 | RedM, peptide synthase | Streptomyces coelicolor A3(2) NP_630012 (69%, 77%) |
| marL | 2253 | RedL, polyketide synthase | Streptomyces coelicolor A3(2) NP_630013 (60%, 69%) |
| marK | 355 | RedK, oxidoreductase | Streptomyces coelicolor A3(2 NP_630014 (72%, 81%) |
| marJ | 277 | RedJ, thioesterase | Streptomyces coelicolor A3(2) NP_630015 (67%, 73%) |
| marI | 346 | RedI, methyltransferase | Streptomyces coelicolor A3(2) NP_630016 (71%, 84%) |
| marH | 900 | RedH, phosphoenolpyruvate-utilizing enzyme | Streptomyces coelicolor A3(2) NP_733678 (70%, 80%) |
| marG | 451 | RedG, oxidase | Streptomyces coelicolor A3(2) NP_630017 (64%, 80%) |
| marE | 317 | Hypothetical protein, Putative acyltransferase | Streptomyces coelicolor A3(2) NP_630019 (78%, 87%) |
| marAa | 155 | Premarineosin reductase | Streptomyces hygroscopicusYP_006247081 (56%, 73%) |
Annotation is based on gene replacement experiments detailed in this study.
The striking similarity between the putative mar cluster and the red cluster and in particular the presence of homologues of every biosynthetic enzyme raised the question of whether it encoded marineosin biosynthesis (rather than undecylprodiginine biosynthesis), and if so, whether all of the necessary genes were present. To investigate this, we sought to move the entire mar gene cluster into a heterologous host to determine the products encoded by the cluster. To facilitate this transfer, the bla gene of 8A7 cosmid was replaced with attP attachment site, integrase gene of phage ΦC31, and aac(3)IV gene (conferring resistance to apramycin). The resulting pMAR cosmid was capable of site-specific integration into another streptomycete. The pMAR cosmid was transformed into E. coli ET12537 harboring pUZ8002 plasmid to be used in conjugation with wild type S. venezuelae, to ultimately provide the apramycin resistant strain S. venezuelae JND2 (Supplementary Figure S3). The engineered JND2 strain grew and sporulated in a manner similar to the wild type S. venezuelae but was characterized by a distinctive red color (Figure 3). The marineosins, by virtue of the partially reduced middle pyrrole, do not have a red color. The JND2 strain was grown under standard shake flask conditions for production of the natural products pikromycin and methymycin.21 An MS profile of the acetone extract of JND2 strain showed accumulation of not only these natural products but also a compound not present in wild type S. venezuelae with [M + H] m/z 410.28, identical to that reported for marineosin15 (Supplementary Figure S5). Analysis by HPLC revealed that the compound has the same retention time (Supplementary Figure S6) and UV profiles (λmax 320 nm) as a marineosin standard. The compound was further purified, and its identity was confirmed as marineosin A (5) by spectroscopic analysis including MS/MS and 1H NMR (Supplementary Figures S7–S9). The marineosin A (5) yields from the JND2 strain were approximately 5 mg/L. Yields of marineosins from Streptomyces CNQ-617 in contrast were reported as 0.5 mg/L.15 A compound with the same mass [M + H] m/z 410.3 was also observed in a very minor quantity after the peak of marineosin A (5) (Figure 4) and was not purified and characterized but is likely marineosin B (6). The production of marineosin A (5) and B (6) in the JND2 strain provides unequivocal evidence that the mar cluster encodes marineosin biosynthesis and that this pathway proceeds in a manner analogous at many steps to that of other prodiginines. In addition to marineosins, the LC–MS/UV profile of the JND2 strain (Figure 4) indicates the accumulation of a myriad of pathway intermediates and, possibly, shunt-metabolites. There were no reports of additional compounds being observed in the streptomyces CNQ-617 host, and the presence of these may be a result of either higher production yields or other factors associated with expression in S. venenzuelae.
Figure 3.
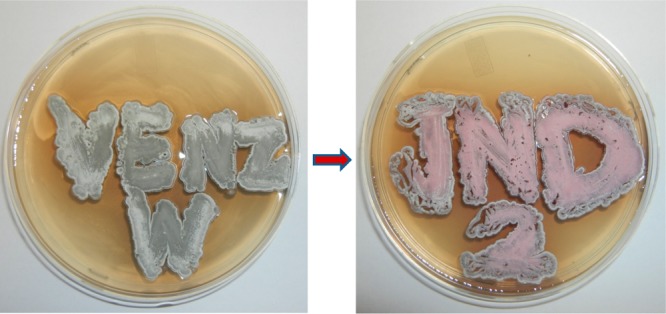
SPA agar plates showing change in phenotype of wild type S. venezuelae (left) from gray to red color as a result of introduction of the mar gene cluster, yielding the JND2 strain (right).
Figure 4.
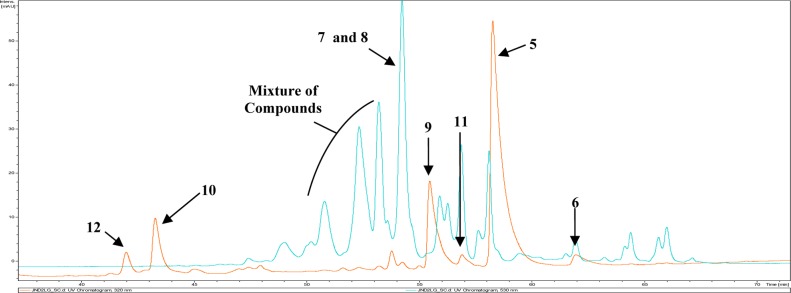
LC–MS/UV profile of S. venezuelae JND2. Two groups of compounds are accumulated due to expression of the marineosin gene cluster. The first group consists of red-colored compounds (7, 8, and mixture of other compounds) with λmax of 530 nm (blue chromatogram), while the second group consists of marineosin A (5), B (6), premarineosin A and B (9 and 11, respectively), and shunt metabolites (10 and 12) with λmax of 320 nm (orange chromatogram).
The manner in which key structural differences of marineosins arise was not clear from an examination of the mar gene cluster alone. To answer these questions, we looked at the additional compounds generated by the JND2 strain and by derivatives of the JND2 strain in which key mar genes had been replaced. Two groups of compounds were identified in the JND2 strain, a group of conjugated red-colored compounds with λmax of 530 nm characteristic to prodiginines and a group of nonconjugated compounds with λmax of 320 nm similar to that of marineosin (Figure 4). We posited that if the red-colored compounds in the JND2 strain are pathway intermediates (rather than shunt metabolites), the reduction of middle pyrrole (Ring B) and hence loss of aromatic ring conjugation of marineosin would be one of the final steps in the biosynthetic process, and thus the biosynthesis of marineosin would proceed via an intermediate analogue of 2. The main compounds within this group were 7 and 8 (Figure 5) with [M + H] m/z 410.28, and m/z 408.26, respectively. Compounds with the same mass and retention time were subsequently purified from a JND2ΔG mutant strain (see below) and shown to be 23-hydroxyundecylprodiginine (7) and 23-ketoundecylprodiginine (8), respectively. No detectable levels of production of undecylprodiginine (2) were observed in extracts of JND2 fermentations. The group of compounds characterized with λmax of 320 nm comprises marineosin A (5) and B (6) and the four other analogues 9, 10, 11, and 12 (Figure 5), with [M + H] m/z 408.26, 422.24, 408.26, and 424.26 with corresponding molecular formulas of C25H34O2N3, C25H32O3N3, C25H34O2N3, and C25H34O3N3. Compounds with the same mass and retention time as 9 and 10 were subsequently isolated from a JND2ΔA mutant and were shown to be premarineosin A (9) and a 16-ketopremarineosin A (10), respectively. Compounds 11 and 12 were not purified and characterized but are likely premarineosin B and 16-hydroxypremarineosin A, respectively (Figure 5).
Figure 5.

Structures of isolated compounds 7–10 and expected compounds 11 and 12 from S. venezuelae JND2ΔG and JND2ΔA.
MarA Is Required for the Last Step in Marineosin Biosynthesis, Selective Reduction of Middle Pyrrole (Ring B)
The most notable difference between the mar and red gene clusters is the presence of marA. We posited that this was required for marineosin biosynthesis, and that MarA might catalyze formation of the spiroaminal ring via an intramolecular hydroalkoxylation reaction. To test this hypothesis we replaced marA in pMAR cosmid with the spectinomycin resistance aadA gene. The resulting pMarΔA was transformed into E. coli ET12537/pUZ8002 and then used in conjugation with wild type S. venezuelae to provide S. venezuelae JND2ΔA. This strain is both apramycin- and spectinomycin-resistant and has a fainter red color when compared to either the JND2 or JND2ΔG strains (see below). Marineosin production was not observed in this mutant demonstrating that marA is essential for biosynthesis of marineosin (Supplementary Figure S10a). However, LC–MS analysis of the acetone extract of JND2ΔA strain (Supplementary Figure S10b) indicates the accumulation of two major and one minor compounds with λmax 320 nm, premarineosin A (9), 16-ketopremarineosin A (10), and premarineosin B (11) with [M + H] m/z 408.2660, m/z 422.2450, and m/z 408.26 and corresponding molecular formulas C25H34O2N3, C25H32O3N3 and C25H34O2N3 respectively (Supplementary Figure S11–14). These three compounds were also observed in the acetone extract of JND2 strain (Figure 4).
Larger scale fermentations of S. venezuelae JND2ΔA permitted both 9 and 10 to be purified and characterized by IR, MS, and extensive 2D NMR experiments. The mass spectral analysis of 9 indicated two fewer hydrogens than the marineosins (5 and 6), while 10 has four fewer hydrogens and additional oxygen. The IR spectra of 9 and 10 are almost identical except for the presence of a peak at 1705 cm–1 in the IR spectrum of 10 indicating the presence of a carbonyl (C=O) group (Supplementary Figure S15). The NMR experiments clearly demonstrated that both compounds had an intact tripyrrole skeleton. One of the significant differences in the 1H NMR spectra of 9, 10, and marineosin is the appearance of six aromatic protons in both 9 and 10 instead of the five protons in marineosin. COSY and TOCSY correlations among aromatic protons at C-1, C-2, and C-3 as well as those at C-11 and C-12 confirm that the six protons form the tripyrrole nucleus (Figure 5). The COSY correlations between protons at C-9 and C-21 that are extended to C-20 and C-22 in the TOCSY of both 9 and 10 confirm the presence of a macrocyclic ring between C-9 and C-21 (Figure 5). The same macrocyclic ring in 10 was confirmed by the HMBC correlations between the proton at C-9 and carbons C-7, C-8, C-10, C-11, C-20, C-21, and C-22 (Figure 6). The HSQC data (Supplementary Figure S19) suggest that compounds 9 and 10 have eight and seven methylenes in macrocyclic and spiroaminal rings, respectively, which suggests the keto group of 10 has replaced one of the methylene carbons of 9. In compound 10, HMBC correlations between a carbonyl carbon at δC 211.3 ppm and protons at C-17 and C-14 suggested that the carbonyl group observed in the IR spectrum of 10 exists in the vicinity of C-17 and C-14 (Figure 6). The TOCSY spectrum of 10 shows strong correlations between protons at C-14 and C-15, but these correlations are not extended to any other proton in the macrocyclic skeleton of 10, suggesting that C-15 is next to either a heteroatom or a quaternary carbon. Also, protons at C-15 and C-17 in 10 are slightly shifted downfield relative to their counterparts in 9 (Supplementary Table S14). Therefore, the keto group of 10 was assigned at C-16 (Figure 6). The methoxy group of 9 and 10 is connected to C-7 on the basis of HMBC correlations from OCH3-25 to C-7 and H-9 to C-7. The C-7 quaternary carbon appears in the most downfield region, which suggests the double bond exists between C-6 and C-7 (Ring B is not reduced); this is the only significant difference between marineosins (5 and 6) and premarineosin A (9). The proton at C-9 demonstrated HMBC correlations to a quaternary carbon at C-8, suggesting the presence of a “spiro carbon”, and the oxygen exists between C-8 and C-23 in both 9 and 10. Full spectral data and correlations of 9 and 10 are presented in detail in Supporting Information (Figures S11–24 and Tables S11–14). Collectively this work is consistent with a structure for 9 identical to that of marineosin A (5), but with a double bond present between C-6 and C-7. A compound (11) with the same mass [M + H] m/z 408.26 (Figure 5) was observed in a very minor quantity after the peak of 9 in the LC–MS/UV of both JND2ΔA (Supplementary Figure S10b) and JND2 strain (Figure 4). Although compound 11 was not purified due to low quantity, it is likely to be a diasteriomer of 9. The production of 9 clearly demonstrates that MarA is not required for formation of the spiroaminal ring. Rather, MarA is likely required for reduction of the C6–C7 olefin (although a clear NADH/NADPH binding site could not be identified from the predicted primary MarA sequence) as the last step in the marineosin biosynthetic pathway and led us to name these compounds premarineosin A (9) and premarineosin B (11) (Scheme 2). The presence of diasteromeric premarineosins indicate that the alternative stereochemical configuration at the spiroaminal carbon (C8) is established before the final biosynthetic step. Efforts thus far to obtain a soluble recombinant MarA to verify this activity in vitro have been unsuccessful. The analyses also show that compound 10 is a 16-ketopremarineosin A, which probably arises from C16-hydroxylation and oxidation of premarineosin A. Compound 12 (Figures 4 and 5) was also not purified due to low quantity but is likely the 16-hydroxypremarineosin A. Both of these compounds are likely shunt metabolites of the main marineosin pathway and may arise either from the action of Mar proteins or enzymes present in the S. venezuelae host.
Figure 6.

Selected 1H→13C HMBC correlations for 8 and 10.
Scheme 2. Marineosin Biosynthetic Pathway.

MarG Is Required for Conversion of a 23-Hydroxyundecylprodiginine (7) to Premarineosins (9 and 11)
The formation of premarineosins (9 and 11) with loss of marA demonstrated that another gene product must be responsible for formation of the spiroaminal ring. Furthermore, this step, formation of the C8–O–C23 bond, and introduction of the hydroxyl group must occur at an earlier step in the pathway. We hypothesized that MarG would likely be responsible for one if not several of these steps. This hypothesis was based on the observation that MarG has homology (64%) to RedG, which catalyzes the oxidative cyclization in the streptorubin B pathway (in this case a cyclization between C-11 and C-20 of 2), and was consistent with an earlier hypothesis that a RedG homologue introduces the hydroxyl group in the marineosin structure.18 Thus we sought the replacement of marG gene in the pMAR cosmid with aadA gene, which confers resistance to spectinomycin via Red/ET-mediated recombination, to generate the pMARΔG cosmid. The pMARΔG cosmid was subsequently transformed into protoplasts of S. venezuelae to generate the JND2ΔG strain. The new strain is both apramycin- and spectinomycin-resistant and is characterized by a dark red color when grown on both solid and liquid media.
MS analysis of the acetone extract of JND2ΔG strain indicates the accumulation of two major red-colored compounds, 23-hydroxyundecylprodiginine (7) and 23-ketoundecylprodiginine (8) (Figure 5), with [M + H] m/z 410.2807 and 408.2622, which correspond to the molecular formulae C25H36O2N3 and C25H34O2N3 respectively. Compounds 7 and 8 were also observed in the acetone extract of JND2 strain (Figure 4 and Supplementary Figure S25). Marineosin production in this mutant was not observed, which demonstrates that MarG, like MarA, is essential for marineosin biosynthesis. Premarineosins (9 and 11) and 16-ketopremarineosin A (10) were also not observed in the JND2ΔG strain. Indeed, LC–MS/UV analysis of JND2ΔG strain demonstrates the accumulation of only red-colored metabolites (Supplementary Figure S26). These observations provide clear evidence that the biosynthesis of marineosin proceeds as a conjugated tripyrrole intermediate that is reduced in a step subsequent to MarG-catalyzed cyclization.
The pure compounds 7 and 8 were obtained by consecutive chromatography on preparative HPLC and preparative TLC. Extensive 1D, 2D NMR studies and side by side comparison with 2 were carried out to elucidate the structures of 7 and 8 (Figure 5). The 1H NMR spectra of 7 and 8 show seven protons at the aromatic region and a singlet at δH 3.91 ppm (3H) and δH 4.12 ppm (3H), respectively. This is similar to 1H NMR data of 2 (Supplementary Table S15), where these proton signals are components of three pyrrole rings with OMe on the middle pyrrole. One of the significant differences in the 1H NMR spectra of 7 and 2 is the appearance of an extra multiplet at δH 3.67 ppm (1H) and one doublet at δH 1.09 ppm (3H) in 7, instead of the triplet of terminal methyl in 2 (Supplementary Figure S31 and Table S15). The multiplet at δH 3.67 ppm (C-23) has correlations with δH 1.09 ppm (C-24) and δH 1.34 ppm (C-22). The δH 1.09 ppm (C-24) doublet has only one correlation with 3.67 ppm (C-23) in the COSY spectrum of 7 (Supplementary Figure S32), supporting the position of the -OH group at C-23. TOCSY correlations among protons at C-21, C-22, C-23, and C-24 also supported the position of the -OH group at C-23 (Supplementary Figure S33). The significant difference in the 1H NMR spectra of 8 and 2 is the appearance of one new triplet at δH 2.46 ppm (2H), and one singlet at δH 2.04 ppm (3H) in 8, instead of the triplet of terminal methyl in 2 (Supplementary Tables S15 and S16). The HSQC spectra confirmed the new methylene at 42.0 ppm and terminal methyl at 26.9 ppm. In the HMBC spectrum, δH 2.46 ppm (2H) protons show correlations with C-24 (δH 2.04, δC 26.9 ppm), C-23 (carbonyl carbon, δC 207.1 ppm), and C-21 (δH 1.30, δC 23.6 ppm); conversely, δH 2.04 ppm (3H) protons show a strong correlation with carbonyl carbon δC 207.1 (C-23) (Figure 6) (Supplementary Table S16). In addition, the 1H–1H correlations between protons of C-22 and C-24 in COSY and TOCSY are missing, which strongly supports the carbonyl functional group position at C-23 (Supplementary Figures S36 and S37). Together these data support the structure assignment of 23-hydroxyundecylprodiginine (7) and 23-ketoundecylprodiginine (8). In addition, total syntheses of 7 and 8 were also undertaken and these compounds were shown to be spectroscopically and chromatographically identical (to be reported elsewhere).
The accumulation of 23-hydroxy- (7), and 23-keto-undecylprodiginines (8) (Figure 5) as a result of marG gene deletion indicates that MarG is essential for the formation of macrocyclic and spiro-tetrahydropyran-aminal rings but is not responsible for the introduction of the oxygen of the spiro-tetrahydropyran ring as postulated by Snider and co-workers.18 Sequence alignments of MarG with other C–C bond-forming non-heme iron Rieske oxygenases such as RedG, McpG, and RphG involved in the biosynthesis of natural products 3, 4, and roseophilin, respectively, together with hydroxylating non-heme iron Rieske oxygneases show that all have conserved N-terminal CXH and CXXH motifs. However, MarG and its homologues have an EXHX4H motif at their C-terminal end rather than the DXHX4H motif characteristic to Rieske hydroxylases (Supplementary Figure S40).14 An aspartate to glutamate mutation of the DXHX4H motif of naphthalene dioxygenase yields an inactive enzyme.22 It might be that the EXHX4H motif in MarG and its homologues is essential to avoid side hydroxylation reactions and achieve selective C–H bond activation and specific C–C bond formation. No detectable levels of undecylprodiginine (2) were observed in this mutant.
The lack of spiroaminal ring-containing intermediates in the JND2ΔG strain demonstrates that the spiroaminal ring formation occurs in the last steps of biosynthesis and must either be part of a reaction catalyzed by MarG or requires the MarG catalyzed formation of C9–C21 macrocycle prior to spiroaminal ring closure (Scheme 2).
Preliminary experiments feeding 7 to a S. venezuelae host with a marG expression plasmid have demonstrated production of 9 and 10. Additionally these products are deuterated when [23-d]-7 is used. This data supports the role of MarG in catalyzing formation of premarineosins directly from 7 and indicates 8 is a shunt metabolite (the deuterium label would be lost in a pathway that proceeded through 8) (Supplementary Figure S41).
An Experimentally Supported Scheme for the Final Steps of Marineosin Biosynthesis
Determination of the major compounds that accumulate in S. venezuelae JND2 and the mix of these that are found in the JND2ΔG and JND2ΔA strains provide the first experimental evidence to delineate key steps in the marineosin biosynthetic pathway. The data do not support either previous hypotheses, in which the pathway either passes through an enone analogue of undecylprodiginine15 or involves a hydroxylation of undecylprodiginine by a RedG homologue (MarG).18 Rather the pathway involves formation of 23-hydroxyundecylprodiginine (7), in a step that precedes the reactions catalyzed by MarG. The hydroxyl (-OH) group of 7 can be either introduced early in the pathway due to the hydroxylation of the 2-UP subunit by an unidentified oxidase (Route 1, Scheme 2), via recruitment of 2-hydroxybutyric acid starter unit by MarP (Route 2, Scheme 2) or introduced later via hydroxylation of 2 (Route 3, Scheme 2). Compound 2 was never detected in any of the strains generated in this study. This observation does not prove that incorporation of the hydroxyl group of 7 occurs early in the pathway as it may be that hydroxylation of 2 occurs faster than its formation (so the intermediate cannot be detected). In any case, the gene product(s) required for introduction of the hydroxyl group remain unknown. The 23-ketoundecylprodiginine (8) is an oxidative product of 7 and is a shunt metabolite. The marineosin pathway proceeds from 7 to premarineosins (9 and 11) in a step that requires MarG (Scheme 2). The pathway most likely proceeds from 7, after an oxidative C9–C21 cyclization (analogous to the oxidative cyclization reaction catalyzed by RedG). In this case there would be a subsequent intramolecular hydroalkoxylation at C-8 (which may be spontaneous or catalyzed by MarG). The 16-keto analogue, 10, of 9 is most likely a shunt metabolite that comes from hydroxylation and oxidation of 9. A 16-keto analogue of either 5 or 7 was not observed in any of the strains. In the proposed pathway the stereochemistry at C-8 is established during the conversion of 7 to 9 and 11. If the proposed pathway (intramolecular hydroalkoxylation at C-8) proceeds enzymatically (catalyzed by MarG), then the premarineosin A (9) might be the intermediate of marineosins A and B. It is noteworthy that the premarineosin B (11) most likely arises from an inversion of the aminal nitrogen of premarineosin A (9) under pathway conditions where the two isomers differ only in stereochemistry at the spiro aminal carbon (C-8). The premarineosin A (9) exists in higher ratio owing to an anomeric effect as demonstrated for marineosin A (5),15 and this trend follows in the higher production of marineosin A (5) in the JND2 strain. The final step of the pathway is a MarA catalyzed reduction of premarineosins (9 and 11) (Scheme 2).
Premarineosin A (9) Exhibits Potent and Selective in Vitro Antimalarial Activity against Multiple-Drug-Resistant P. falciparum Strains
The generation of marineosin pathway intermediates and shunt metabolites provided new compounds to test for biological activity. Additional compounds may be generated via established methodologies such as mutasynthesis and creation of hybrid pathways. Our interest has been the antimalarial activity of prodiginine-type compounds.4 In a continuation of this work we determined the biological activity of 9, 10, and marineosin A (5). The compounds were evaluated for antimalarial activity against the chloroquine-sensitive (CQS) D6 and the chloroquine-resistant (CQR) Dd2 and 7G8 strains of P. falciparum with chloroquine (CQ) as a reference drug. Premarineosin A (9) showed potent antimalarial activity against D6 (IC50 = 2.3 nM), Dd2 (IC50 = 12 nM), and 7G8 (IC50 = 1.5 nM) strains and was more effective than chloroquine (D6 (IC50 = 14.3 nM), Dd2 (IC50 = 110 nM), and 7G8 (IC50 = 131 nM)) or other tested compounds. The activity of 9 compares favorably with the most potent synthetic and naturally occurring prodiginines.4 The activity of marineosin A (5) against D6 (IC50 = 138 nM), Dd2 (IC50 = 127 nM), and 7G8 (IC50 = 199 nM) was poorer than that of 9, demonstrating that the oxidized middle pyrrole ring is required for maximal activity. The presence of a keto group in the macrocyclic ring of 10 also decreases the antimalarial activity (D6 (IC50 = 138 nM), Dd2 (IC50 = 127 nM), and 7G8 (IC50 = 199 nM)) by ∼100 times when compared to 9. The ability of 9 to inhibit the hepatocellular HepG2 cancer cell line was used to assess its cytotoxicity and hence selectivity.27 Compound 9 has an IC50 = 4169 nM against HepG2 cells, giving it a large therapeutic index ranging from 347 (for the Dd2 strain) to 2779 (for the 7G8 strain), indicating very selective antimalarial activity.
Conclusions
The final steps of the marineosin biosynthetic pathway have now been experimentally established. The genes and tools for an in-depth investigation of a new class of reduced prodiginines as well as spiroaminal natural products have been discovered and developed. Notable in this regard is the class of C–C bond-forming non-heme Rieske iron monoxygenases such as MarG, which could be involved in the biosynthesis of other spiroaminal natural products. New marineosin and undecylprodiginine analogues have been identified, characterized, and purified, and routes to obtain additional compounds are possible. The generation of marineosin pathway absent the final MarA catalyzed step has led to the isolation and identification of a novel spiroaminal prodiginine with potent antimalarial activities.
Acknowledgments
We thank Dr. William Fenical (Scripps Institute of Oceanography, San Diego, CA) for providing the Streptomyces sp. CNQ-617 and marineosin standard. We also thank Dr. David Peyton (Portland State University, Portland, OR) for NMR data acquisition and Dr. Jane Xu Kelly (Portland State University, Portland, OR) for performing the in vitro antimalarial activity. This work was supported by grant GM077147 from the National Institutes of Health.
Supporting Information Available
List of cosmids, strains, and primers, and detailed spectroscopic data, and mass, IR, and NMR of isolated compounds 5, 7, 8, 9, and 10. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fürstner A. Angew Chem., Int. Ed. 2003, 42, 3582–3603. [DOI] [PubMed] [Google Scholar]
- Magae J.; Miller M. W.; Nagai K.; Shearer G. M. J. Antibiot. 1996, 49, 86–90. [DOI] [PubMed] [Google Scholar]
- Nguyen M.; Marcellus R. C.; Roulston A.; Watson M.; Serfass L.; Madiraju S. R. M.; Goulet D.; Viallet J.; Bélec L.; Billot X.; Acoca S.; Purisima E.; Wiegmans A.; Cluse L.; Johnstone R. W.; Beauparlant P.; Shore G. C. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 19512–19517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papireddy K.; Smilkstein M.; Kelly J. X.; Shweta; Salem S. M.; Alhamadsheh M.; Haynes S. W.; Challis G. L.; Reynolds K. A. J. Med. Chem. 2011, 54, 5296–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessio R.; Bargiotti A.; Carlini O.; Colotta F.; Ferrari M.; Gnocchi P.; Isetta A.; Mongelli N.; Motta P.; Rossi A.; Rossi M.; Tibolla M.; Vanotti E. J. Med. Chem. 2000, 43, 2557–2565. [DOI] [PubMed] [Google Scholar]
- Cerdeño A. M.; Bibb M. J.; Challis G. L. Chem. Biol. 2001, 8, 817–829. [DOI] [PubMed] [Google Scholar]
- Thomas M. G.; Burkart M. D.; Walsh C. T. Chem. Biol. 2002, 9, 171–184. [DOI] [PubMed] [Google Scholar]
- Stanley A. E.; Walton L. J.; Kourdi Zerikly M.; Corre C.; Challis G. L. Chem. Commun. 2006, 38, 3981–3983. [DOI] [PubMed] [Google Scholar]
- Mo S.; Sydor P. K.; Corre C.; Alhamadsheh M. M.; Stanley A. E.; Haynes S. W.; Song L.; Reynolds K. A.; Challis G. L. Chem. Biol. 2008, 15, 137–148. [DOI] [PubMed] [Google Scholar]
- Haynes S. W.; Sydor P. K.; Stanley A. E.; Song L.; Challis G. L. Chem. Commun. 2008, 1865–1867. [DOI] [PubMed] [Google Scholar]
- Gerber N. N.; McInnes A.; Smith D.; Walter J.; Wright J.; Vining L. Can. J. Chem. 1978, 56, 1155–1163. [Google Scholar]
- Wasserman H. H.; Skles R. J.; Peverada P.; Shaw C. K.; Cushley R. J.; Lipsky C. R. J. Am. Chem. Soc. 1973, 95, 6874–6875. [DOI] [PubMed] [Google Scholar]
- Bentley S. D.; Chater K. F.; Cerdeño-Tárraga A. M.; Challis G. L.; Thomson N. R.; James K. D.; Harris D. E.; Quail M. A.; Kieser H.; Harper D.; Bateman A.; Brown S.; Chandra G.; Chen C. W.; Collins M.; Cronin A.; Fraser A.; Goble A.; Hidalgo J.; Hornsby T.; Howarth S.; Huang C. H.; Kieser T.; Larke L.; Murphy L.; Oliver K.; O’Neil S.; Rabbinowitsch E.; Rajandream M. A.; Rutherford K.; Rutter S.; Seeger K.; Saunders D.; Sharp S.; Squares R.; Squares S.; Taylor K.; Warren T.; Wietzorrek A.; Woodward J.; Barrell B. G.; Parkhill J.; Hopwood D. A. Nature 2002, 417, 141–147. [DOI] [PubMed] [Google Scholar]
- Sydor P. K.; Barry S. M.; Odulate O. M.; Barona-Gomez F.; Haynes S. W.; Corre C.; Song L.; Challis G. L. Nat. Chem. 2011, 3, 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonlarppradab C.; Kauffman C. A.; Jensen P. R.; Fenical W. Org. Lett. 2008, 10, 5505–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christmann M.; Bräse S.. Asymmetric Synthesis: The Essentials, 2nd ed.; Wiley-VCH: Weinheim, 2008. [Google Scholar]
- John Pal A. P.; Kadigachalam P.; Mallick A.; Doddi V. R.; Vankar Y. D. Org. Biomol. Chem. 2011, 9, 809–819. [DOI] [PubMed] [Google Scholar]
- Cai X. C.; Wu X.; Snider B. B. Org. Lett. 2010, 12, 1600–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich L. N.; Dawson E. S.; Lindsley C. W. Org. Lett. 2010, 12, 1048–1051. [DOI] [PubMed] [Google Scholar]
- Labonte J. W.; Townsend C. A. Chem. Rev. 2013, 11332182–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Beyer N. J.; Borisova S. A.; Liu H. W. Biochemistry 2003, 42, 14794–14804. [DOI] [PubMed] [Google Scholar]
- Parales R. E.; Parales J. V.; Gibson D. T. J. Bacteriol. 1999, 181, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gust B.; Challis G. L.; Fowler K.; Kieser T.; Chater K. F. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J.; Russell D. W.. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Woodbury, NY, 2001. [Google Scholar]
- Kieser T.; Bibb M. J.; Buttner M. J.; Chater K. F.; Hopwood D. A.. Practical Streptomyces Genetics; John Innes Foundation: Norwich, U.K., 2000. [Google Scholar]
- Gust B. Methods Enzymol. 2009, 458, 159–180. [DOI] [PubMed] [Google Scholar]
- Pešić D.; Starčević K.; Toplak A.; Herreros E.; Vidal J.; Almela M. J; Jelić D.; Alihodžić S.; Spaventi R.; Perić M. J. Med. Chem. 2012, 55, 3216–3227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.