

Abstract
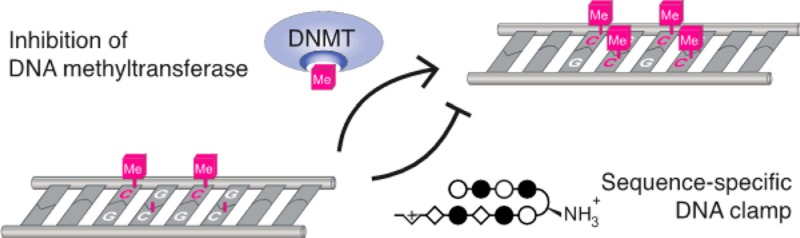
The CpG dyad, an important genomic feature in DNA methylation and transcriptional regulation, is an attractive target for small molecules. To assess the utility of minor groove binding oligomers for CpG recognition, we screened a small library of hairpin pyrrole-imidazole polyamides targeting the sequence 5′-CGCG-3′ and assessed their sequence specificity using an unbiased next-generation sequencing assay. Our findings indicate that hairpin polyamide of sequence PyImβIm-γ-PyImβIm (1), previously identified as a high affinity 5′-CGCG-3′ binder, favors 5′-GCGC-3′ in an unanticipated reverse binding orientation. Replacement of one β alanine with Py to afford PyImPyIm-γ-PyImβIm (3) restores the preference for 5′-CGCG-3′ binding in a forward orientation. The minor groove binding hairpin 3 inhibits DNA methyltransferase activity in the major groove at its target site more effectively than 1, providing a molecular basis for design of sequence-specific antagonists of CpG methylation.
Introduction
The role of epigenetic dysregulation in cancer has motivated interest in DNA methylation and methods for its modulation.1,2 In mammals, DNA methylation occurs in the major groove of DNA at the 5′ position of both cytosine residues in the palindromic CG dyad (CpG). CpGs are rare in the genome and 70% methylated, with nearly all unmethylated CpGs clustered in G,C-rich regions called “CpG islands”.3 Approximately 60% of RNA Polymerase II transcribed human genes contain CpG islands,4 and their methylation causes transcriptional repression.5 In cancer, for example, otherwise functional tumor suppressor genes can be silenced by hypermethylation in their associated CpG island.6 Importantly, inhibition of DNA methylation at tumor suppressor genes has been shown to reactivate apoptotic pathways and sensitize cancer cells to previously ineffective chemotherapy.7,8
The most effective demethylation agents are cytidine analogues such as 5-aza-deoxycytidine which find limited use due to significant side effects.1 These cytidine analogues are suicide inhibitors incorporated into DNA to form covalent methyltransferase-DNA adducts.9 The methyltransferase is sequestered and unavailable to methylate CpGs resulting in genome-wide demethylation. DNA binding molecules, such as the bis-intercalating natural product echinomycin,10 can disrupt CpG methylation in vitro but have dose-limiting toxicities that have abrogated further clinical advancement.11 While other CpG methylation inhibitors are under investigation,12−14 none of these agents have demonstrated the ability to inhibit DNA methylation in a sequence-specific fashion.
Hairpin pyrrole-imidazole (Py-Im) polyamides are a class of sequence-specific oligomers that bind in the minor groove of DNA.15−20 Programmable sequence preference is accomplished by side-by-side pairings of aromatic amino acids that distinguish the edges of the four Watson–Crick base pairs.15−20 Referred to as the pairing rules, Im/Py codes for G•C base pair, Hp/Py codes for T•A base pairs, and Py/Py binds both T•A/A•T in preference to G•C/C•G. Eight-ring hairpin oligomers linked by a central aliphatic γ-aminobutyric acid unit have affinities for match sites with Ka ∼108 to 1010 M–1.16,21 These binding energetics are comparable to natural transcription factors and, like natural DNA binding proteins, are sensitive to differences in the sequence-dependent microstructure of DNA. To relax the curvature of all ring hairpins, β alanine (β) can be substituted for Py-rings in some cases such that β/β pairs replace Py/Py for T•A/A•T specificity, and Im/β replaces Im/Py pairs in strategic locations while retaining specificity for G•C base pair.22−26 Hairpin Py-Im polyamides usually bind with the N-to-C terminus aligned in the 5′-to-3′ direction of DNA, referred to as “forward orientation”.27 This modest forward binding preference can be enforced by substitution of the prochiral α position in the γ-turn, i.e., replacement of γ-aminobutyric acid by (R)-2,4-diaminobutyric acid.28 Hairpin architectures containing β/β pairs and β/ring pairs have been found in some cases to prefer the N to C terminus aligned in a 3′-to-5′ direction of DNA.29 While adhering to the pairing rules, this reverse hairpin orientation would bind a different DNA sequence. Recently, we used massively parallel sequencing methods in conjunction with biotin-tagged hairpins, termed Bind-n-Seq, to scan genome-size DNA sequence space for hairpin high affinity sites.30 Although the canonical pairing rules are remarkably predictive of polyamide DNA binding specificity, we identified high affinity DNA binding sites in the reverse orientation for several polyamides containing β/Im pairs.30
Eight-ring hairpin Py-Im polyamides have been shown to discriminate 5′-GGGG-3′, 5′-GCGC-3′, and 5′-GGCC-3′ with appropriate arrangement of four Im/Py pairs.31 From experience, sequences with CpG steps such as 5′-CGCG-3′ are not as readily accessed for reasons not well understood. In an effort to improve the affinity of an eight-ring hairpin polyamide for the sequence 5′-CGCG-3′, Sugiyama and co-workers replaced two Im/Py pairs with Im/β pairs. A change from PyImPyIm-γ-PyImPyIm (S1) to PyImβIm-γ-PyImβIm (S2) afforded a 65-fold increase in affinity for 5′-CGCG-3′.32 Both hairpins conform to the pairing rules and would bind 5′-CGCG-3′ in the forward orientation. In this study, we employ a high-throughput sequencing assay of polyamide-DNA association to revisit targeting the 5′-CGCG-3′ sequence. Our findings indicate that hairpin polyamides of sequence PyImβIm-γ-PyImβIm S2 favor 5′-GCGC-3′, a reverse binding mode. The issue of designing a hairpin polyamide sequence that prefers 5′-CGCG-3′ to 5′-GCGC-3′ remains to be solved. Using Bind-n-Seq methods30 as our screen for a library of polyamide–biotin conjugates, we find that replacement of one β alanine with Py to afford PyImPyIm-γ-PyImβIm restores the preference for forward binding 5′-CGCG-3′. Recent structural work has shown that a cyclic Py-Im polyamide binding in the minor groove causes significant widening of the minor groove width of DNA,33,34 and provides a mechanistic rationale for disruption of DNA-binding proteins in the major groove. We demonstrate the ability of our 5′-CGCG-3′ specific minor groove binding hairpin polyamides to inhibit enzymatic CpG methylation in the major groove of a 5′-CGCG-3′ sequence.
Results
Sequence Based Analysis of PyImβIm-γ-PyImβIm Specificity
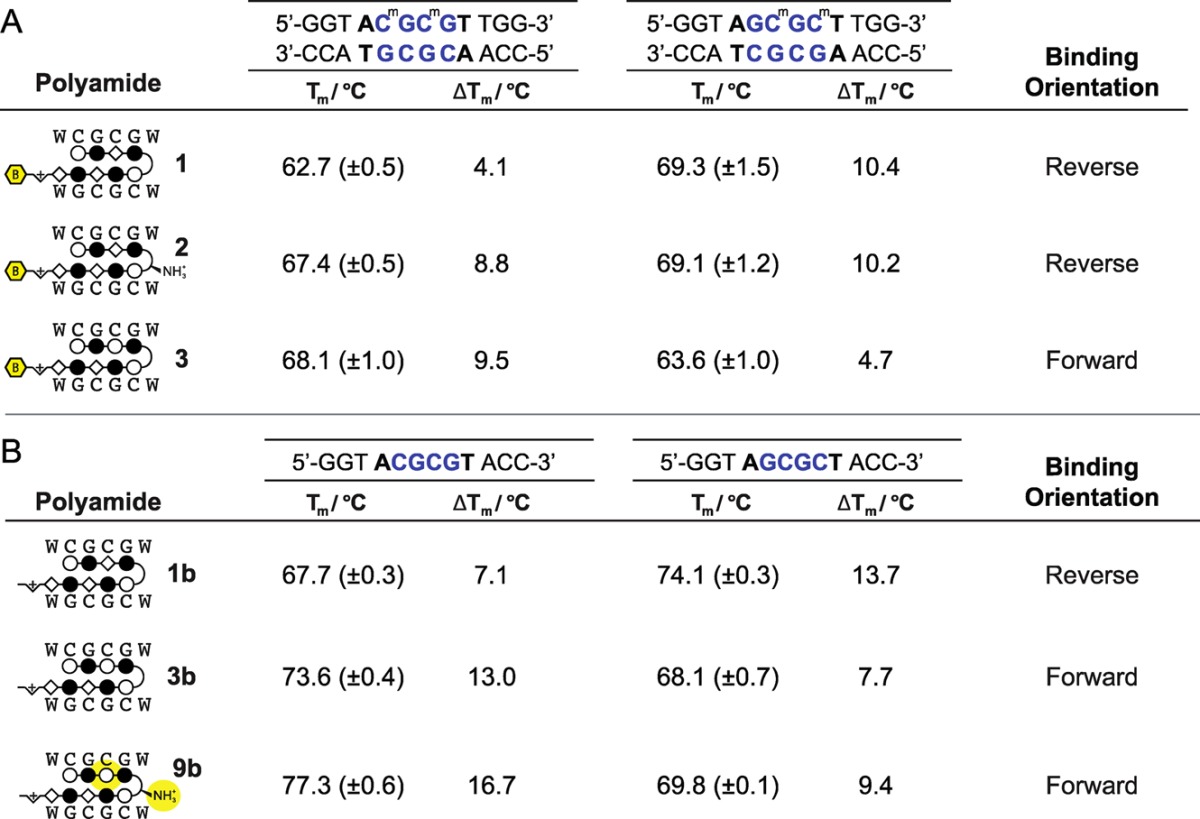
The 5′-CGCG-3′ sequence is a compelling DNA target for an 8-ring hairpin Py-Im polyamide because it is one of the least represented 6-bp sequence patterns in the human genome, potentially promoting greater genomic specificity.30 Minoshima and co-workers have previously targeted this sequence and shown that polyamide S2 (Figure 1) can bind the fully methylated sequence.32 In their study, the substitution of two β’s for Py moieties resulted in improved affinity for 5′-CGCG-3′ over the eight-aromatic ring architecture S1 (SI Figure S1). In light of recent Bind-n-Seq studies, however, we wondered whether these changes may have also had the unintended effect of reducing the preference of the polyamide for binding in the forward orientation.30 Bind-n-Seq is a high-throughput sequencing method that allows facile identification of high affinity binding sites of biotin-labeled Py-Im polyamides by affinity purification followed by sequencing (Figure 2A).30 As a first step, we synthesized an analogue of S2 and examined polyamide–biotin conjugate 1 of sequence PyImβIm-γ-PyImβIm (Figure 2B), which has a biotin affinity tag appended at the C-terminus of the heterocyclic oligomer. (Full structures of all polyamides are in SI Figure S1.) Polyamide–biotin conjugate 1 was incubated at 50 nM in a library of all possible 21 base pair DNA sequences, enriched, and sequenced to identify polyamide-bound sequences. This data set was then analyzed by the DREME algorithm to construct a motif logo summarizing the highest affinity sequences. A binding preference for 5′-GCGC-3′ was revealed, suggestive of a reverse binding mode (Table 1).
Figure 1.

Structure of Py-Im polyamide S2 previously reported to bind methylated 5′-CGCG-3′ oligonucleotide duplex.32 Legend for ball-and-stick notation.
Figure 2.
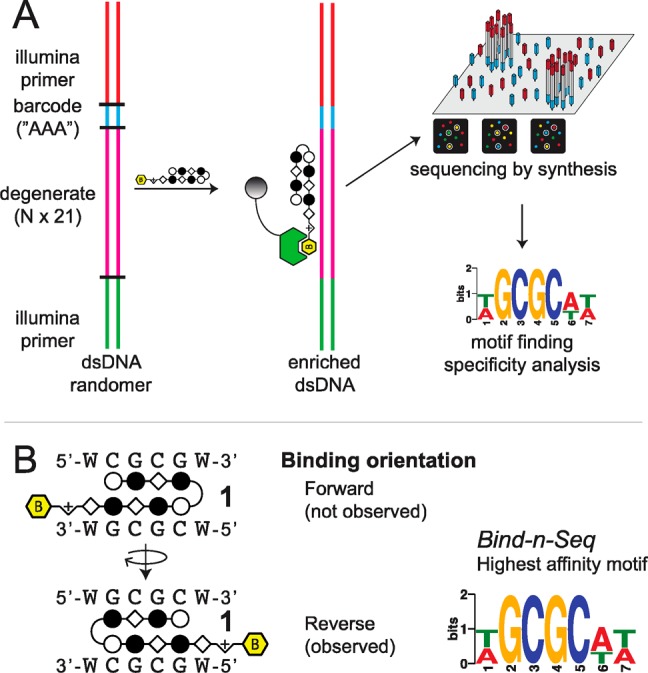
(a) Scheme of Bind-n-Seq method.30 Polyamide–biotin conjugate is incubated in a genome-sized library of all possible 21mers, enriched, sequenced, and the resulting data set analyzed with motif-finding software.30 (b) Polyamide 1 could potentially bind in the forward orientation or the reverse orientation. The highest affinity binding sequence of 1 is the reverse orientation binding 5′-GCGC-3′.
Table 1. Preferred Binding Orientations of Polyamides 1–8 Were Queried with Bind-n-Seq to Generate the Highest Affinity Sequence Motifa.
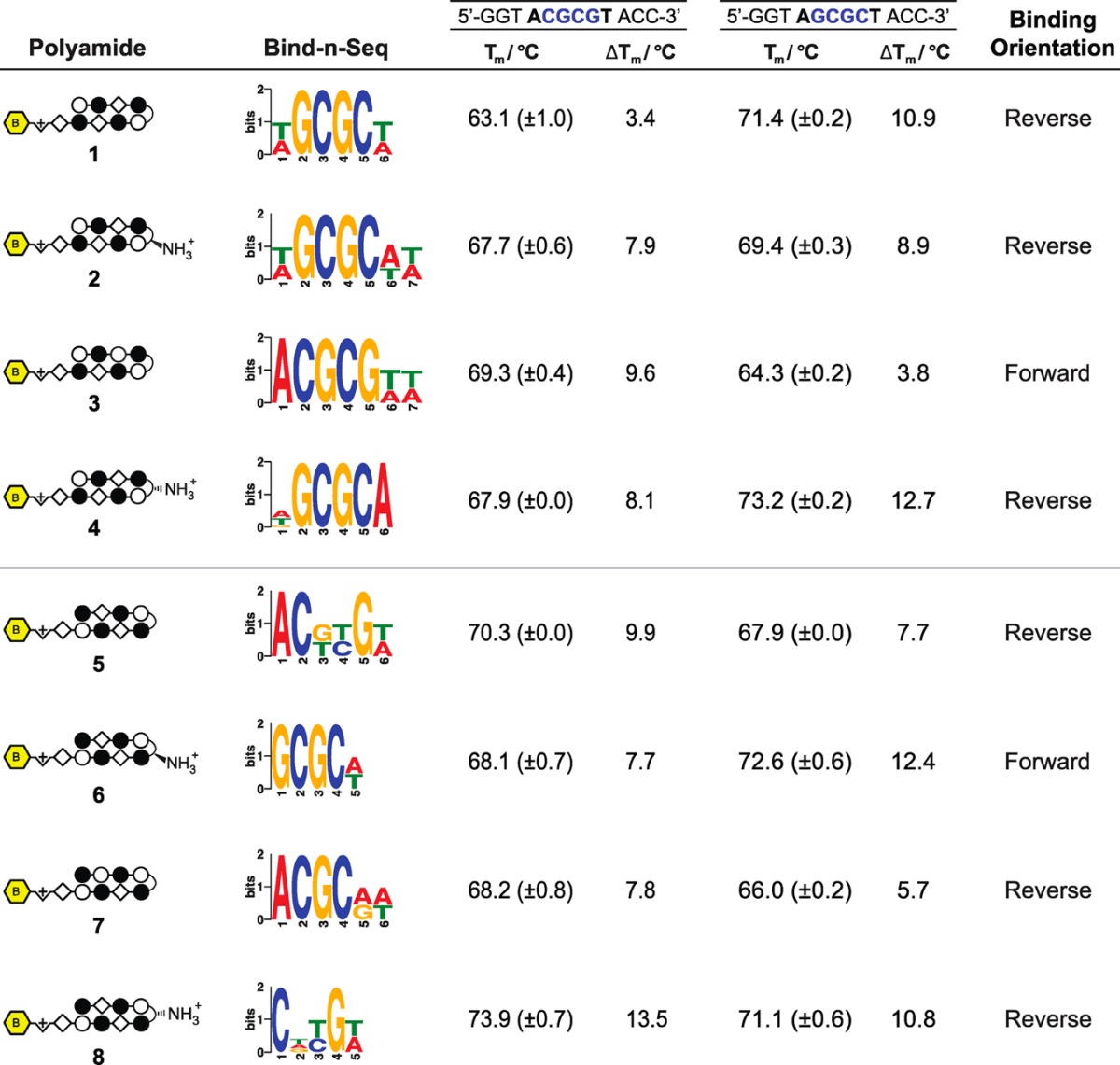
Polyamide-mediated thermal stabilization (ΔTm) of 12 base pair oligonucleotides of the forward (5′-CGCG-3′) and reverse (5′-GCGC-3′) sequences were used to validate the revealed motifs. Melting temperatures reflect the mean and standard deviation of quadruplicate measurements.
Redesign Hairpin for CGCG versus GCGC Preference
In order to restore the preference for binding 5′-CGCG-3′ in the forward orientation, we considered two possible points of modification (Figure 3A). First, we made a single modification to 1 at the turn unit, replacing the GABA turn to a chiral α-amino GABA, affording 2 (Figure 3B). The α-amino GABA turn has previously been shown to restore forward orientation and increase affinity, including in β-containing polyamides.28−30 This effect is thought to arise from a steric interaction with the floor of the minor groove when the chiral α-amino GABA turn unit is bound in the reverse orientation.23,28 Assessment of polyamide 2 by Bind-n-Seq found that this modification improved the reverse/forward ratio but was insufficient to restore a preferred forward orientation binding preference (Table 1). To confirm the high-throughput sequencing findings, we performed a thermal DNA denaturation study, as previous studies have shown that thermal stabilization (ΔTm) of duplex DNA by Py-Im polyamides correlates well with binding affinity.35 Assays were performed with DNA oligonucleotides differing only in the central binding sequence (5′-CGCG-3′ versus 5′-GCGC-3′) to directly test the binding orientations identified by the Bind-n-Seq logos (Table 1). This analysis substantiated a reverse orientation binding preference for polyamide 1, with a ΔTm of 10.9 °C in the reverse direction as compared to 3.4 °C in the forward direction. Modification at the turn to the α-amino GABA in polyamide 2 resulted in increased stabilization of the forward 5′-CGCG-3′ oligomer by 4.5 °C; stabilization by polyamide 2 in the reverse 5′-GCGC-3′ orientation was diminished by 2.0 °C. This indicated an improved forward preference for 5′-CGCG-3′. Nonetheless, the relative magnitudes of the ΔTm support an overall modest energetic preference for reverse orientation binding.
Figure 3.
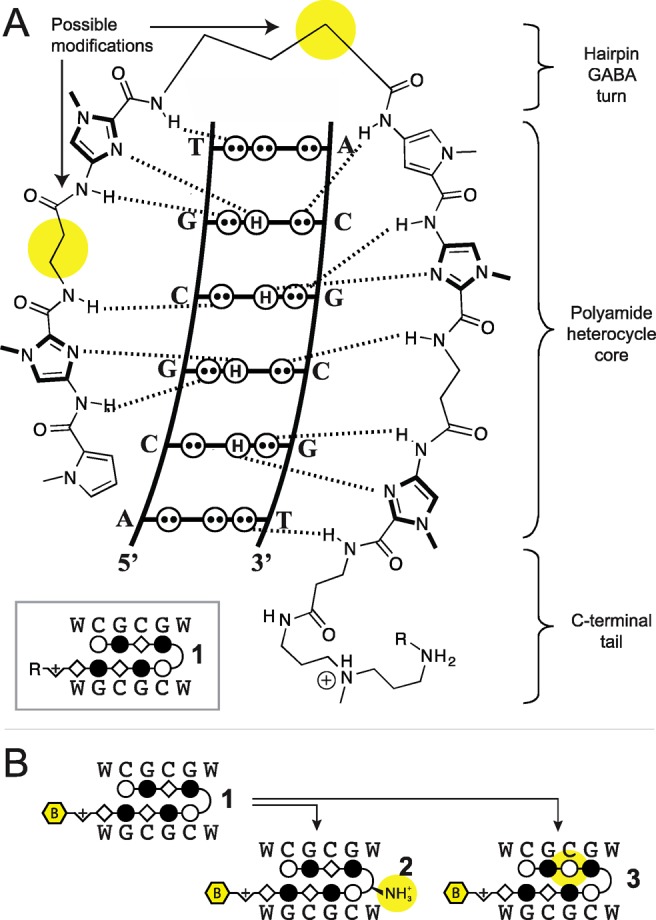
(a) Scheme of Py-Im polyamide binding in the minor groove of DNA. (b) Single position changes made to hairpin polyamide 1 to afford 2 and 3. Positions are highlighted in yellow.
The inability of the α-amino GABA turn to enforce forward orientation binding led us to investigate alternative solutions for the molecular recognition of 5′-CGCG-3′ (Figure 3). We posited that reverse binding is abetted by the flexibility afforded by the two β units in the core binding region, as had been similarly noted in polyamides containing a β/β pair.29 We thus considered whether removing one β residue might reinstitute sufficient rigidity in one of the polyamide strands to limit reverse binding while retaining the specificity and affinity provided by the other β. Of the two β moieties in the core of polyamide 1, the C-terminal β in the core binding region was retained based on previous studies that have shown it is necessary for high affinity recognition of the 5′ C•G base pair.24 To isolate the effect of each modification, we returned to parent polyamide 1 and replaced the N-terminal β with a Py while retaining the achiral GABA turn, to provide polyamide 3 (Figure 3B). The assessment of 3 by Bind-n-Seq followed by DREME analysis generated a high affinity motif consistent with forward binding 5′-CGCG-3′ (Table 1). This was corroborated by ΔTm measurements showing considerable preference for the forward 5′-CGCG-3′ direction.
We further examined whether a hairpin polyamide designed to target a reverse orientation sequence may productively bind CpGs with high specificity. To test this, we expanded the library of compounds to include polyamides 4–8, single modifications targeting the 5′-GCGC/CGCG-3′ core (Figure 4). In contrast to our findings with 5′-CGCG-3′ targeting polyamide 2, we confirmed that the incorporation of an α-amino GABA turn in polyamide 6 restores forward orientation binding for the 5′-GCGC-3′ sequence.30 This difference is striking given that the two polyamides are composed of nearly identical amino acid sequences. Bind-n-Seq data and Tm assays of polyamides 4, 5, 7, and 8 together suggest that all other modifications preferentially bind the reverse orientation, and 5, 7, and 8 do so with poor specificity (Table 1). Indeed, among all variations tested of both 5′-CGCG-3′ forward binding and 5′-GCGC-3′ reverse binding cores, polyamide 3 displayed the highest specificity for the 5′-CGCG-3′ sequence (Table 1).
Figure 4.
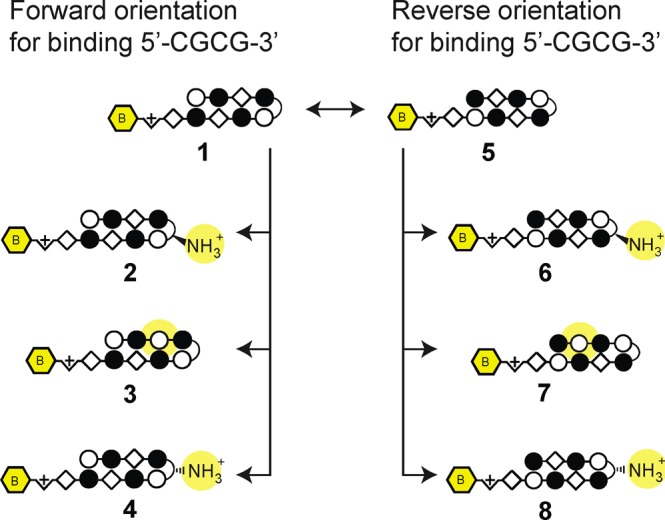
Panel of polyamides synthesized for assessment by Bind-n-Seq and DNA thermal stabilization for binding the 5′-CGCG-3′ sequence. According to the pairing rules, polyamides 1–4 target 5′-CGCG-3′ in the forward orientation and polyamides 5–8 target 5′-CGCG-3′ in the reverse orientation. Structural modifications are highlighted in yellow.
Sequence-Specific Binding Hemi-Methylated DNA
Next, we considered the potential for minor groove binding hairpin Py-Im polyamides to prevent DNA methylation undergoing DNA replication. To do so, they must be able to bind the hemi-methylated DNA of daughter strands that have not yet undergone maintenance methylation. DNA thermal stabilization analysis was used to pursue evidence of the above trends of binding orientation with hemi-methylated DNA sequences. The sense strands of each of the 12 base pair oligomers containing 5′-CGCG-3′ or 5′-GCGC-3′ cores were methylated on both cytosines, whereas the antisense strands were left unmethylated. Flanking sequences were modified to lack self-complementarity and enforce hemi-methylated duplex formation. Analysis of ΔTm of the hemi-methylated DNA oligomers confirmed the above magnitudes of stabilization and trends of reverse and forward binding modes for 1, 2, and 3 (Table 2A).
Table 2. (A) Tm Study with Hemi-Methylated DNA Duplex; (B) Tm Study of Three Generations of 5′-CGCG-3′ Methylation Inhibitors without Biotin Affinity Tags.
Inhibition of Methyltransferase
With a specific polyamide capable of binding hemi-methylated DNA in hand, we evaluated its application as a sequence-specific inhibitor of DNA methyltransferases. The biotin enrichment tag was deleted from the C-terminus by resynthesis to afford parent hairpins 1b and 3b. Melting temperature analyses confirmed that these molecules show comparable binding preference to biotin conjugates 1 and 3, respectively (Table 2B). We developed an in vitro assay to probe the methylation state of specific sites employing the methylation-sensitive restriction enzyme MluI to compare sequence specific effects of 1b, 3b, and AT-binding distamycin D as a control (SI Figure S2A). In this assay, we measured the ability of these compounds to inhibit the methylation activity of M.SssI, a robust prokaryotic methyltransferase that operates in a processive manner like human methyltransferases and shares structural similarities with the catalytic core of human DNMT1.36 We employed the methylation-sensitive enzyme MluI, which cleaves at seven 5′-ACGCGT-3′ sites,37 to interrogate methylation of the λ-phage DNA (48.5 kb), of which 5 bands were visualized by agarose gel electrophoresis. Both 1b and 3b were titrated from increasing concentrations 1 nM to 1 μM, while D was dosed 10-fold higher from 10 nM to 10 μM. Full digestion of the DNA by MluI indicates a lack of CpG methylation at 5′-ACGCGT-3′ restriction sites, and is demonstrated by positive control lane 2 (SI Figure S2B). In contrast, full methylation would protect DNA from MluI digestion, as in lane 1 where no compound was added to DNA prior to exposure to M.SssI for methylation.
Consistent with our biophysical characterization of the compounds, polyamide 3b showed the most robust inhibition of CpG methylation (SI Figure S2B, lanes 7–10) at 5′-ACGCGT-3′ sites. In lane 10, full MluI digestion comparable to positive control lane 2 was observed at 1 μM of 3b, indicating this concentration was sufficient to block all methylation at the cognate binding sites. Further, incomplete protection was evidenced at 100 nM of 3b by additional, partially digested bands in lane 9. In contrast, polyamide 1b showed weak inhibition of M.SssI and was active only at the highest concentration (SI Figure S2B, lanes 3–6). This reflects its weaker affinity for the 5′-CGCG-3′ forward binding orientation, also observed by thermal duplex denaturation analysis. Inhibition by 1b at 1 μM, however, is reduced relative to that observed at 100 nM of 3b, consistent with the binding preferences of the two molecules. There was no inhibition by distamycin D at all concentrations tested, even at the highest concentration of 10 μM, underscoring the importance of CpG specificity of Py-Im polyamides in preventing CpG methylation. To enable quantitation of enzyme activity inhibition, the substrate DNA was changed to a 7.5 kb fragment containing a single 5′-ACGCGT-3′ site (Figure 5A).
Figure 5.

(a) Scheme of in vitro DNA methyltransferase (DNMT) inhibition assay. Generic polyamide shown in ball-and-stick notation and CpG sites represented by red squares. DNA (7.5 kb) with a single MluI restriction site was incubated at 50 pM with inhibitor and subjected to methylation by M.SssI. DNA was isolated for restriction digest by MluI to reveal methylation at the target site. (b) Single changes in polyamides 2 and 3 that promote forward orientation binding were combined in designing 9b as a third generation candidate for improved methylation inhibition. (c) Representative gel image of the polyamides 1b, 3b, and 9b in the assay described in (a), suggestive of differential inhibitory activity. Dose ranges of compounds were adjusted in relation to their DNA binding affinities. (d) IC50 values of 5′-CGCG-3′ targeting polyamides. Values are determined from band intensities in the in vitro as assay shown in (c) and normalized against maximal methylation with no inhibitor. IC50 values were calculated from at least three replicates and fit to a four-variable, dose–response model.
We were encouraged by these results to consider the design of an improved methylation antagonist at 5′-CGCG-3′. We revisited the single modifications to 1 in polyamide 2 and 3 that had promoted forward orientation binding. We combined the α-amino modification at the GABA turn that had encouraged 2 to bind in the forward orientation, albeit insufficiently, with the Py substitution in the top strand, as in 3, to afford 9b (Figure 5B). Analysis by thermal denaturation assays revealed that the effects of the modifications were additive, and 9b displayed increased affinity and preference for forward orientation binding (Table 2B).
We then sought to determine IC50 values for the three generations of 5′-CGCG-3′ methylation inhibitors: 1b, 3b, and 9b. With consideration for their DNA binding affinities, compounds 1b, 3b, and 9b were titrated from 10 nM to 33 μM, 330 pM to 10 μM, and 33 pM to 1 μM, respectively (Figure 5C). It is worth noting that an additional SDS wash step was necessary in this assay to remove the higher affinity 9b from the DNA before resolution by the MluI restriction enzyme. Prior to the addition of this SDS incubation, inhibition was maximally revealed to approximately 40%, due to polyamide inhibition of the MluI restriction enzyme. Overnight incubation of DNA in 2% SDS removed additional polyamide and improved the revealed inhibition, suggesting the compressed inhibitory range is an artifact of this method and the high affinity of 9b. The IC50 values of 1b, 3b, and 9b were determined to be 2.2 μM (95% confidence: 1.2–3.9 μM), 117 nM (95% confidence: 65–210 nM), and 2.6 nM (95% confidence: 1.0–6.7 nM), respectively (Figure 5D). This is in good correlation with the iterative improvement shown in the biophysical analyses of these compounds, as well as the previous qualitative in vitro assay. Polyamide 9b shows nearly 1000-fold improvement over 1b as a 5′-CGCG-3′ methylation antagonist.
Discussion
Design of Antagonists of CpG Methylation
This study provides a basis for design of sequence-specific DNA-binding molecules for targeted inhibition of CpG methylation. The disparity in methyltransferase inhibition between AT-binding distamycin D and hairpin polyamide 3b suggests that the specific CpG-binding capability and widening of the minor groove by bound Py-Im polyamides are critical for disrupting DNA methylation in the major groove. At the same time, applying the pairing rules demands caution in the design of imidazole and β-rich polyamides as the inherent conformational flexibility of the β subunit can support unintended reverse DNA-binding modes. While previous studies have shown an α-amino GABA turn unit can be used to restore the forward orientation binding preference of β-containing polyamides, we found that 5′-CGCG-3′ binding Py-Im polyamides required an alternative solution. Specifically, restoring the rigidity of the N-terminal strand via substitution of its β-subunit with a Py appears necessary to resolve the undesired reverse-binding of these architectures.
Potential Mechanism for Inhibition of CpG Methylation
The lack of inhibition of CpG methylation by the reverse binding 1b as compared to 3b at the interrogated 5′-CGCG-3′ sites suggests that inhibition of the preceding M.SssI enzyme is a sequence-specific, localized event. The M.SssI methyltransferase, like eukaryotic methyltransferases, is a “flipase” that swings the target cytosine out of the double helix and into its catalytic core.38 All known CpG methyltransferases operate by this conserved mode of action. Structural studies of mouse DNMT1, the relevant mammalian methyltransferase for maintenance methylation, show that enzyme residues enter the double helix from both the major and minor grooves in an intercalative-manner around the target CpG.39 These residues disrupt local base pairing and rotate the substrate cytosine around the sugar–phosphate backbone and into the catalytic core of the enzyme. A Py-Im polyamide bound to the target DNA site likely acts as a stabilizing clamp in the minor groove and prevents the intrusion of these residues. The increased DNA stability disallows the conformational reorganization of the CpG substrate necessary for catalysis and results in the inhibition of methyltransferase activity.
Conclusion
In this study, we examined programmable Py-Im polyamides targeting the 5′-CGCG-3′ sequence as a model for sequence-specific inhibition of CpG methylation. The unbiased Bind-n-Seq method was critical for revealing unanticipated binding modes of the polyamides. Through deliberate, incremental synthetic modifications, we were able to discern structure activity relationships that guided improved design of CpG methylation antagonists. Further work will be necessary to understand whether this represents a more general solution for controlling Py-Im polyamide orientation or is specific to the 5′-CGCG-3′ sequence. This study demonstrates that high affinity minor groove binding Py-Im polyamides can inhibit major groove CpG methylation by methyltransferase in a sequence-specific manner. It will be the focus of future research to assess these molecules as antagonists of CpG methylation in cells and its utility in the desilencing of specific genes. It will be of interest whether the intrinsic rarity of the CpG dinucleotide sequence and noncovalent binding of polyamides will reduce off-target effects.
Experimental Section
Py-Im Polyamide Synthesis
Polyamides were synthesized by microwave-assisted, solid-phase synthesis on PAM resin (Peptides International) according to previously described protocols.40,41 The polyamides were cleaved from resin with 3,3′-diamino-N-methyldipropylamine and purified by reverse phase HPLC. For biotin-conjugated polyamides, the free amine at the C-terminus was allowed to react with 2 equiv of preactivated PEG4-biotin NHS ester (Thermo Scientific) and 4 equiv of DIEA for 1 h at 55 °C in DMF. The product was purified by reverse phase HPLC and lyophilized. Purity and identity of compounds were verified by analytical HPLC and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry.
Bind-n-Seq of Polyamide–Biotin Congugates
Sequence motif logos of the highest affinity DNA binding sites of polyamide–biotin congugates 1–8 were determined according to previously reported methods.30 Each Py-Im polyamide–biotin conjugate was equilibrated at 50 nM concentration for 15 h with a uniquely barcoded library of all possible 21mers. DNA associated with polyamide–biotin conjugates were affinity purified with streptavidin magnetic beads (M-280 Dynabeads) and eluted. Isolated DNA was amplified by touchdown PCR and sequenced at the California Institute of Technology Millard and Muriel Jacobs Genetics and Genomics Laboratory on an Illumina HiSeq 2000 Genome Analyzer. The generated data set was then distributed by barcode using scripts in the MERMADE pipeline and a fasta file of a random 25% of sequences for each compound submitted for DREME motif analysis.30
DNA Thermal Denaturation Assay
Unmethylated DNA duplexes and hairpin polyamides were mixed to a final concentration of 2 and 3 μM, respectively, for polyamides 1–8, 1b, and 3b in 1 mL total volume. For experiments with hemi-methylated oligonucleotides, DNA duplexes and hairpin polyamides were mixed to a final concentration of 1 and 1.5 μM, respectively. An aqueous solution of 10 mM sodium cacodylate, 10 mM KCl, 10 mM MgCl2, and 5 mM CaCl2 at pH 7.0 was used as analysis buffer. All oligonucleotides (100 μM solutions dissolved in 10 mM Tris-Cl, 0.1 mM EDTA, pH 8.0) were purchased from Integrated DNA Technologies. The assay was conducted on a Varian Cary 100 spectrophotometer equipped with a thermocontrolled cell holder with a cell path length of 1 cm. Samples were heated to 90 °C and cooled to a starting temperature of 25 °C prior to heating at a rate of 0.5 °C/min to 90 °C. Denaturation profiles were recorded at λ = 260 nm and melting temperatures were defined as the maximum of the first derivative of the denaturation profile. Reported data represents the average of four measurements.
In Vitro Inhibition of CpG Methylation Assay
In PCR tubes, serially diluted concentrations of polyamides 1b, 3b, and distamycin D control were incubated in 96 μL of 10 pM unmethylated λ-phage DNA (Promega) and 1× NEB2 buffer (New England Biolabs) in DEPC-treated water (USB) for 12 h at 25 °C. Two additional samples of DNA in buffer without compound were kept for controls. After incubation, S-adenosyl methionine (New England Biolabs) and M.SssI (New England Biolabs) or water was added to all samples to a final concentration of 320 μM and 0.25 Units, respectively, to afford 100 μL of total solution. Samples were then incubated for 3 h at 37 °C on a Biorad MyCycler thermal cycler and heat inactivated for 15 min at 65 °C. DNA was ethanol precipitated in a centrifuge at 4 °C for 15 min with the addition of 10 μL of 3 M NaOAc, 1 μL of glycogen, and 2.5 volumes of ethanol at −20 °C. DNA was washed once with 75% aqueous ethanol at −20 °C and allowed to air-dry for 30 min. Samples were dissolved in 35 μL of water and 15 μL taken for MluI restriction enzyme digestion. Samples were prepared in PCR tubes per manufacturer’s protocol with 1 Unit of MluI per sample and incubated at 37 °C for 1 h. Blue loading buffer 6× (New England Biolabs) was added to samples and 20 μL added to a 0.7% agarose gel in 0.5× TBE buffer. DNA was visualized with SYBR gold (Invitrogen) and a Typhoon FLA9000 Scanner (GE Healthcare).
Determination of IC50
The in vitro assay was run as described above with 1b, 3b, and 9b at concentrations titrated at 10-fold and 3-fold intervals ranging from 10 nM to 33 μM, 330 pM to 10 μM, and 33 pM to 1 μM, respectively, and DNA at 50 pM. The substrate DNA fragment (7.5 kb) was PCR amplified from PTYB21 (New England Biolabs) after linearization with BamHI (New England Biolabs). Primers 5′-ACTTTTCGGGGAAATGTGCG-3′ and 5′-TTAGAGGCCCCAAGGGGTTA-3′ (IDT DNA) were used for amplification with the Expand Long Template PCR System (Roche). The amplicon was isolated with QIAquick PCR Purification Kit (Qiagen), and the amplicon size was verified by agarose gel electrophoresis. After the ethanol precipitation step which follows methylation, the DNA pellet was dissolved in 100 μL of 2% SDS and incubated overnight at 55 °C to wash off residual polyamide. The high affinity of 9b made this additional wash step necessary prior to MluI digestion. To the solution, 10 μL of 2 M NaCl followed by 2.5 volumes of ethanol were added to reprecipitate the DNA. The pellet was washed twice with cold 75% ethanol before submission to MluI digest, as described above. Digested samples were run on 1% agarose gels and visualized with SYBR Gold. Gels were scanned on a Typhoon FLA Scanner (GE Healthcare) and the bands quantitated using ImageQuant Software (GE Healthcare). Percentage inhibition was normalized against maximal methylation in the presence of no inhibitor.
IC50 curves and 95% confidence intervals were determined using GraphPad Prism by variable-slope, nonlinear regression fit to a dose response model with a bottom constraint of 0. At least three replicates of each concentration were used.
Acknowledgments
Sequencing was conducted at the Millard and Muriel Jacobs Genetics and Genomics Laboratory at the California Institute of Technology. Mass spectrometry analyses were performed in the Mass Spectrometry Laboratory of the Division of Chemistry and Chemical Engineering at the California Institute of Technology. Gels were scanned in the Center for the Chemistry of Cellular Signaling at the California Institute of Technology. National Institutes of Health (grant numbers GM51747 and GM27681); Tobacco-Related Disease Research Program (award number 20DT-0037 to J.S.K., dissertation research award); American Cancer Society (grant number PF-10-015-01-CDD to J.L.M., postdoctoral fellowship). Funding for open access charge: National Institutes of Health (grant number GM51747).
Supporting Information Available
Full chemical structures of polyamides, methylation inhibition assay with λ-phage DNA, Bind-n-Seq logos with E-values, analytical data for polyamides. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Jones P. A.; Baylin S. B. Nat. Rev. Genetics 2002, 3, 415–428. [DOI] [PubMed] [Google Scholar]
- Rountree M. R.; Bachman K. E.; Herman J. G.; Baylin S. B. Oncogene 2001, 20, 3156–3165. [DOI] [PubMed] [Google Scholar]
- Bird A. P. Nature 1986, 321, 209–213. [DOI] [PubMed] [Google Scholar]
- Ballestar E.; Wolffe A. P. Eur. J. Biochem. 2001, 268, 1–6. [DOI] [PubMed] [Google Scholar]
- Merlo A.; Herman J. G.; Mao L.; Lee D. J.; Gabrielson E.; Burger P. C.; Baylin S. B.; Sidransky D. Nat. Med. 1995, 1, 686–692. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen R. A.; Baylin S. B.; Herman J. G. Blood 1999, 93, 4347–4353. [PubMed] [Google Scholar]
- Plumb J. A.; Strathdee G.; Sludden J.; Kaye S. B.; Brown R. Cancer Res. 2000, 60, 6039–6044. [PubMed] [Google Scholar]
- Dammann R.; Li C.; Yoon J. H.; Chin P. L.; Bates S.; Pfeifer G. P. Nat. Genet. 2000, 25, 315–319. [DOI] [PubMed] [Google Scholar]
- Christman J. K. Oncogene 2002, 21, 5483–5495. [DOI] [PubMed] [Google Scholar]
- Waring M. J.; Wakelin L. P. G. Nature 1974, 252, 653–657. [DOI] [PubMed] [Google Scholar]
- Adams R. L.; Rinaldi A. FEBS Lett. 1987, 215, 266–268. [DOI] [PubMed] [Google Scholar]
- Stresemann C.; Brueckner B.; Musch T.; Stopper H.; Lyko F. Cancer Res. 2006, 66, 2794–2800. [DOI] [PubMed] [Google Scholar]
- Fagan R. L.; Cryderman D. E.; Kopelovich L.; Wallrath L. L.; Brenner C. J. Biol. Chem. 2013, 288, 23858–23867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi A.; Rajavelu A.; Ragozin S.; Sénamaud-Beaufort C.; Bashtrykov P.; Testa N.; Dali-Ali H.; Maulay-Bailly C.; Amand S.; Guianvarch D. ACS Chem. Biol. 2013, 8, 543–548. [DOI] [PubMed] [Google Scholar]
- Wade W. S.; Mrksich M.; Dervan P. B. J. Am. Chem. Soc. 1992, 114, 8783–8794. [Google Scholar]
- Trauger J. W.; Baird E. E.; Dervan P. B. Nature 1996, 382, 559–561. [DOI] [PubMed] [Google Scholar]
- White S.; Baird E. E.; Dervan P. B. Chem. Biol. 1997, 4, 569–578. [DOI] [PubMed] [Google Scholar]
- White S.; Szewczyk J. W.; Turner J. M.; Baird E. E.; Dervan P. B. Nature 1998, 391, 468–471. [DOI] [PubMed] [Google Scholar]
- Kielkopf C. L.; Baird E. E.; Dervan P. B.; Rees D. C. Nat. Struct. Biol. 1998, 5, 104–109. [DOI] [PubMed] [Google Scholar]
- Kielkopf C. L.; White S.; Szewczyk J. W.; Turner J. M.; Baird E. E.; Dervan P. B.; Rees D. C. Science 1998, 282, 111–115. [DOI] [PubMed] [Google Scholar]
- Mrksich M.; Parks M. E.; Dervan P. B. J. Am. Chem. Soc. 1994, 116, 7983–7988. [Google Scholar]
- Trauger J. W.; Baird E. E.; Mrksich M.; Dervan P. B. J. Am. Chem. Soc. 1996, 118, 6160–6166. [Google Scholar]
- Swalley S. E.; Baird E. E.; Dervan P. B. Chem.—Eur. J. 1997, 3, 1600–1607. [Google Scholar]
- Trauger J. W.; Baird E. E.; Dervan P. B. J. Am. Chem. Soc. 1998, 120, 3534–3535. [Google Scholar]
- Turner J. M.; Swalley S. E.; Baird E. E.; Dervan P. B. J. Am. Chem. Soc. 1998, 120, 6219–6226. [Google Scholar]
- Wang C. C.; Ellervik U.; Dervan P. B. Bioorg. Med. Chem. 2001, 9, 653–657. [DOI] [PubMed] [Google Scholar]
- White S.; Baird E. E.; Dervan P. B. J. Am. Chem. Soc. 1997, 119, 8756–8765. [Google Scholar]
- Herman D.; Baird E.; Dervan P. B. J. Am. Chem. Soc. 1998, 120, 1382–1391. [Google Scholar]
- Rucker V.; Melander C.; Dervan P. Helv. Chim. Acta 2003, 86, 1839–1851. [Google Scholar]
- Meier J. L.; Yu A. S.; Korf I.; Segal D. J.; Dervan P. B. J. Am. Chem. Soc. 2012, 134, 17814–17822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swalley S. E.; Baird E. E.; Dervan P. B. J. Am. Chem. Soc. 1997, 119, 6953–6961. [Google Scholar]
- Minoshima M.; Bando T.; Sasaki S.; Fujimoto J.; Sugiyama H. Nucleic Acids Res. 2008, 36, 2889–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenoweth D. M.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 13175–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenoweth D. M.; Dervan P. B. J. Am. Chem. Soc. 2010, 132, 14521–14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilch D. S.; Poklar N.; Gelfand C. A.; Law S. M.; Breslauer K. J.; Baird E. E.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A 1996, 93, 8306–8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueckner B.; Garcia Boy R.; Siedlecki P.; Musch T.; Kliem H. C.; Zielenkiewicz P.; Suhai S.; Wiessler M.; Lyko F. Cancer Res. 2005, 65, 6305–6311. [DOI] [PubMed] [Google Scholar]
- McClelland M.; Nelson M. Nucleic Acids Res. 1992, 20Suppl2145–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darii M. V.; Cherepanova N. A.; Subach O. M.; Kirsanova O. V.; Raskó T.; Slaska-Kiss K.; Kiss A.; Deville-Bonne D.; Reboud-Ravaux M.; Gromova E. S. Biochim. Biophys. Acta 2009, 1794, 1654–1662. [DOI] [PubMed] [Google Scholar]
- Song J.; Teplova M.; Ishibe-Murakami S.; Patel D. J. Science 2012, 335, 709–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird E. E.; Dervan P. B. J. Am. Chem. Soc. 1996, 118, 6141–6146. [Google Scholar]
- Puckett J. W.; Green J. T.; Dervan P. B. Org. Lett. 2012, 14, 2774–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.