

Abstract
Rearranging hydrogen bonding groups adds nucleobases to an artificially expanded genetic information system (AEGIS), pairing orthogonally to standard nucleotides. We report here a large-scale synthesis of the AEGIS nucleotide carrying 2-amino-3-nitropyridin-6-one (trivially Z) via Heck coupling and a hydroboration/oxidation sequence. RiboZ is more stable against epimerization than its 2′-deoxyribo analogue. Further, T7 RNA polymerase incorporates ZTP opposite its Watson–Crick complement, imidazo[1,2-a]-1,3,5-triazin-4(8H)one (trivially P), laying grounds for using this “second-generation” AEGIS Z:P pair to add amino acids encoded by mRNA.
One of many accomplishments of synthetic biology over the past two decades has been the generation of DNA systems that have additional nucleotide “letters” that form additional nucleobase pairs.1 These include artificially expanded genetic information systems (AEGIS),1e,2 species of DNA that use nonstandard hydrogen bonding patterns to form up to six mutually exclusive nucleobase pairs with standard Watson–Crick geometry, four more than are found in natural DNA and RNA (Figure 1). Other approaches to add nucleotides to the genetic alphabet diverge in structure more severely.1
Figure 1.
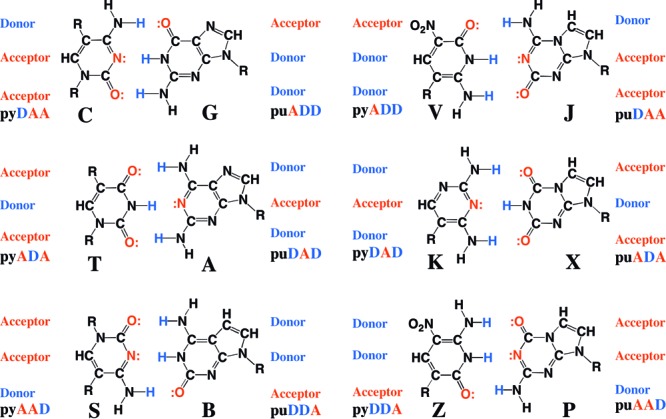
Second-generation artificially expanded genetic information system (AEGIS) uses alternative arrangements of hydrogen bond donor and acceptor groups to create a total of 12 nucleotides forming six mutually exclusive nucleobase pairs.
Because of their orthogonality, “first-generation” AEGIS pairs are today used widely. In the clinic, AEGIS DNA is used to monitor the load of viruses in the blood of patients infected with human immunodeficiency and hepatitis C viruses,3a detect mutations that cause cystic fibrosis,3b and detect viruses causing respiratory diseases.3c,3d,3f In the laboratory, AEGIS is supported by a developing molecular biology, including polymerases that perform AEGIS PCR4 and procedures to sequence AEGIS DNA.5 These have allowed AEGIS to support in vitro evolution that generates AEGIS-containing aptamers that bind to cancer cells3e (inter alia).
One “second-generation” pair of AEGIS nucleotides has performed especially well with enzymes as part of an expanded genetic system. The components of this pair carry the 2-aminoimidazo[1,2-a]-1,3,5-triazin-4(8H)one (trivially named P) and 6-amino-5-nitro-2(1H)-pyridone (trivially named Z) heterocycles (Figure 1). Implementing (respectively) the hydrogen bonding “acceptor–acceptor–donor” and “donor–donor–acceptor” patterns, these replace a first-generation AEGIS nucleobase analogue (based on a pyrazine ring system6a) that implemented the donor–donor–acceptor hydrogen bonding pattern but was susceptible to epimerization via specific-acid catalysis.6b
The Z:P pair has also proven to perform well with natural DNA polymerases.3d,4a As a hypothesis accounting for this, both the Z and P heterocycles place electron density in the minor groove, the first from the exocylic oxygen of the pyrimidine analogue and the second from N3 of the purine analogue. This density may interact with hydrogen-bond-donating amino acid side chains of polymerases.7
Should these successes with DNA be transferred to RNA, many applications can be envisioned. For example, AEGIS nucleotides might increase the number of codons in the genetic code, allowing ribosomes to synthesize proteins containing additional amino acids.8−11 With the development of cells able to replicate plasmids containing GACTZP DNA, RNA polymerases able to synthesize Z- and P-RNA intracellularly might allow for the expression of proteins with more than the 20 standard amino acids. This was shown already 20 years ago for the first-generation AEGIS pair between isocytidine and isoguanosine (implementing, respectively, the acceptor–acceptor–donor and donor–donor–acceptor hydrogen bonding patterns).8
This vision motivated us to undertake the synthesis of Z- and P-ribonucleosides, one carrying the Z heterocycle, the other carrying the P heterocycle. The synthesis of the P-ribonucleoside having a β configuration by a standard sequence coupling a preformed nucleobase to a perbenzoylated ribose proved not to be problematic.12,13 Standard chemistry did not yield the Z-ribonucleoside, however. While Heck coupling gave the 2′-deoxyribonucleoside, the analogous strategy did not generate the analogous species with a 2′-hydroxyl group.
The literature does report the synthesis of the ribonucleoside carrying an unsubstituted 2-aminopyridin-6-one, which presents the same donor–donor–acceptor hydrogen bonding pattern as Z. However, this heterocycle is prone to oxidation. We previously reported that, as deoxyribonucleosides, the nitro-14 and cyano15-substituted aminopyridin-6-ones were both stable to oxidation and slower to epimerize. Since the nitro species was both more stable and better accepted as a substrate for DNA polymerases, we chose the nitro substituent for development as an RNA component.
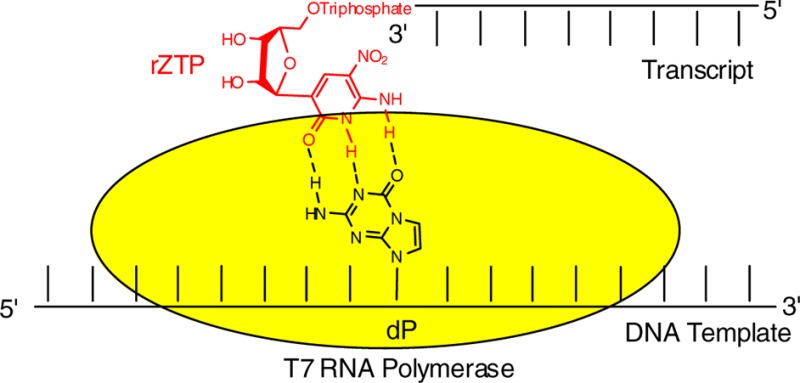
We report here a route for the large-scale synthesis of the ribonucleoside analogue of 2-amino-3-nitropyridin-6-one (riboZ). We also report physical properties of the nucleoside, including its susceptibility to epimerization. Finally, we provide evidence that rZTP is incorporated by T7 RNA polymerase opposite dP in a template and vice versa, the first examples where a potential mRNA containing the Z:P pair has been prepared by transcription.
To synthesize riboZ, we initially explored a method frequently used to prepare C-nucleosides that involves condensation of ribonolactone with a lithiated base. Unfortunately, all of our efforts to obtain the lithiated 2-amino-3-nitropyridin-6-one in protected form were not successful, apparently due to incompatibility of the protecting group and the conditions for lithiation.
We then considered condensation of 2,6-dichloropyridine and ribonolactone as a precursor of 2-amino-3-nitropyridin-6-one nucleoside (Scheme 1). Here, the condensation product was obtained with good yield. However, deoxygenation of the product with BF3·OEt2/Et3SiH16 gave mostly the α nucleoside. The reduction of the product with l-Selectride/ZnCl217 followed by Mitsunobu reaction gave the α nucleoside as a major product in low yield (35%). Further, the conversion of dinitropyridine to 2-amino-3-nitropyridin-6-one was problematic.
Scheme 1. Attempted Synthesis of C-Ribonucleoside with 2,6-Dichloropyridine.

We then exploited the manipulation of the silyl enol ether moiety in the Heck product, the intermediate in our synthesis of the 2′-deoxyribonucleoside analogue.14 The literature reports various transformations of silyl enol ethers of furanose nucleosides, including their oxidation with osmium tetroxide18 or dimethyldioxirane19 to give the hydroxyketone, and hydroboration/oxidation20 to make diols. In this system, oxidation of the silyl enol ether of the furanose nucleoside (8 in Scheme 2) with OsO4 gave a mixture of diastereomers with low yields. Also, treatment with dimethyldioxirane also did not yield the desired products.
Scheme 2. Synthesis of the Ribonucleoside of Z.

However, hydroboration/oxidation of compound 8(15) gave the desired trans-diol 9 exclusively and in good yield (>80%) (Scheme 2). This intermediate was suitable for further elaboration to give the desired nucleoside analogue. Thus, acetylation of 9 followed by desilylation gave 11. The configuration of the 3′-OH group of 11 was then inverted by a two-step oxidation/reduction sequence, where the stereochemical course of the reduction was controlled by the 5′-hydroxyl group. Acetyl migration gave two compounds (13a and 13b) as a mixture.
Removal of acetate groups in both versions of 13 by treatment with ammonia gave 14. Subsequent removal of the nitrophenylethyl (NPE) group (DBU in CH3CN) gave ribonucleoside 15. This was converted into its triphosphate 17 (rZTP) using the Ludwig–Eckstein procedure21 applied to 16, prepared from 13 with 5′-DMTr protection, acetylation, and 5′-DMTr deprotection (Scheme 3).
Scheme 3. Triphosphate Synthesis of the Ribonucleoside of Z.

The pKa of the Z-heterocycle of the ribonucleoside 15 was estimated from a series of UV spectra collected at 380 nm (λmax for protonated 15) and 400 nm (λmax for deprotonated 15) as the pH of the aqueous solution was adjusted by adding dilute aqueous solutions of HCl and NaOH. A plot of the ratio of absorbance at 380 nm/400 nm (Supporting Information Figure S1) gave a pKa value 7.9 ± 0.1. This value is similar to that measured for 2-deoxyribonucleoside of Z, implying (as expected) that a hydroxyl group in the 2′-position does not affect the pKa of the Z base.
A special feature of C-nucleosides is their susceptibility to epimerization. This phenomenon was described for pseudouridine several decades ago22 and for other C-nucleosides since.23 2′-Deoxyribosides where pyrazine heterocycles present a donor–donor–acceptor hydrogen bonding pattern also epimerize, via specific-acid catalysis, to give mixtures of furanose and pyranose isomers.6 However, electron-withdrawing groups (nitro and cyano) placed at the 3-position of the 2-aminopyridin-6-one system (analogous to the 5-position of a pyrimidine) slow epimerization.14
These results prompted us to ask whether the presence of the 2′-hydroxyl group in riboZ also slowed the epimerization. Epimerization is indeed slower in the ribo species than in the 2′-deoxyribo species (Table 1). While about half of a sample of dZ was epimerized at pH 7 and 90 °C in 6 h, the epimerization of riboZ under these conditions was less than 10%. At pH 2 and 20 °C, dZ showed significant epimerization (∼30%) in 6 h, while epimerization of riboZ was less than 1% in this condition. This is consistent with the specific acid-catalyzed mechanism observed for epimerization for an analogous systems, a mechanism requiring initial protonation of the ribose ring oxygen. The electron-withdrawing nature of the 2′-hydroxyl group is expected to lower the pKa of the protonated ribose ring oxygen, making this mechanism less effective at pH values approaching neutrality.
Table 1. Remaining Amount of dZ and rZ Nucleosides at pH 2 and 7a.
| 0 h | 0.5 h | 1 h | 2 h | 4 h | 6 h | ||
|---|---|---|---|---|---|---|---|
| pH 7, 90 °C | dZ | 100 | 90 | 81.7 | 70.4 | 58.5 | 52.5 |
| rZ | 100 | 99.8 | 98.4 | 96.2 | 94.2 | 91.2 | |
| pH 2, 20 °C | dZ | 100 | 94.7 | 89.9 | 84.8 | 74.0 | 68.3 |
| rZ | 100 | 100 | 99.9 | 99.9 | 99.6 | 99.4 |
The loss of the nucleoside was caused by epimerization at the given pH and temperature.
The availability of riboZTP allowed us to explore the molecular biology that might be enabled by the Z:P pair. Several DNA molecules with dP downstream from a T7 RNA polymerase promoter segment were synthesized (Table S1).
Transcription reactions using standard and P-containing templates and T7 RNA polymerase (MC T7 RNA Pol, having the wild-type sequence, purified in-house) with or without rZTP were performed. Without rZTP, transcription of the template having one dP provided full-length product without significant pausing. This showed that the polymerase could mistmatch a single standard ribonucleotide (presumably C) efficiently opposite a single template dZ. However, substantial pausing was observed in the transcription of increasing numbers of dPs in the template. When the transcription mixture contained rZTP, as well, however, no pausing was observed; the transcription gave only full-length product (Figure 2). Analogous results with oligonucleotides containing dZ in the template and dPTP are shown in the Supporting Information; these show less overall processivity.
Figure 2.

PAGE (20%, with 7 M urea) of transcription products with four RNA length standards (left, 42, 25, 14, 5 nt). Transcription of “standard template” shows unreacted 32P-GTP (large band at bottom), typical “stutter” bands, full-length transcript (30 nt), and small amounts of very long products (unassigned). Remaining bands are with indicated templates containing from 1 to 3 dPs (Table S1) obtained without rZTP (left cluster) and with rZTP (right cluster). Full-length product is made in the absence of rZTP by misincorporation of a standard nucleotide (presumably rC) opposite dP, with increasing pausing with increasing numbers of template dPs. Pausing disappears when rZTP is added.
The availability of the ribonucleoside of Z and its triphosphates in substantial quantities, its stability to the acid-catalyzed epimerization, and its ability to be incorporated opposite its complementary purine analogue in a template by T7 RNA polymerase advance the in vitro molecular biology for the Z:P system. In parallel work, we described tools to sequence DNA containing dZ and/or dP, polymerases that PCR-amplify dZ and dP-containing DNA, and in vitro evolution from libraries containing dZ and dP.3e In future work, we hope to move the Z:P system into living cells, where it can be used to encode additional amino acids and where the power of GACTZP nucleic acids to evolve can be coupled to the ability of cells to evolve.
Experimental Section
N,N-Bis[(tert-butoxy)carbonyl]-5-[3′,5′-di-O-(tert-butyldiphenylsilyl)-β-d-2,5-dihydrofuranosyl]-3-nitro-6-[2-(4-nitrophenyl)ethoxy]-2-pyridinamine (8)
To a mixture of compound 7 (183 g, 279 mmol) and imidazole (34.4 g, 505 mmol) in dichloromethane (2 L) was added TBDPSCl (72.2 mL, 278 mmol). The mixture was stirred at room temperature for 1 h and washed with H2O, dried over Na2SO4, and filtered. The solution was concentrated by rotary evaporation, and the residue was resolved by flash chromatography (silica, ethyl acetate/hexanes = 1:3 to 1:2). The fractions containing the product were combined, concentrated, and dried. The residue was then redissolved in dichloromethane (2L), to which were then added DMAP (1.5 g), triethylamine (130 mL), and di-tert-butyl dicarbonate (120 g). The mixture was stirred at room temperature overnight and washed with brine, dried over Na2SO4, and filtered. The solution was concentrated by rotary evaporation, and the residue was resolved by flash chromatography (silica, ethyl acetate/hexanes = 1:4 to 1:3) to give 8 as a pale yellow solid (235 g, 77%): mp 56–58 °C; 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 8.01 (d, 2H, J = 8.7 Hz), 7.62–7.76 (m, 8H), 7.26–7.49 (m, 12H), 7.19 (d, 2H, J = 8.4 Hz), 5.59 (d, 1H, J = 3.0 Hz), 4.83 (m, 1H), 4.35–4.58 (m, 3H), 3.85–4.10 (m, 2H), 2.85–3.05 (m, 2H), 1.38 (s, 18H), 1.04 (s, 9H), 0.98 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 160.6, 151.5, 150.1, 147.0, 145.7, 143.2, 136.5, 135.9, 135.8, 135.6, 134.4, 133.4, 133.3, 131.5, 131.4, 130.6, 129.9, 128.2, 128.1, 127.9, 127.9, 126.7, 123.9, 101.3, 84.5, 84.0, 78.1, 67.4, 65.2, 35.1, 28.0, 27.0, 26.5, 19.43, 19.39; HRMS (TOF- ESI) m/z calcd for C60H70N4O12Si2Na (M + Na)+ 1117.4421, found 1117.4405.
N,N-Bis[(tert-butoxy)carbonyl]-5-[3′,5′-di-O-(tert-butyldiphenylsilyl)-β-d-xylofuranosyl]-3-nitro-6-[2-(4-nitrophenyl)ethoxy]-2-pyridinamine (9)
To a solution of the compound 8 (65 g, 59.3 mmol) in THF (700 mL) was added diborane in THF (1 M, ∼200 mL, 3.4 equiv) at 0 °C. The mixture was slowly warmed to room temperature and stirred overnight. It was quenched with 70 mL of H2O for 5 min and treated with sodium perborate (100 g) in water (600 mL). The mixture was vigorously stirred at room temperature for 1 h. THF was removed by rotary evaporation, and the residue was extracted with ethyl acetate. The organic layer was then dried over Na2SO4 and filtered. The solution was concentrated by rotary evaporation, and the residue was resolved by flash chromatography (silica, ethyl acetate/hexanes = 1:4 to 1:3) to give 9 as a pale yellow solid (61 g, 92%): mp 71–73 °C; 1H NMR (300 MHz, CDCl3) δ 8.62 (s, 1H), 8.10 (d, 2H, J = 8.7 Hz), 7.64–7.74 (m, 4H), 7.14–7.50 (m, 18H), 4.68 (s, 1H), 4.60 (t, 2H, J = 6.3 Hz), 4.00–4.28 (m, 4H), 3.64 (br s, 1H), 3.09 (t, 2H, J = 5.4 Hz), 1.42 (s, 18H), 1.09 (s, 9H), 0.81 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 160.2, 150.3, 147.1, 145.6, 143.3, 136.7, 136.0, 135.9, 135.8, 134.2, 133.9, 133.7, 133.4, 132.0, 130.2, 129.95, 129.86, 128.1, 127.9, 125.0, 124.0, 84.2, 83.7, 81.9, 79.4, 67.3, 62.8, 35.0, 28.1, 27.2, 26.9, 19.5, 19.2; HRMS (TOF-ESI) m/z calcd for C60H72N4O13Si2Cl (M + Cl)− 1147.4328, found 1147.4365; C62H75N4O15Si2 (M + CH3COO)− 1171.4773, found 1171.4825.
N,N-Bis[(tert-butoxy)carbonyl]-5-[3′,5′-di-O-(tert-butyldiphenylsilyl)-2′-O-acetyl-β-d-xylofuranosyl]-3-nitro-6-[2-(4-nitrophenyl)ethoxy]-2-pyridinamine (10)
A mixture of compound 9 (128 g, 115 mmol), acetic anhydride (20 mL), pyridine (30 mL), and DMAP (0.2 g) in dichloromethane (1200 mL) was stirred at room temperature for 2 h. The mixture was washed with 0.5 N HCl solution and brine. The organic layer was concentrated by rotary evaporation. The residue was purified by flash chromatography (silica, ethyl acetate/hexanes = 1:4) to give a light yellow solid (128 g, 96%): mp 74–76 °C; 1H NMR (300 MHz, CDCl3) δ 8.70 (s, 1H), 8.15 (d, 2H, J = 8.7 Hz), 7.55–7.75 (m, 4H), 7.15–7.45 (m, 18H), 4.99 (s, 1H), 4.86 (s, 1H), 4.45–4.65 (m, 2H), 4.05–4.2 (m, 3H), 3.81 (dd, 1H, J = 10.2, 3.3 Hz), 3.05 (t, 2H, J = 6.9 Hz), 1.90 (s, 3H), 1.41 (s, 18H), 1.07 (s, 9H), 0.79 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 168.7, 160.2, 150.2, 147.1, 145.6, 143.7, 136.6, 136.1, 135.9, 135.8, 134.7, 133.6, 133.4, 132.7, 132.0, 130.1, 130.0, 129.9, 129.8, 127.9, 124.2, 124.0, 84.4, 84.1, 81.6, 79.4, 77.1, 67.3, 63.3, 34.8, 28.1, 27.1, 27.0, 21.0, 19.4, 19.2; HRMS (TOF-ESI) m/z calcd for C62H74N4O14Si2Na (M + Na)+ 1177.4632, found 1177.4620.
N-Acetyl-5-(2′-O-acetyl-β-d-xylofuranosyl)-3-nitro-6-[2-(4-nitrophenyl)ethoxy]-2-pyridinamine (11)
To a solution of compound 10 (128 g, 110.8 mmol) in dichloromethane (1100 mL) was added trifluoroacetic acid (33 mL) at room temperature. The mixture was stirred for 3 h, neutralized by aqueous NaHCO3, and mixed with water and extracted with dichloromethane. The combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was then resolved by flash chromatography (silica, ethyl acetate/hexanes = 1:2) to give the NH2 (Boc removed) product as a pale yellow solid; 1H NMR (300 MHz, CDCl3) δ 8.62 (d, 1H, J = 0.9 Hz), 8.15 (d, 2H, J = 8.7 Hz), 7.55–7.7 (m, 4H), 7.1–7.5 (m, 18H), 5.02 (s, 1H), 4.77 (s, 1H), 4.4–4.55 (m, 2H), 4.05–4.15 (m, 3H), 3.86 (dd, 1H, J = 13.8, 7.2 Hz), 3.04 (t, 2H, J = 6.8 Hz), 1.89 (s, 3H), 1.07 (s, 9H), 0.79 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 168.9, 162.3, 153.1, 147.1, 145.9, 136.2, 136.1, 135.9, 135.8, 135.4, 133.7, 133.5, 133.1, 132.1, 130.0, 129.9, 129.8, 128.0, 127.9, 127.7, 124.0, 122.8, 114.4, 84.1, 81.8, 79.4, 77.4, 66.7, 63.4, 34.9, 27.2, 26.9, 21.1, 19.4, 19.2.
The product was then dissolved in pyridine (600 mL). DMAP (4 g) and acetic anhydride (60 mL) were added to the solution, and the mixture was stirred for 5 h at 60 °C. Pyridine was then removed by rotary evaporation, and the residue was dissolved in ethyl acetate. The mixture was treated with 0.5 N HCl and stirred vigorously. The organic phase was separated, dried, and purified by column chromatography (silica gel, ethyl acetate/hexanes) to give a pale yellow solid, which was a mixture of the N-acetyl and N,N-diacetyl compounds. Spectral data for N-acetyl compound: 1H NMR (300 MHz, CDCl3) δ 10.64 (s, 1H), 8.72 (d, 1H, J = 0.9 Hz), 8.15 (d, 2H, J = 8.7 Hz), 7.6–7.7 (m, 4H), 7.1–7.5 (m, 18H), 5.03 (s, 1H), 4.83 (s, 1H), 4.55–4.65 (m, 2H), 4.05–4.2 (m, 3H), 3.85 (dd, 1H, J = 10.2, 3.3 Hz), 3.12 (t, 2H, J = 6.9 Hz), 2.45 (s, 3H), 1.92 (s, 3H), 1.08 (s, 9H), 0.76 (s, 9H).
The mixture of N-acetyl and N,N-diacetyl compounds was mixed with triethylamine trihydrofluoride (160 mL), triethylamine (140 mL), and THF (1400 mL) then stirred at room temperature for 24 h. The mixture was poured into aqueous NaHCO3 and extracted with ethyl acetate. The combined organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography (silica, ethyl acetate/hexanes = 3:1 to ethyl acetate 100%) to give the product as a pale yellow solid (49 g, this is a mixture of mono- and diacetyl compounds and the monoacetyl is major, 84%): mp 52–54 °C; 1H NMR (300 MHz, CDCl3) δ 10.52 (s, 1H), 8.73 (d, 1H, J = 0.6 Hz), 8.16 (d, 2H, J = 8.7 Hz), 7.43 (d, 2H, J = 8.7 Hz), 5.03 (dd, 1H, J = 3.0, 1.8 Hz), 4.91 (dd, 1H, J = 2.7, 0.9 Hz), 4.6–4.8 (m, 2H), 4.3 (m, 1H), 4.05–4.2 (m, 3H), 3.9–3.95 (m, 1H), 3.22 (t, 2H, J = 6.9 Hz), 2.95–3.05 (m, 1H), 2.44 (s, 3H), 2.06 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 170.2, 169.3, 161.6, 147.1, 145.6, 145.5, 135.9, 130.0, 126.1, 124.0, 118.2, 83.5, 81.0, 78.5, 77.4, 68.1, 61.5, 34.9, 26.5, 21.1; HRMS (TOF-ESI) m/z calcd for C22H24N4O11Na (M + Na)+ 543.1334, found 543.1331.
N-Acetyl-5-(3′-deoxy-3′-oxo-2′-O-acetyl-β-d-ribofuranosyl)-3-nitro-6-[2-(4-nitrophenyl)ethoxy]-2-pyridinamine (12)
A mixture of compound 11 (23.4 g, 45.0 mmol), trityl chloride (13.8 g, 49.5 mmol), DMAP (0.4 g) in triethylamine (12.5 mL), and dichloromethane (350 mL) was stirred at room temperature for 2 h. It was washed with water, and the organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography (silica, ethyl acetate/hexanes = 2:1) to give a light yellow solid: 1H NMR (300 MHz, CDCl3) δ 10.59 (s, 1H), 8.73 (d, 1H, J = 0.6 Hz), 8.16 (d, 2H, J = 8.7 Hz), 7.2–7.5 (m, 17H), 5.08 (dd, 1H, J = 2.1, 0.9 Hz), 4.92 (dd, 1H, J = 2.1, 0.9 Hz), 4.6–4.8 (m, 2H), 4.15–4.3 (m, 2H), 3.65 (d, 2H, J = 4.5 Hz), 3.22 (t, 2H, J = 3.9 Hz), 3.08 (d, 1H, J = 3.0 Hz), 2.44 (s, 3H), 2.07 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 169.6, 168.9, 161.4, 147.1, 145.6, 145.5, 143.3, 135.7, 130.0, 128.7, 128.3, 127.6, 126.1, 124.0, 118.4, 87.9, 82.7, 80.9, 79.2, 79.9, 68.0, 62.2, 35.0, 26.5, 21.1.
The product was then dissolved in dichloromethane (350 mL) and treated with Dess-Martin periodinane (28 g, 66 mmol) at room temperature and stirred for 4 h. It was filtered through Celite, and the filtrate was concentrated. The residue was purified by flash chromatography (silica, ethyl acetate/hexanes = 1:1) to give a light yellow solid. It was then dissolved in dichloromethane (600 mL) and treated with trifluoroacetic acid (30 mL) and stirred at room temperature for 3 h. The mixture was neutralized by aqueous sodium bicarbonate and mixed with water and extracted with dichloromethane. The combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography (silica, ethyl acetate/hexanes = 2:1) to give a light yellow solid (15.9 g, 68%): mp 66–68 °C; 1H NMR (300 MHz, CDCl3) δ 10.64 (s, 1H), 8.65 (s, 1H), 8.17 (d, 2H, J = 9.0 Hz), 7.47 (d, 2H, J = 8.7 Hz), 5.22 (d, 1H, J = 9.6 Hz), 5.09 (d, 1H, J = 9.3 Hz), 4.6–4.9 (m, 2H), 4.25 (t, 1H, J = 2.7 Hz), 3.98 (m, 2H), 3.26 (t, 2H, J = 6.9 Hz), 2.44 (s, 3H), 2.07 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 207.5, 169.6, 168.4, 162.8, 147.3, 146.5, 145.2, 137.3, 130.1, 125.7, 124.1, 115.5, 81.4, 76.4, 75.0, 68.7, 61.6, 35.0, 26.6, 20.5; HRMS (TOF-ESI) m/z calcd for C22H22N4O11Na (M + Na)+ 541.1177, found 541.1181.
5-(2′-O-Acetyl-β-d-ribofuranosyl)-3-nitro-6-[2-(4-nitrophenyl)ethoxy]-2-pyridinamine (13a) and 5-(3′-O-Acetyl-β-d-ribofuranosyl)-3-nitro-6-[2-(4-nitrophenyl)ethoxy]-2-pyridinamine (13b)
To a solution of compound 12 (29.0 g, 51.7 mmol) in acetic acid (200 mL) and acetonitrile (200 mL) was added sodium triacetoxyborohydride (21.9 g, 103 mmol) at 0 °C and stirred for 1 h. It was poured into water and extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was resolved by flash chromatography (silica, ethyl acetate/hexanes = 2:1 to 100% ethyl acetate) to give 13a/13b as a pale yellow solid (20.5 g, 70%): mp 164–165 °C; HRMS (TOF-ESI) m/z calcd for C22H24N4O11Na (M + Na)+ 543.1334, found 543.1333.
5-(β-d-Ribofuranosyl)-3-nitro-6-[2-(4-nitrophenyl)ethoxy]-2-pyridinamine (14)
A mixture of compound 13 (15 g, 28.8 mmol) and ammonium hydroxide (100 mL) in methanol (500 mL) was stirred at room temperature overnight. The solution was then concentrated in vacuo, and the residue was resolved by flash chromatography (silica, ethyl acetate/methanol = 15:1) to give a light yellow solid (11.3 g, 90% for two steps): mp 155–156 °C; 1H NMR (300 MHz, CD3OD) δ 8.52 (d, 1H, J = 0.6 Hz), 8.18 (d, 2H, J = 9.0 Hz), 7.60 (d, 2H, J = 8.7 Hz), 4.82 (d, 2H, J = 3.6 Hz), 4.6–4.7 (m, 2H), 3.65–3.95 (m, 5H), 3.24 (t, 2H, J = 6.3 Hz); 13C NMR (75 MHz, CD3OD) δ 163.6, 154.0, 147.0, 146.8, 135.2, 130.3, 123.4, 121.7, 113.5, 83.4, 79.5, 75.9, 70.9, 67.0, 61.9, 34.7; HRMS (TOF-ESI) m/z calcd for C18H20N4O9Na (M + Na)+ 459.1122, found 459.1118.
6-Amino-3-(β-d-ribofuranosyl)-5-nitro-2(1H)-pyridone (15)
To a solution of compound 14 (30 mg, 0.069 mmol) in acetonitrile (5 mL) was added DBU (0.1 mL); the mixture was then stirred overnight at room temperature. The mixture was then concentrated in vacuo, and the residue was resolved by flash chromatography (silica, CH2Cl2/MeOH = 5:1 to 5:2) to give a yellow solid (15 mg, 75%): mp 200–202 °C;1H NMR (300 MHz, D2O) δ 8.14 (s, 1H), 4.59 (d, 1H, J = 4.8 Hz), 4.03 (t, 1H, J = 4.8 Hz), 3.96 (t, 1H, J = 6.0 Hz), 3.8–3.9 (m, 1H), 3.72 (dd, 1H, J = 12.6, 3.0 Hz), 3.59 (dd, 1H, J = 12.6, 4.5 Hz); 13C NMR (75 MHz, D2O) δ 163.3, 150.9, 135.4, 116.7, 115.0, 83.1, 80.3, 74.3, 70.7, 61.5; HRMS (TOF-ESI) m/z calcd for C10H12N3O7 (M – H)− 286.0681, found 286.0681.
N-Acetyl-5-[2′,3′-O-diacetyl-β-d-ribofuranosyl]-3-nitro-6-[2-(4-nitrophenyl)ethoxy]-2-pyridinamine (16)
To a solution of compound 13 (500 mg, 0.961 mmol), triethylamine (0.27 mL), and DMAP (10 mg) in dichloromethane (10 mL) was added DMTr chloride (358 mg, 1.057 mmol). The mixture was stirred at room temperature for 1 h then washed with water. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography (silica, ethyl acetate) to give a light yellow solid. This was dissolved in dichloromethane (20 mL) and pyridine (0.5 mL) and treated with acetic anhydride (0.18 mL) and DMAP (10 mg). The mixture was stirred at room temperature for 3 h then washed with water. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was then resolved by flash chromatography (silica, ethyl acetate/hexanes = 2:1) to give product as a light yellow solid: 1H NMR (300 MHz, CDCl3) δ 10.61 (s, 1H), 8.66 (s, 1H), 8.17 (d, 2H, J = 8.7 Hz), 7.15–7.5 (m, 11H), 6.81 (d, 4H, J = 8.1 Hz), 5.49 (t, 1H, J = 4.8 Hz), 5.32 (dd, 1H, J = 12.3, 3.6 Hz), 5.01 (d, 1H, J = 4.2 Hz), 4.69 (t, 2H, J = 6.6 Hz), 4.2–4.3 (m, 1H), 3.77 (s, 6H), 3.35–3.45 (m, 2H), 3.21 (t, 2H, J = 6.6 Hz), 2.44 (s, 3H), 2.06 (s, 3H), 2.04 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 169.8, 169.5, 168.7, 161.9, 158.7, 147.1, 145.9, 145.5, 144.7, 135.9, 135.8, 135.7, 130.3, 130.1, 128.3, 128.1, 127.1, 125.9, 124.0, 117.0, 113.4, 86.7, 80.2, 77.6, 74.8, 71.2, 68.1, 62.5, 60.6, 55.4, 35.0, 26.6, 20.8, 20.7.
The product was dissolved in dichloromethane (30 mL) and treated with dichloroacetic acid (0.6 mL). The mixture was stirred at room temperature for 1 h then washed with water. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was resolved by flash chromatography (silica, ethyl acetate/hexanes = 2:1 to ethyl acetate 100%) to give 16 as a light yellow solid (400 mg, 74%): mp 160–161 °C; 1H NMR (300 MHz, CDCl3) δ 10.60 (s, 1H), 8.73 (s, 1H), 8.17 (d, 2H, J = 8.7 Hz), 7.47 (d, 2H, J = 8.7 Hz), 5.2–5.4 (m, 2H), 4.96 (d, 1H, J = 4.5 Hz), 4.74 (t, 2H, J = 6.3 Hz), 4.1–4.2 (m, 1H), 3.95–4.05 (m, 1H), 3.74–3.84 (m, 1H), 3.26 (t, 2H, J = 7.2 Hz), 2.44 (s, 3H), 2.23 (t, 1H, J = 5.7 Hz), 2.10 (s, 3H), 2.06 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 170.3, 169.6, 168.7, 162.1, 147.2, 146.1, 145.4, 136.7, 130.1, 125.8, 124.1, 116.6, 81.6, 78.4, 74.7, 70.5, 68.3, 61.5, 35.0, 26.6, 20.8; HRMS (TOF-ESI) m/z calcd for C24H26N4O12Na (M + Na)+ 585.1439, found 585.1436.
6-Amino-3-(5′-O-triphosphate-β-d-ribofuranosyl)-5-nitro-2(1H)-pyridone (17)
To a solution of compound 16 (60 mg, 0.107 mmol) in pyridine (1 mL) and dioxane (2 mL) was added a solution of 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one (32.4 mg, 0.16 mmol) in dioxane (1.0 mL) at room temperature. After 15 min, a mixture of tributylammonium pyrophosphate in DMF (0.2 M, 1.6 mL, 0.32 mmol) and tributylamine (0.17 mL) was added. After 20 min, a solution of iodine (40.6 mg, 0.16 mmol) and water (0.063 mL) in pyridine (3.1 mL) was added. After 30 min, the reaction was quenched by the addition of aqueous Na2SO3 (5%, until color disappears). The pyridine and dioxane were removed in vacuo. The residue was dissolved in acetonitrile (10 mL) and water (10 mL) and kept at room temperature overnight. It was purified by reverse-phase prep HPLC (25 mM TEAA to 25 mM TEAA/CH3CN = 30:70 in 20 min, running time 30 min), then the collected fraction was lyophilized (12.9 min in the analytical HPLC, 50 mM TEAB to 60% CH3CN + 40% 50 mM TEAB in 20 min). The lyophilized residue was dissolved in CH3CN (5 mL) and treated with DBU (0.1 mL). The mixture was stirred at room temperature for 24 h. Volatiles were removed by rotary evaporation. To the residue was added ammonium hydroxide (5 mL), and the mixture was stirred at room temperature for 1 h. Ammonia was removed by rotary evaporation, and the residue was diluted with water and lyophilized. The residue was dissolved in water, filtered, and resolved by ion exchange HPLC (water to 0.5 M ammonium bicarbonate in 25 min). The collected fraction was lyophilized to give a yellow solid (0.042 mmol, 39%): 1H NMR (300 MHz, D2O) δ 8.17 (s, 1H), 4.0–4.2 (m, 6H); 31P NMR (121 MHz, D2O) δ −7.9 (d, 1P, J = 20 Hz), −10.0 (d, 1P, J = 19 Hz), −21.6 (t, 1P, J = 19 Hz); HRMS (TOF-ESI) m/z calcd for C10H15N3O16P3 (M – H)− 525.9671, found 525.9666.
Measuring the pKa of 15
An aqueous solution (30 mL) of 15 (∼0.5 mg) was acidified to pH 4 by the addition of dilute aqueous HCl (10 mM). The pH of the solution was then varied by addition of various amounts of dilute aqueous NaOH (10 or 100 mM). UV scans (220–500 nm) were taken at the various pH values. The pKa was determined by plotting the pH versus the quotient of absorption at two different wavelengths (400 nm/380 nm).
Measuring the Rate of Epimerization of 15
Compound 15 was dissolved in a dilute HCl aqueous solution (pH 2.0) and solution of 25 mM Et3N–HOAc buffer (pH 7.0). The concentration of 15 was 0.4 mM. The solutions were incubated at room temperature (pH 2) or 90 °C (pH 7). At time intervals, aliquots (10 μL) were removed, neutralized with aqueous triethylammonium bicarbonate buffer (50 mM; pH 8; 0.1 mL), and analyzed by analytical rp-HPLC (Sunfire C18 5 μm, 3.0 × 150 mm, eluent A = 50 mM TEAB, eluent B = 35% CH3CN and 65% 50 mM TEAB, gradient from 100% A to 70% A, 30% B in 30 min, flow rate 0.5 mL/min).
Transcription Reaction
Transcription templates were prepared by independently combining equimolar ratios of top strand (NLT1) and bottom strand (NL standard, NLP1, NLP2, NLP3, or NLP4) in 1× transcription buffer (20 mM NaCl, 40 mM Tris pH 7.8, 6 mM MgCl2, 2 mM spermidine, and 10 mM DTT), heating to 95 °C, and then cooling to room temperature.
Transcription reactions contained a final concentration of 0.2 μM template DNA (NLT1 and various bottom strands as indicated in Figure 2), 1× transcription buffer (20 mM NaCl, 40 mM Tris pH 7.8, 6 mM MgCl2, 2 mM spermidine, and 10 mM DTT), 1 μCi/μL α32P-GTP, MC T7 RNA Pol RNA polymerase (0.05 units/μL final), and 0.5 mM each rNTP in the minus experiment or 0.5 mM each rNTP and rZTP in the plus experiment. Reactions were incubated at 37 °C for 40 min and 2 h. Reactions were quenched with three-fold formamide quench buffer, and samples were resolved on a 20% PAGE.
Acknowledgments
We thank Dr. Matthew Carrigan for providing polymerase (MC T7 RNA Pol). We are also indebted to HDTRA1-13-1-0004 from the Defense Threat Reduction Agency, DARPA under its “Foundries” program C13K11520, the Department of Defense under W911NF-12-C-0059, and the NIAID under its program to analyze chronic and recent HIV infections (R01AI098616).
Supporting Information Available
Epimerization and pKa measurement of compound 15, and 1H, 13C, and 31P NMR spectra for compounds 8, 9, 10, 11, 12, 13, 14, 15, 16, and 17, and data on incorporation of rPTP opposite template dZ. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Two authors are named as coinventors on patent applications covering this technology.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Malyshev D. A.; Dhami K.; Quach H. T.; Lavergne T.; Ordoukhanian P.; Torkamani A.; Romesberg F. E. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 12005. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hirao I.; Kimoto M.; Yamashige R. Acc. Chem. Res. 2012, 45, 2055. [DOI] [PubMed] [Google Scholar]; c McMinn D. L.; Ogawa A. K.; Wu Y.; Liu J.; Schultz P. G.; Romesberg F. E. J. Am. Chem. Soc. 1999, 121, 11585. [Google Scholar]; d Matsuda S.; Henry A. A.; Schultz P. G.; Romesberg F. E. J. Am. Chem. Soc. 2003, 125, 6134. [DOI] [PubMed] [Google Scholar]; e Benner S. A. Acc. Chem. Res. 2004, 37, 784. [DOI] [PubMed] [Google Scholar]; f Hirao I.; Ohtsuki T.; Fujiwara T.; Mitsui T.; Yokogawa T.; Okuni T.; Nakayama H.; Takie K.; Yabuki T.; Kigawa T.; Kodama K.; Yokogawa T.; Nishikawa K.; Yokogawa S. Nat. Biotechnol. 2002, 20, 177. [DOI] [PubMed] [Google Scholar]; g Hirao I.; Mitsui T.; Kimoto M.; Yokoyama S. J. Am. Chem. Soc. 2007, 129, 15549. [DOI] [PubMed] [Google Scholar]; h Leconte A. M.; Matsuda S.; Hwang G. T.; Romesberg F. E. Angew. Chem., Int. Ed. 2006, 45, 4326. [DOI] [PubMed] [Google Scholar]
- Geyer C. R.; Battersby T. R.; Benner S. A. Structure 2003, 11, 1485. [DOI] [PubMed] [Google Scholar]
- a Collins M. L.; Irvine B.; Tyner D.; Fine E.; Zayati C.; Chang C.; Horn T.; Ahle D.; Detmer J.; Shen L.-P.; Kolberg J.; Bushnell S.; Urdea M. S.; Ho D. D. Nucleic Acids Res. 1997, 25, 2979. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Johnson S. C.; Marshall D. J.; Harms G.; Miller C. M.; Sherrill C. B.; Beaty E. L.; Lederer S. A.; Roesch E. B.; Madsen G.; Hoffman G. L.; Laessig R. H.; Kopish G. J.; Baker M. W.; Benner S. A.; Farrell P. M.; Prudent J. R. Clin. Chem. 2004, 50, 2019–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Arens M. G.; Buller R. S.; Rankin A.; Mason S.; Whetsell A.; Agapov E.; Lee W.-M.; Storch G. A. J. Clin. Microbiol. 2010, 48, 2387–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lee W. M.; Grindle K.; Pappas T.; Marshall D. J.; Moser M. J.; Beaty E. L.; Shult P. A.; Prudent J. R.; Gern J. E. J. Clin. Microbiol. 2007, 45, 2626–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Sefah K.; Yang Z.; Bradley K. M.; Hoshika S.; Jimeneza E.; Zhu G.; Shanker S.; Yu F.; Tan W.; Benner S. A. Proc. Natl. Acad. Sci. U.S.A. 2013, 10.1073/pnas.1311778111. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Nolte F. S.; Marshall D. J.; Rasberry C.; Schievelbein S.; Banks G. G.; Storch G. A.; Arens M. Q.; Butler R. S.; Prudent J. R. J. Clin. Microbiol. 2007, 45, 2779–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yang Z.; Chen F.; Chamberlin S. G.; Benner S. A. Angew. Chem., Int. Ed. 2010, 49, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sismour A. M.; Lutz S.; Park J.-H.; Lutz M. J.; Boyer P. L.; Hughes S. H.; Benner S. A. Nucleic Acids Res. 2004, 32, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sismour A. M.; Benner S. A. Nucleic Acids Res. 2005, 33, 5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Chen F.; Alvarado J. B.; Benner S. A. J. Am. Chem. Soc. 2011, 133, 15105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Voegel J. J.; Benner S. A. J. Am. Chem. Soc. 1994, 116, 6929. [Google Scholar]; b von Krosigk U.; Benner S. A. J. Am. Chem. Soc. 1995, 117, 5361. [Google Scholar]; c Voegel J. J.; Benner S. A. Helv. Chim. Acta 1996, 79, 1863. [Google Scholar]
- Hendrickson C. L.; Devine K. G.; Benner S. A. Nucleic Acids Res. 2004, 32, 2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bain J. D.; Switzer C.; Chamberlin A. R.; Benner S. A. Nature 1992, 356, 537. [DOI] [PubMed] [Google Scholar]; b Switzer C.; Moroney S. E.; Benner S. A. J. Am. Chem. Soc. 1989, 111, 8322–8323. [Google Scholar]; c Switzer C. Y.; Moroney S. E.; Benner S. A. Biochemistry 1993, 32, 10489–10496. [DOI] [PubMed] [Google Scholar]
- a Mehl R. A.; Anderson C.; Santoro S. W.; Wand L.; Martin A. B.; King D. S.; Horn D. M.; Schultz P. G. J. Am. Chem. Soc. 2003, 125, 935. [DOI] [PubMed] [Google Scholar]; b Seo Y. J.; Matsuda S.; Romesberg F. E. J. Am. Chem. Soc. 2009, 131, 5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann I. U.; Rovner A. J.; Aerni H. R.; Rogulina S.; Cheng L.; Olds W.; Fischer J. T.; Söll D.; Isaacs F. J.; Rinehart J. FEBS Lett. 2012, 586, 3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai H. W. Anal. Bioanal. Chem. 2012, 403, 2089. [DOI] [PubMed] [Google Scholar]
- Kim S.-H.; Bartholomew D. G.; Allen L. B. J. Med. Chem. 1978, 21, 883. [DOI] [PubMed] [Google Scholar]
- Lu J.; Li N.-S.; Koo S. C.; Piccirilli J. A. J. Org. Chem. 2009, 74, 8021. [DOI] [PubMed] [Google Scholar]
- Hutter D.; Benner S. A. J. Org. Chem. 2003, 68, 9839. [DOI] [PubMed] [Google Scholar]
- Kim H. J.; Chen F.; Benner S. A. J. Org. Chem. 2012, 77, 3664. [DOI] [PubMed] [Google Scholar]
- Piccirilli J. A.; Krauch T.; Macpherson L. J.; Benner S. A. Helv. Chim. Acta 1991, 74, 397. [Google Scholar]
- Hanessian S.; Machaalani R. Tetrahedron Lett. 2003, 44, 8321. [Google Scholar]
- Alonso D.; Caballero E.; Medarde M.; Tome F. Tetrahedron Lett. 2007, 48, 907. [Google Scholar]
- Paquette L. A.; Zhao M. J. Am. Chem. Soc. 1998, 120, 5203. [Google Scholar]
- Falck J. R.; Yadagiri P. J. Org. Chem. 1989, 54, 5851. [Google Scholar]
- Ludwig J.; Eckstein F. J. Org. Chem. 1989, 54, 631. [Google Scholar]
- Cohn W. E. J. Biol. Chem. 1960, 235, 1488. [PubMed] [Google Scholar]
- Chambers R. W.; Kurkov V.; Shapiro R. Biochemistry 1963, 2, 1192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.