

Abstract
Anisotropic colloidal hybrid nanoparticles exhibit superior optical and physical properties compared to their counterparts with regular architectures. We herein developed a controlled, stepwise strategy to build novel, anisotropic, branched, gold nanoarchitectures (Au-tripods) with predetermined composition and morphology for bioimaging. The resultant Au-tripods with size less than 20 nm showed great promise as contrast agents for in vivo photoacoustic imaging (PAI). We further identified Au-tripods with two possible configurations as high-absorbance nanomaterials from various gold multipods using a numerical simulation analysis. The PAI signals were linearly correlated with their concentrations after subcutaneous injection. The in vivo biodistribution of Au-tripods favorable for molecular imaging was confirmed using small animal positron emission tomography (PET). Intravenous administration of cyclic Arg-Gly-Asp-d-Phe-Cys (RGDfC) peptide conjugated Au-tripods (RGD-Au-tripods) to U87MG tumor-bearing mice showed PAI contrasts in tumors almost 3-fold higher than for the blocking group. PAI results correlated well with the corresponding PET images. Quantitative biodistribution data revealed that 7.9% ID/g of RGD-Au-tripods had accumulated in the U87MG tumor after 24 h post-injection. A pilot mouse toxicology study confirmed that no evidence of significant acute or systemic toxicity was observed in histopathological examination. Our study suggests that Au-tripods can be reliably synthesized through stringently controlled chemical synthesis and could serve as a new generation of platform with high selectivity and sensitivity for multimodality molecular imaging.
Introduction
Current biomedical nanotechnology has been aimed at effectively imaging and characterizing abnormal biological processes at the cellular or even molecular level in living subjects.1−5 Successful progresses in the fundamental molecular imaging research including instrumentations and image construction/registration techniques greatly motivate the innovations of imaging probes. As a relatively new molecular imaging technique, photoacoustic imaging (PAI) has recently attracted significant research interests.6 PAI takes advantage of individual strengths of both optical and acoustic imaging while largely overcoming the weaknesses associated with each modality, providing functional and molecular information of abnormalities with deep tissue penetration, high sensitivity, and excellent spatial resolution.7 In addition to several endogenous contrasts (such as melanin and hemoglobin),8 various light-absorbing nanoparticles (including Au-based nanostructures, carbon nanotubes, and nanodroplets) have been developed for PAI contrast enhancement.9−25 Although there are some nanoparticle-based exogenous agents particularly promising for PAI,20,22 their potential toxicity is still under investigation, and the improvement of their in vivo behavior remains a challenge since most attempts to enhance the contrast effect compromise their pharmacokinetic profile.
As one class of strong optical-absorption nanomaterials, colloidal metallic nanostructures (NPs) have recently attracted significant attention from diverse disciplines for many biomedical applications.26−28 Many synthetic methods have been developed for the construction of a variety of plasmonic nanostructures with controllable size and uniform shape (for instance, triangles, prisms, rods, cubes, shells, stars, and cages),9−25,29 largely because the effect of induced-shape anisotropy of these nanostructures results in the splitting of the underlying surface plasmon enhancement (SPR) into several shape-dependent modes, which could be accordingly tuned within the near-infrared window (NIR, 650–950 nm).30−36 As representative anisotropic nanogeometries, Au-nanorods and -nanocages have shown great promise as PAI contrasts; however, they generally have relatively large particle sizes (∼50 nm), which could result in unfavorable in vivo behavior and severely limit their application for targeted cancer imaging.7,11,12,21,37 By making a compromise by trading contrast effects for the ability to have preferable in vivo behaviors, strenuous efforts have been made recently to improve their targeting efficiency, and to control pharmacokinetics and biodistributions through variations of their sizes, shapes, and surface properties. Although some metallic nanostructures with complex shapes showed desired tumor targeting efficiency without compromising optical properties, great challenges still remain in the precise control of their geometries and monodispersity.28−32
Recently, many colloidal hybrid nanostructures with sophisticated architectures have been constructed from multiple functional components either assembled using linkers or fused together by solid-state interfaces.38 In order to build novel hybrid nanostructures with optimal structure and morphology for SPR, we herein developed a synthetic strategy to construct a series of anisotropic gold-based nanomultipods, including dipods, tripods, and tetrapods (Figure 1), with improved yield and excellent quality in a predictable, controlled, and stepwise manner. Among them, the gold tripods (Au tripods) have relatively small sizes with a narrow size distribution and display stringently controlled morphology and, more importantly, have well-defined absorptions in visible and NIR regions. As a proof of concept, we further conjugated αvβ3 integrins targeting the peptide, cyclic c(RGDfC) peptide,39 to PEGylated Au-tripods (RGD-Au-tripods) and used this novel nanoprobe as a PAI contrast agent to image tumor angiogenesis. The tumor targeting efficacy and in vivo profile of PEGylated Au-tripods (modified with different sizes of PEGs and/or RGD) labeled with radionuclide 64Cu were evaluated in a subcutaneous αvβ3-positive U87MG glioblastoma xenograft model using small animal positron emission tomography (PET). Finally, PAI was performed to investigate the targeting and imaging performance of RGD-Au-tripods as photoacoustic contrast agents.
Figure 1.

Construction of gold multipods (including Au–Pt dumbbell NPs, Au-dipods, Au-tripods and Au-tetrapods). (a) Schematic showing the stepwise syntheses of various gold multipods via a set of known nucleation reactions and epitaxial growth processes. Various gold multipods are modeled by Lumerical FDTD Solution (Lumerical Solution Inc.). Considering the regioselectivity, several possible regioisomers are shown in the bottom-left panel. (b) TEM images of the resultant gold multipods at different magnifications. HRTEM images of representative gold multipods are shown in the bottom-right panels.
Results
Construction and Characterization of Au-Tripods
By sequentially applying a set of nucleation reactions and epitaxial growth processes, we successfully built up four gold-based nanostructures, comprising Au–Pt dumbbell, dipods, tripods, and tetrapods, with predetermined composition and morphology (Figure 1a). In this structure-guided approach, we chose colloidal platinum (Pt) NPs as core seeds (or starting materials) because they are highly monodisperse and their sizes can be facilely tuned from 3 to 7 nm,40,41 and more importantly, they have definite and rigid cubic shapes (Supporting Information [SI], Figure S4). In order to tune the sizes of Pt nanocrystals, we applied stepwise seed-mediated growth processes to obtain monodisperse Pt nanocubes (SI section C.1.1. and Figure S4). As-synthesized 4.9 nm Pt NPs with truncated cubic shapes could preferably grew into thermodynamically more stable morphologies (cubic 6.5 nm Pt NPs) after an extra seed-mediated growth process. Similarly, 7.5 nm Pt NPs were obtained when 5.8 nm Pt NPs were used as seeds. All Pt nanocubes have a very narrow size and shape distribution (Figure S5 and Table S2). Due to excellent lattice coherence between Pt and Au crystals in the fcc phase, the epitaxial growth of gold preferentially occurs at the vertices of cubic Pt seeds (SI section C.1.2), resulting in the formation of various Au heterostructures (Figure 1). Typically, the epitaxial growth of Au on 4.9 nm Pt seeds produced dumbbell-like Au–Pt NPs. Use of 5.8 nm Pt NPs as seeds resulted in Au-dipods. There are two geometrical isomers; one is linear and the other is bent, corresponding to para and meta configurations, respectively. Interestingly, we found that, unlike 7.5 nm Pt NPs as seeds resulting in a mixture of tripods and tetrapods, 6.5 nm Pt seeds exclusively led to the formation of tripods with a narrow distribution of size and shape (Figure 1b). The statistical analysis suggested that a majority of resultant NPs were Au-tripods (SI Figure S7–S9). To the best of our knowledge, it is the first time for constructing novel Au tripods with high yield and improved quality by sequentially applying a set of known nucleation reactions and epitaxial growth processes.
There are two geometrical isomers identified in the resultant tripods; namely one with C3v symmetry is called as tripod-A and the other with C2v symmetry is tripod-T (Figure 1a). The steric accessibility determines the spatial distribution of Au crystals on the Pt nanocubes (SI Scheme S2 and S3). There are two meta positions favorable for the nucleation of incoming Au crystals when an intermediate has a bent shape, thereby resulting in the formation of tripod-A; there are six nucleation positions equal to the incoming Au crystals in the case of a linear intermediate, producing the other type of tripods (tripod-T). The statistical analysis results showed that an amount of tripod-T was finally obtained slightly higher than that of tripod-A (60% vs 40% in the resultant tripods, n = 300, Figure 1a). The sizes and shapes of various Au-multipods and each component within NPs were determined by dynamic light scattering (DLS) and TEM (SI Table S3 and Figure S9). The Au-tripods showed the lowest circularity among the branched NPs, largely due to their anisotropic nanostructures and surface roughness.
The representative TEM and scanning transmission electron microscope (STEM) images (Figure 2b–e) of Au-tripods showed that well-crystallized interfaces were formed between the Pt seeds and grown Au branches. The lattice fringes between Pt seeds and Au branches shown in SI Figure S11 are 0.23 nm and related to (111) planes of either Pt or Au in the fcc phase [2.260 Å for (111) plane of Pt, 2.355 Å for (111) plane of Au]. STEM images clearly showed that each Au branch epitaxially grew at one of the vertices of cubic Pt seeds. Two types of tripods, tripod-T and tripod-A, were definitively identified in the STEM images (Figure 2d–e). The forward and inverse fast Fourier transforms (FFT) were further applied to their HR-TEM images to distinguish Au branches and Pt core. The right panel of Figure 2 shows the HR-TEM images of three typical tripods (the first two belong to the type ‘tripod-T’; the third one is ‘tripod-A’). The inset in diffractogram patterns shows the splits of the (222) and (331) peaks into two spots: one (blue) for Pt and the other (black) for Au crystal, because of slightly different planar distances of the Pt and Au crystals. Inverse FFT reconstructions of the Pt (bottom) or Au (top) NPs using only their own reflections, [222], [331], [220], and [111], provided the real-spatial distributions of Pt and Au in tripods, respectively. Therefore, the lattice images coupled with the forward and inverse fast Fourier transform further confirmed the spatial configuration of Au branches and Pt core within Au-tripods.
Figure 2.

HRTEM and STEM images of Au-tripods, and their Fourier transform and inverse Fourier transform analyses. (a) TEM image of typical Au-tripods. There are two types of tripods: tripod-A (b) and tripod-T (c). (d,e) STEM images of two types of tripods. (f–h) HRTEM images of typical tripod-T and its fast Fourier transform (FFT) and inverse fast Fourier transform (inverse FFT) analyses. The insets in (g) show the splits of the (222) and (331) peaks into two spots: one for Pt and the other for Au crystal. Inverse FFT reconstructions of the Pt (bottom) or Au (top) NPs using only the superlattice reflections, [222], [331], [220], and [111], are shown in (h). (i–k) HRTEM, FFT, and inverse FFT reconstruction of the other tripod-T with different orientation. Inverse FFT reconstruction of the Pt (bottom) or Au (top) NPs using the superlattice reflections [222], [331], [220], and [111]. (l–n) HRTEM, FFT, and inverse FFT reconstruction of tripod-A. The split of the (222) peaks is attributed to the difference between Pt and Au crystals and is shown in the inset of (m). Inverse FFT reconstructions of the Pt (bottom) or Au (top) NPs using only the superlattice reflections [222], [220], and [111] are shown in (n).
Optical Properties of Au-Tripods and Simulation
The PAI requires contrast agents with relatively large absorption cross section. A conventional UV–vis–NIR spectrometer can measure the extinction spectra of nanostructures, which comprise two components: scattering and absorption, eventually providing the extinction cross section (σe, σe = σa + σs, where σa is absorption cross section and σs is scattering cross section). As seen in Figure 3a, the Au-tripods have a much stronger extinction peak in the NIR region, compared with the other Au-multipods. There are two plasmon resonances at the 540 and 700 nm peaks in the region of 400 to 1000 nm. The 540 nm feature corresponds to a quadrupole resonance out of the plane of the gold tripods, and the 700 nm feature is attributed to a dipole resonance in the plane of the Au-tripods.20
Figure 3.

Optical properties of Au-tripods, and measurement and simulation of optical absorption cross sections of tripods. (a) UV–vis extinction curves of various gold multipods (including Au–Pt dumbbell NPs, Au-dipods, Au-tripods, and Au-tetrapods) at the same sample weight (based on ICP-MS). (b) UV–vis extinction curves of gold nanospheres, cubic platinum NPs, and gold nanorods (54 nm length and 18 nm diameter, more information in the SI, sections C.2 and C.3). (c) The calculated absorption cross section of tripod-T as a function of ωx (the incident beam is polarized along the z-axis, and the tripod-T is rotated around the x-axis. ωx is the angle between the e-field and the long axis of tripod-T. (d) The calculated absorption cross section of tripod-A as a function of ωx (the incident beam is polarized along the z-axis, and the tripod-A is rotated around the x-axis. ωx is the angle between the e-field and the side of tripod-A. Polarization dependence of the average electric field intensity of tripod-T (e) and tripod-A (f). Electric field intensity contours in xz plane, xy plane, and yz plane at 700 nm were obtained from the FDTD calculations on both tripod-T and tripod-A. The long axis of tripod-T is parallel to the z-axis; one side of tripod-A is parallel to the z-axis. The excitation polarization relative to the z-axis is 0°. x and y represent the horizontal and vertical lengths of the calculated area.
The σa of Au-tripods was calculated using combined UV–vis–NIR spectrometer and photoacoustic measurements.42 While UV–vis–NIR spectrometer measures the total extinction coefficient, in photoacoustic imaging, the detected signal is directly proportional to the absorption coefficient (μa) of the NPs. Because the SPR peak of the tripods is tuned to 700 nm, methylene blue is suitable as a reference dye to obtain the calibration curve. On the basis of the linear relationship between μa and photoacoustic signal amplitude and the known σa of methylene blue (SI Figure S15), the photoacoustic signals from Au-tripods with different concentrations were converted into μa, and the σa of Au-tripods was calculated by dividing the μa by the corresponding concentration of the Au-tripods. In the calculation, the σa and σe of the tripods at 670 nm were 2.02 ± 0.03 × 10–16 and 2.06 ± 0.03 × 10–16 m–2, respectively (σa/σe = 0.98). As a result, on a per-weight basis, the Au-tripods are generally able to generate more contrast (33%) on PA images within the NIR region compared to gold rods (SI Table S4).
To gain more insight into the localized surface plasmon resonance (LSPR) spectra and to obtain better structural optimization, we performed numerical analysis on various gold multipods using a commercial finite difference time domain (FDTD) simulation package (Lumerical Solution Inc., Canada). The Au-multipods are modeled as a cubic Pt core with Au spheres (see detailed modeling in the SI sections B.3 and C.4). The corresponding geometrical parameters were obtained from the previous results in SI Tables S2 and S3. The contributions of absorption cross sections of two types of Au-tripods (tripod-T and tripod-A) as a function of the rotated angle around the x-axis were obtained using the FDTD simulation and are shown in c and d of Figure 3. The calculated absorption cross section maxima of tripod-T were at 530 and 710 nm, in good agreement with experimental data (a and c of Figure 3), while theoretical absorption maximum peaks of tripod-A occurred at 520 and 900 nm (very broad peak centered at 900 nm). Using the FDTD method, we have further studied the near-field optical properties of all gold multipods, gold rods, and nanospheres (SI section C.4, Figures S16–S21). The electric field intensity contours were calculated at the excitation wavelength of 700 nm as the excitation polarization was varied gradually from the longitudinal to transverse directions. Similar to gold rods, the Au-tripods have a strong polarization-dependent cross section. It was clear that under the resonance excitation, the maximum field enhancement regions were observed to rotate away from vertical tripod as the excitation polarization rotated around the y-axis (SI Figure S18). It is worth noting that the electric fields on the surface of Au-tripods (especially Au-tripod-T) are ∼2–3 orders of magnitude higher when compared to fields around spherical gold nanoparticles at the same weight in the NIR range. As seen in the Figure 3e,f and SI Figure S18, the edges of tripods and the junctions between two Au–Au NPs on the Pt cores are locations of enhanced fields (also called hot spots), due to the occurrence of coupled plasmons.
Surface Modification of Au-Tripods
In order to stabilize the Au-tripods in the aqueous solution and provide the capability for subsequent surface modification, we developed a facile, versatile PEGylation strategy which can significantly increase in vivo circulation time of resultant NPs and reduce their reticuloendothelial system (RES) accumulation versus uncoated counterparts. The PEGylation of Au-tripods involved the formation of gold–thiolate bonds at the gold–sulfur interface and the self-assembly of a monolayer on gold surface (SI Scheme S1).43 The bidentate thiol-terminated polyethylene glycol (PEG) chains facilitated subsequent immobilization of various biological molecules via bioconjugation chemistry. The c(RGDfC) was efficiently and site-specifically conjugated on the maleimide-terminated NPs in an oriented and homogeneous fashion. In order to track the RGD-Au-tripods in vivo by PET, the radioactive metal chelator, 2-(4-isothiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid (p-SCN-Bn-NOTA) was further conjugated to the surface of RGD-Au-tripods in a well-defined manner for 64Cu radiolabeling. The hydrodynamic size change and the corresponding change in ζ potentials of modified Au-tripods clearly confirmed the efficient conjugation of the c(RGDfC) peptide to the nanoparticles (SI Figure S25 and Table S5–S8). RGD-Au-tripods also showed excellent stability under physiological conditions. There were no significant size changes or aggregation in the presence of the mouse serum after 48 h at 37 °C (SI Figure S27). We also validated the targeting ability of RGD-Au-tripods to αvβ3-positive U87MG cells in vitro by determining their cellular uptake using TEM and inductively coupled plasma mass spectrometry (ICP-MS) analysis (SI Figures S28 and S29).
In order to obtain a better understanding of the in vivo behavior of Au-tripods and study their potential toxicity, we carried out a pilot preclinical animal toxicity study to assess the potential toxicity of tripods to pave the way for their clinical translation. Both hematology and serum biochemistry analyses, and histologic and microscopic examination revealed that no evidence of significant acute toxicity was observed and the tripods are likely highly biocompatible in small living subjects (SI Figures S35–S37).
Pharmacokinetics and Biodistribution of Au-Tripods
The pharmacokinetics, biodistribution, and tumor-targeting ability of Au-tripods were investigated in small living animals. As described in the previous section, the water-soluble Au-tripods were coated with a layer of functional PEG chains. Quantification of grafted PEG density on PEGylated Au-tripods was performed using spectrophotometric analyses of free amines on the PEGylated Au-tripods (SI Table S6). Typically, the density of amine groups on Au-tripod (PEG 3400 coating) was 0.966 ± 0.07 number/nm2; the ratio of amine groups to a single tripod was 757 ± 56. The chelating agent NOTA was attached to the terminal of PEG chains for PET radionuclide 64Cu. Importantly, the 64Cu radiolabels on the tripods remained intact on the tripods even though they were incubated in the mouse serum over 24 h.
The U87MG tumor-bearing mice (n = 4) were tail-vein injected with 64Cu–Au-tripod, followed by small animal PET scans at different time points (1, 2, 4, 24, and 48 h). PET imaging results revealed nonspecific uptake of PEGylated tripods by the liver, spleen, and even kidney (Figure 4a and SI Figures S30–S32), but minimum accumulation in the muscle or other major organs. Although most of the tripods were eliminated through hepatic excretion, the kidney retention of radio-labeled tripods over time suggested that the renal excretion could be an additional clearance route for tripods in mice. Since the heart acted as a cardiac blood pool, the signals from the heart were used as indicators to calculate the blood circulation time. The PEGylated tripods showed slow systemic clearance because of a long blood circulation time (t1/2 = 2.29 h), making themselves more available for distribution to target tissues via the enhanced permeability and retention (EPR) effect.
Figure 4.

Small animal PET images and PET quantification of intravenous injected tripods with different surface functionalization in mice bearing the U87MG human glioblastroma tumor. (a–c) Targeting of integrin αvβ3-postitive U87MG tumor in mice by RGD-functionalized tripods. Decay-corrected whole-body coronal PET images of nude mice bearing human U87MG tumors at 1, 4, 24, and 48 h after injection of 3.7 MBq of 64Cu-RGD-Au-tripod, 64Cu-RGD-Au-tripod with a blocking dose of c(RGDfC) (21 μmol of c(RGDfC)/kg of mouse body weight), and 64Cu–Au-tripod (200 pmol/kg of mouse body weight, or 2 mg/kg of mouse body weight). (d,f) PET quantification of tumors and major organs after intravenous injection to mice bearing subcutaneous U87MG glioma xenografts (n = 4 per group, data represent means ± SD). (g–i), Comparison of tumor and major organ uptake of 64Cu-RGD-Au-tripod, 64Cu-RGD-Au-tripod with a blocking dose of c(RGDfC), and 64Cu–Au-tripod for a time period up to 48 h after intravenous injection to U87MG tumor-bearing mice (n = 4 per group). Data represent mean ± SD ** P < 0.01, *P < 0.05 (two-sided Student’s t-test).
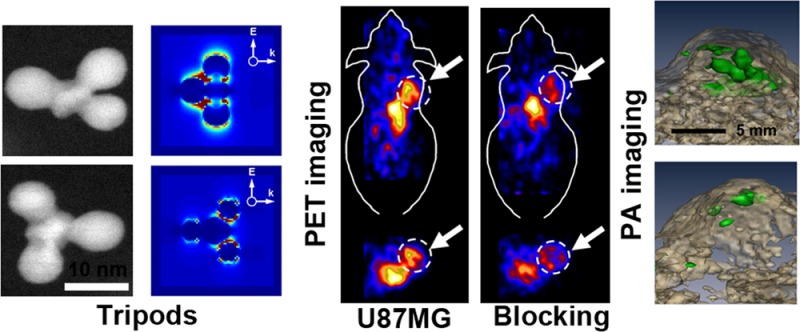
Targeting Characteristics of RGD-Au-Tripods
In order to target the integrin αvβ3 for imaging tumor angiogenesis and metastasis, the Au-tripods were conjugated with both p-SCN-Bn-NOTA for 64Cu labeling and c(RGDfC) for tumor targeting. The numbers of NOTA on the RGD-tripod were determined by an isotope dilution assay and typically equaled 54.5 ± 3.3.44 The conjugated RGD was actually determined by measuring the differences in concentration of RGD using HPLC before and after addition of Au-tripods during the coupling reaction. The ratio of RGD to NOTA on Au-tripods was 8.2 ± 0.5. The targeting ability and specificity of RGD-Au-tripods for the αvβ3 integrin were evaluated in the U87MG tumor-bearing mice (n = 4). The representative coronal and transverse PET images of a mouse acquired at 1, 4, 24, and 48 h after tail vein injection of 64Cu-RGD-Au-tripods, 64Cu–Au-tripods, or 64Cu-RGD-Au-tripods coinjected with a blocking dose of c(RGDfC) are shown in Figure 4. Quantitative analysis showed that tumor uptake of 64Cu-RGD-Au-tripods gradually accumulated in the tumor between 1 and 24 h, reaching a plateau at about 24 h post-injection, and then leveled off in the next 24 h. The 64Cu-RGD-Au-tripods exhibited a significantly high tumor uptake of ∼7.9% ID/g after 24 h post-injection, which was more than 3 times higher than that of plain 64Cu–Au-tripods (2.6% ID/g). Such high tumor accumulation was attributed to the tumor specific binding affinity of RGD functionalization on the tripods and their long blood circulation time. In the control group, the tumor uptake was significantly blocked when a blocking dose of c(RGDfC) was injected into tumor-bearing mice along with 64Cu-RGD-Au-tripods. The reduced tumor uptake was observed (3.8% ID/g after 24 h post-injection), which is significantly lower than that of unblocked one (P < 0.05). Interestingly, compared to plain tripods, 64Cu-RGD-Au-tripods displayed much longer blood circulation time (t1/2 = 10.3 h for 64Cu-RGD-Au-tripods vs t1/2 = 6.4 h for 64Cu–Au-tripods) and less RES uptake. 64Cu-RGD-Au-tripods were also found to be excreted by both hepatobiliary and renal routes, as indicated by the deposition of radioactivity in the kidney and the gastrointestinal tract (b and e of Figure 4), which were also confirmed by the tissue histology following in vivo imaging (SI Figure S40).
To confirm the photoacoustic detection of tripods in tumor cells, we incubated U87MG cells with RGD-Au-tripods for 2 h under standard conditions. The cells with internalized RGD-Au-tripods after incubation were washed and placed in agarose phantom at increasing cell concentrations from 11.5 × 103 to 368 × 103 cells per well (n = 3 per group). Quantitative analysis of the photoacoustic signals from the phantom revealed that the minimum detectable number of tumor cells exposed to RGD-Au-tripods could be as low as 11.5 × 103 (a and b of Figure 5, and Figure S17 of SI). We observed a linear correlation between the number of RGD-Au-tripod-loaded cells and the corresponding photoacoustic signal. The photoacoustic signal from the phantom when excited at 700 nm provided the best sensitivity to detect tumor cells. Even at 900 nm, the signal was still detectable for 50 × 103 of tumor cells.
Figure 5.

High sensitivity of Au-tripods for photoacoustic molecular imaging. (a) The top view of three-dimensional (3D) volume rendering of photoacoustic images of an agarose phantom containing decreasing number of U87MG cancer cells exposed to RGD-Au-tripod at different wavelengths (670, 700, 725, 750, 800, 850, 900 nm). The inhomogeneous signal inside wells is due to possible aggregation of cells. (b) Quantitative analysis of the photoacoustic signal (relative to the background signal) from the phantom (n = 3). (c) RGD-Au-tripod ranging in concentrations from 390 pM to 12.5 nM were injected subcutaneously into the flank of living mice (n = 3) and scanned with photoacoustic instrument. (d,e) Picomolar photoacoustic detection of tripods in living mice. The coronal view (d) of 3D volume rendering of photoacoustic images of subcutaneous inclusions. The skin is visualized in the ultrasound image (gray-scale images), which is overlaid with photoacoustic images (green-scale images). (f,g) Three-dimensional volume rendering of photoacoustic images (green) and ultrasound images (brown) of subcutaneous inclusion. a.u. = arbitrary units. (h) Photoacoustic signals recorded in vivo increased linearly with the tripod concentration (R2 = 0.96, n = 3 mice, data represent mean ± SD). The background level represents the endogenous signal measured from tissues. (i) Quantitative analysis of the photoacoustic signal (relative to the background signal) (mice n = 3).
To test the tripod’s PAI sensitivity in living mice, we subcutaneously injected the right hind side of a mouse with 30 μL of RGD-Au-tripods mixed with Matrigel (n = 3) at increasing concentrations of 0.39, 0.78, 1.56, 3.125, 6.25, and 12.5 nM. After the incision was solidified in place, the mouse was placed on its left side (left lateral recumbent) and partially embedded in the agarose gel covered with a water bath, and was then scanned under the photoacoustic system. While the ultrasound signals were used to reconstruct the mouse anatomy, including skin and inclusion edges, the photoacoustic signals showed the tripods contrast in the mice (Figure 5c–h). Quantitative analysis of the photoacoustic signals from each inclusion using a 3D region of interest drawn over the inclusion revealed a linear correlation between the tripod concentration and the corresponding photoacoustic signal. Compared to the tissue background, about 200 pM of RGD-Au-tripods extrapolated from the signal-concentration curve provided the equivalent photoacoustic signal as the tissue background.
Similar to PET imaging, the PAI imaging ability of RGD-Au-tripods to αvβ3 integrin-positive tumor was evaluated in the U87MG tumor-bearing mice (n = 3) (see Figure 6). Before the injection, the photoacoustic and ultrasound images of the mice were taken. Photoacoustic images with lateral step size of 0.25 mm were acquired at 700 nm wavelength. Following the photoacoustic scan, an ultrasound image of the entire tumor area was acquired. The U87MG tumor-bearing mice were then injected with 100–200 μL of RGD-Au-tripods (200 pmol per kg mouse body weight) in PBS through the tail vein. For the receptor-blocking experiment, mice were coinjected with 21 μmol of c(RGDfC)/kg of mouse body weight and 100–200 μL of RGD-Au-tripods (200 pmol per kg mouse body weight) in PBS through the tail vein. After injection, photoacoustic images were acquired at 700 nm at 0.5, 1, 2, and 4 h post-injection. Quantitative analysis of the photoacoustic signal from the tumor was done by drawing three-dimensional (3D) regions-of-interest (ROIs) around tumors on the basis of the ultrasound images. Since there was the background blood signal in each photoacoustic scan, a subtraction image calculated at the 2-h post-injection image minus the preinjection image is shown in SI Figure S39 and was used to subtract out the background hemoglobin signal and examine the differences between groups. Mice injected with RGD-Au-tripods showed significantly higher photoacoustic signal in the tumor compared with the blocking group coinjected with RGD after 2 h post-injection. Quantitative analysis showed that tumor uptake of RGD-Au-tripods gradually accumulated in the tumor between 1 and 4 h, reaching a plateau at about 2 h post-injection, and then leveled off in the next 2 h. The mice injected with RGD-Au-tripods at 2 h post-injection showed more than 3 times higher photoacoustic signal in the tumor than the mice coinjected with RGD and RGD-Au-tripods. The tumor tissue histology combined with silver staining further confirmed the specific targeting ability of RGD-Au-tripods toward U87MG tumors (SI Figure S40).
Figure 6.

Targeting of integrin αvβ3-postitive U87MG tumors in mice by RGD-Au-tripod. (a) The coronal, sagittal, and transverse views of 3D volume rendering of photoacoustic images and ultrasound images of nude mice bearing U87MG tumors were obtained before injection or at 1, 4, 24, and 48 h after intravenous injection of RGD-Au-tripod (200 pmol/kg of mouse body weight, or 2 mg/kg of mouse body weight). (b) The coronal, sagittal, and transverse views of 3D volume rendering of photoacoustic images and ultrasound images of nude mice bearing U87MG tumors were obtained before injection or at 1, 4, 24, and 48 h after coinjection of a blocking dose of c(RGDfC) (21 μmol of c(RGDfC)/kg of mouse body weight) and RGD-Au-tripod (200 pmol/kg of mouse body weight, or 2 mg/kg of mouse body weight). Subtraction images were calculated at the 2-h post-injection image minus the preinjection image (SI Figure S34). (c) Mice injected with RGD-Au-tripod showed significantly higher photoacoustic signal than mice with coinjection of a blocking dose of c(RGDfC) and the same amount of RGD-Au-tripod (p < 0.001, two-sided Student’s t-test). The error bars represent standard error (n = 3 per group). (d) The perspective views of 3D volume rendering of photoacoustic images (green) and ultrasound images (brown) of tumors.
Discussion
We showed the ability to engineer and manipulate constitutional nanocrystals at the nanometer-scale to build novel nanotripods in a predictable and controlled manner. The construction of Au-tripods involved a set of nucleation reactions and epitaxial growth processes, which are controlled by surface diffusion.45,46 As solid-state analogues of multifunctional organic molecules, Pt nanocubes could be used as building blocks to construct sophisticated hybrid architectures.38,41 Although Sun and co-workers provided an important insight in the formation of various branched NPs,41 the stochastic simulation in two dimensions inevitably overlooked certain configurations of constructed nanostructures. The selective growth of a heterogeneous nanocrystal phase onto certain regions of nanocrystal seeds depends on the surface potential and lattice matching.46 We found that regioselectivity of the heterogeneous nucleation on cubic Pt seeds plays an important role on the formation of nanotripods (SI Schemes S1 and S2). Considering the possible nucleation regions and steric effect, 6.5 nm cubic seeds exclusively resulted in the formation of uniform Au-tripods with high yield and improved quality by sequentially applying a set of known nucleation reactions and epitaxial growth processes.
The theoretical calculation coupled with experimental results on such anisotropic tripod nanostructures was used as a guide for the design and fabrication of PAI nanoprobes. We successfully identified tripods with two possible configurations as high absorbance nanomaterials from various gold multipods using a numerical simulation analysis. The enhancement of the electric field on the surface of Au-tripods is due to the occurrence of coupled plasmons at the edges of tripods and the junctions between gold nanocrystals on the Pt cubes. Optimizing the geometrical configurations of gold nanocrystals on the Pt cube could improve the plasmon resonances of Au-tripods. The measured absorption spectrum of Au-tripods was identified by the calculated FDTD spectrum. Due to an obvious increase in the cross section of Au-tripods on a per-weight basis compared to that of the gold rods, Au-tripods could generate more contrast on PA images within the NIR region. Among nanoparticle-based PA contrast agents, Au-tripods exhibit superior optical properties in the NIR region, and more importantly, they have exceptionally small sizes, which are distinctive advantages over traditional gold-based NPs or carbon nanotubes for in vivo molecular imaging.15,24,47
We have successfully validated novel Au-tripods as multimodality probes for in vivo molecular imaging (PAI and PET). The in vivo biodistribution of Au-tripods favorable for living subject imaging was confirmed by 64Cu radiolabeling and imaging their localization over time using PET. After intravenous administration, the Au-tripods accumulated in liver and spleen, suggesting that the hepatic excretion is a major route of elimination of Au-tripods. Interestingly, the kidney retention of Au-tripods over time revealed that the renal excretion could be an additional clearance route for Au-tripods. Considering the anisotropic shape of Au-tripods [the thickness of tripods is close to the renal clearance threshold (less than 7 nm)], it is possible for Au-tripods to be cleared to some extent through the renal system.48,49 The PAI signals were linearly correlated with their concentrations after subcutaneous injection. Intravenous administration of RGD-Au-tripods to U87MG tumor-bearing mice showed remarkably higher contrast in tumors than competitive injection controls even at subnanomolar concentrations. PAI results correlated well with the corresponding PET images. Quantitative biodistribution data revealed that 7.9% ID/g of RGD-Au-tripods accumulated in the U87MG tumor after 24 h post-injection, but an obvious decrease in RES uptake (low liver and spleen accumulation, Figure 4) of RGD-Au-tripods was observed during the imaging. It is clear that their tumor uptake was higher than most of traditional gold-based NPs for in vivo molecular imaging,24,47 probably due to their unique anisotropic shape and relatively small size. A pilot mouse toxicology study confirmed that no evidence of significant acute and systemic toxicity was observed in histopathological examination.
Conclusion
By sequentially applying a set of nucleation reactions and epitaxial growth processes, we herein developed a control and stepwise strategy to build novel anisotropic Au-tripods with predesigned shape, high yield, and excellent quality. These Au-tripods exhibit superior optical and physical properties compared to their counterparts with regular architectures. We showed the ability to tune their shape-dependent plasmon resonances to the NIR window without compromising their pharmacokinetic profile. Our strategy opens up novel ways to enable the creation of sophisticated nanostructures with predetermined optical and physical properties for specific biomedical applications. Moreover, our study suggests that highly selective and sensitive detection of cancer cells in a living subject is possible using molecular specific Au-tripods as PAI contrast agents. The functionalized RGD-tripods showed significantly enhanced photoacoustic contrast effect in both phantom and small animal imaging experiments. Functional and molecular information of the tumor with high spatial resolution was further obtained by PAI, which correlated well with the corresponding PET quantification. Due to their excellent biocompatibility and stability in a biological environment, ease of functionalization, passive and activated targeting capabilities and potential hepatic and renal clearance, the Au-tripods represent a new generation of a nanoplatform for biomedical research and personalized therapy.
Methods and Materials
Materials
The integrin αvβ3 targeting peptide cyclo (Arg-Gly-Asp-d-Phe-Cys) (cRGDfC) was purchased from Peptides International, Inc. (Louisville, KY). The p-SCN-Bn-NOTA was purchased from Macrocyclics, Inc. Hydrogen tetrachloroaurate(III) hydrate (HAuCl4) and platinum(II) acetylacetonate (Pt(acac)2) was ordered from Strem Chemicals, Inc. N-Hydroxysuccinimide (NHS), N-hydroxysulfosuccinimide (sulfo-NHS), succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (sulfo-SMCC), and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide, hydrochloride (EDC) were purchased from Thermo Fisher Scientific. All other chemicals were purchased from Sigma-Aldrich. Other solvents and chemicals were used as received. All buffers and media were purchased from Invitrogen Corp. Deionized water was obtained from a Millipore Milli-DI Water Purification system. The dialysis membrane tubing (MWCO: 12,000–14,000, and 100,000) were purchased from Spectrum laboratories.
Size Tuning of Cubic Pt NPs
The size of various cubic Pt NPs was successfully tuned by adjustment of the reaction conditions in a precise manner. Monodisperse Pt seeds were synthesized by thermal decomposition of platinum precursor [Pt(acetylacetonate)2, Pt(acac)2], in 1-octadecene solution containing a trace amount of iron pentacarbonyl [Fe(CO)5], which facilitates a fast nucleation and improves the homogeneous growth of platinum crystals.40 The shape and size of Pt NPs are dependent on the reaction temperature at which a trace amount of Fe(CO)5 was injected. Multiple stepwise seed-mediated growth processes were applied to obtain monodisperse Pt nanocubes. The detailed synthesis procedure is described in the SI Methods.
Synthesis and Surface Modification of Au-Tripods
The gold precursor (hydrogen tetrachloroaurate, HAuCl4, 100 mg, 0.29 mmol) was dissolved in 20 mL of 1-octadecene (ODE) containing 2 mL of oleylamine under nitrogen protection. After stirring at room temperature for 5 min, the solution was heated up to 80 °C. Twenty milligrams of 6.5 nm freshly synthesized Pt NPs (dispersed in 1 mL of hexane) was quickly injected into the above solution. The resultant mixture was then heated up to 110 °C and kept at this temperature for one hour before it was cooled down to room temperature. The solution finally turned to gray-purplish color, indicating the formation of gold branched nanostructures. The particles were precipitated out by adding 30 mL of isopropanol and collected by a centrifuge (3000 rcf, 5 min). The resultant particles were redispersed in 5 mL of hexane and then precipitated out by adding ethanol. This purification step was repeated twice to remove the extra surfactant and ODE. The final product (Au-tripods) was dispersed in 10 mL of hexane in the presence of 0.01 mL of oleylamine for further use.
Surface PEGylation of Au-tripods is described in detail in the SI sections A.4, B.2, C.5, and C.6. Briefly, the Au-tripods (10 mg) were suspended in 3 mL of a chloroform solution of bidentate thiol-termined PEG chains (O-(3-aminopropyl)polyethylene glycol lipoate amide, LP-PEG-3400-NH2, 0.02 mmol, SI section A.3). After stirring at room temperature for 24 h, PEGylated Au-tripods were precipitated by adding 20 mL of hexane, collected by a brief centrifugation, and dried under the nitrogen gas flow. PEGylated Au-tripods were then dispersed in water, and the unbound PEG and any other excess reagents were removed by dialysis against water or phosphate-buffered saline (PBS) (10 mM, pH 7.4) by dialysis tubing (Spectrum Spectra/Por dialysis membrane tubing, MWCO = 12 KDa). Any impurity or precipitate was removed by a 0.22 μm syringe filter. The final gold or platinum concentration of PEGylated Au-tripods was measured by inductively coupled plasma mass spectrometry (ICP-MS) analysis. Those NPs were further characterized with transmission electron microscopy (TEM), UV–vis absorption spectroscopy, and dynamic light scattering (DLS) (SI sections B.2, B.3, B.5, and sections C.4, C.6).
Conjugation of Au-tripods with cRGD (RGD-Au-tripods) is described in detail in the SI Sections A.5, C.6, and C.7. Typically, The cross-linker solution, the water-soluble Au-tripods (100 nM, 0.5 mL, 5 × 10–11 mol, see the determination of concentrations of tripods and rods in SI section C.3) in 10 mM PBS (pH = 7.2) were incubated with the cross-linker solution (sulfo-SMCC [0.5 mg, 1.5 μmol], was freshly prepared in 15 μL of DMSO) for 2 h at room temperature. After removal of excessive sulfo-SMCC and byproducts using a PD-10 column (GE Healthcare, Piscataway, NJ), the resultant thiol-active Au-tripods were concentrated to the final volume of 0.5 mL with a centrifugal-filter (Amicon centrifugal filter device, MWCO = 30 kDa) and were incubated with the cRGDfC stock solution (50 μL of 5 mM in the degassed water, 0.25 μmol, the final RGD concentration in the mixture was 0.5 mM) with stirring. The conjugation reaction proceeded for 24 h at 4 °C. After the uncoupled RGD and byproducts were removed through PD-10 column, the resultant product, RGD-Au-tripods, was concentrated by a centrifugal-filter (Amicon centrifugal filter device, MWCO = 30 KDa) and stored at 4 °C for one month without losing targeting activity. The final RGD-Au-tripods was reconstituted in PBS and filtered through a 0.22 μm filter for cell and animal experiments. The gold and platinum concentrations of RGD-Au-tripods were measured by ICP-MS analysis.
The process to conjugate the tripods with both RGD and NOTA was similar to the conjugation of tripods and RGD, except that sulfo-SMCC solution was mixed with NOTA aqueous solution in the first step before being added into the tripod solution. Briefly, The sulfo-SMCC (0.5 mg, 1.5 μmol) was dissolved in 15 μL of DMSO and mixed with 4.5 μL of p-SCN-Bn-NOTA solution in the water (33 mM, 0.15 μmol). The ratio of SMCC to NOTA was optimized according to the specific activity of radioactive tracer-labeled NPs (SI section C.7).
Small Animal PET Imaging
PET imaging was carried out on a microPET R4 rodent model scanner (manufactured by CTI Concorde Microsystems, Knoxville, TN) as previously reported.50−52 PET scans were performed at 1, 2, 4, 24, and 48 h post-injection (p.i.). U87MG tumor-bearing mice were divided into several injection groups (4 mice per each group) to evaluate differences in specific targeting and biodistribution of Au-tripods and RGD modified tripods (SI section A.5 and C.7). For the tripod groups, each mice was injected with 3.7 MBq of 64Cu-RGD-Au-tripod (200 pmol/kg of mouse body weight, or 2 mg/kg of mouse body weight, normally 5 pmol per each mouse) in 100–200 μL PBS via the tail vein. For the receptor-blocking experiment, U87MG tumor-bearing mice were coinjected with 12 mg of c(RGDfC)/kg of mouse body weight and 100–200 μL of 64Cu-RGD-Au tripod (200 pmol per kg mouse body weight) in PBS via the tail vein. Prior to imaging, mice were anesthetized with isoflurane (5% for induction and 2% for maintenance in house oxygen at 2 L/min). Mice (4 mice per group) were placed in the prone position and near the center of the field of view (FOV) of the scanner. The 5-min static scans were obtained at the predetermined time points after post-injection. All the PET images were then reconstructed by a two-dimensional ordered-subset expectation maximization (OSEM) algorithm with a spatial resolution of 1.66–1.85 mm.53 PET quantifications were analyzed using both AsiPro and Amide image processing software. PET quantification details are included in the SI.
Au-Tripod-RGD Tumor Targeting in Living Mice
The custom-built photoacoustic instrument was described previously and is shown in SI Figure S2.54−56 The photoacoustic characterizations of Au-tripods were further determined by NEXUS 128 (Endra, MI). Two groups of female nude mice (n = 3 in each group, 6–8 weeks old) were inoculated subcutaneously at their right hind side with 5 × 106 U87MG cells in 50 μL of PBS. The tumors were allowed to grow to a volume of 150–200 mm3. The mouse was placed on its left side (left lateral recumbent) and partially embedded in the agarose gel covered with a water bath (SI Figure S2), and was then scanned under the photoacoustic system. Before the injection, the photoacoustic and ultrasound images of the mice were taken. A photoacoustic image with lateral step size of 0.25 mm was acquired using the 5 MHz transducer at 670, 700, and 725 nm wavelength. Following the photoacoustic scan, an ultrasound image was acquired using the 25 MHz transducer. The U87MG tumor-bearing mouse was then injected with 100–200 μL of RGD-Au-tripods (200 pmol per kg mouse body weight) in PBS through the tail vein using a butterfly catheter to avoid any position change during the injection (50 μL of dead volume) (SI Figure S2B). For the receptor-blocking experiment, mice were coinjected with 21 μmol of c(RGDfC)/kg of mouse body weight and 100–200 μL of RGD-Au-tripods (200 pmol per kg mouse body weight) in PBS through the tail vein. After injection, photoacoustic images (20 mm × 20 mm) were acquired at 670, 700, and 725 nm with step size of 0.25 mm and at 0.5, 1, 2, and 4 h post-injection. Following the photoacoustic scan, the ultrasound images were acquired to confirm the scan area. The photoacoustic and ultrasound images were analyzed, coregistered, and displayed using AMIDE software. Quantitative analysis of the photoacoustic signal from the tumor was done by drawing three-dimensional ROIs around the tumor on the basis of the ultrasound images. After 4 h post-injection, the mice were sacrificed, and tumors and tissues of interest were collected for TEM (SI section B.1), elemental analysis (SI section B.5), and histology study (SI section B.6).
Acknowledgments
This work was partially supported by the Office of Science (BER), U.S. Department of Energy (DE-SC0008397), NIH In vivo Cellular Molecular Imaging Center (ICMIC) grant P50 CA114747, NIH 5R01 HL093172, and NCI Cancer Center Nanotechnology Excellence Grant (CCNE-TR) U54 CA119367, CA151459. The authors thank Dr. Richard Luong and Dr. Donna M. Bouley for necropsies and hematological analysis.
Supporting Information Available
Detailed methodology, characterization, simulation, additional figures, and discussion. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Dr. Sam Gambhir serves on the board of Endra Inc. and serves as a consultant for Visualsonics Inc. Both of these companies develop photoacoustic small animal imaging equipment.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Rudin M.; Weissleder R. Nat. Rev. Drug Discovery 2003, 2, 123. [DOI] [PubMed] [Google Scholar]
- Ferrari M. Nat. Rev. Cancer 2005, 5, 161. [DOI] [PubMed] [Google Scholar]
- Weissleder R. Science 2006, 312, 1168. [DOI] [PubMed] [Google Scholar]
- Willmann J. K.; van Bruggen N.; Dinkelborg L. M.; Gambhir S. S. Nat. Rev. Drug Discovery 2008, 7, 591. [DOI] [PubMed] [Google Scholar]
- James M. L.; Gambhir S. S. Physiol. Rev. 2012, 92, 897. [DOI] [PubMed] [Google Scholar]
- Wang L. V.; Hu S. Science 2012, 335, 1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.; Favazza C.; Wang L. V. Chem. Rev. 2010, 110, 2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C.; Cheng K.; Chen K.; Hu X.; Liu Y.; Lan X.; Zhang Y.; Liu H.; Xu Y.; Bu L.; Su X.; Zhu X.; Meng S.; Cheng Z. Sci. Rep. 2013, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.; Cho E. C.; Chen J.; Song K. H.; Au L.; Favazza C.; Zhang Q.; Cobley C. M.; Gao F.; Xia Y.; Wang L. V. ACS Nano 2010, 4, 4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal A.; Huang S. W.; O’Donnell M.; Day K. C.; Day M.; Kotov N.; Ashkenazi S. J. Appl. Phys. 2007, 102, 064701. [Google Scholar]
- Mallidi S.; Larson T.; Tam J.; Joshi P. P.; Karpiouk A.; Sokolov K.; Emelianov S. Nano Lett. 2009, 9, 2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K. H.; Kim C.; Cobley C. M.; Xia Y.; Wang L. V. Nano Lett. 2008, 9, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Zerda A.; Zavaleta C.; Keren S.; Vaithilingam S.; Bodapati S.; Liu Z.; Levi J.; Smith B. R.; Ma T. J.; Oralkan O.; Cheng Z.; Chen X. Y.; Dai H. J.; Khuri-Yakub B. T.; Gambhir S. S. Nat. Nanotechnol. 2008, 3, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanzha E. I.; Shashkov E. V.; Kelly T.; Kim J. W.; Yang L. L.; Zharov V. P. Nat. Nanotechnol. 2009, 4, 855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. W.; Galanzha E. I.; Shashkov E. V.; Moon H. M.; Zharov V. P. Nat. Nanotechnol. 2009, 4, 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y. D.; Jia C. X.; Huang S. W.; O’Donnell M.; Gao X. H. Nat. Commun. 2010, 1, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehl C. L.; Hafner J. H. J. Mater. Chem. 2008, 18, 2415. [Google Scholar]
- de la Zerda A.; Liu Z. A.; Bodapati S.; Teed R.; Vaithilingam S.; Khuri-Yakub B. T.; Chen X. Y.; Dai H. J.; Gambhir S. S. Nano Lett. 2010, 10, 2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard L. S.; Anwar M. S.; Liu G. L.; Hann B.; Xie Z. H.; Gray J. W.; Wang X. D.; Pines A.; Chen F. F. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wustholz K. L.; Henry A. I.; McMahon J. M.; Freeman R. G.; Valley N.; Piotti M. E.; Natan M. J.; Schatz G. C.; Van Duyne R. P. J. Am. Chem. Soc. 2010, 132, 10903. [DOI] [PubMed] [Google Scholar]
- Yang X. M.; Stein E. W.; Ashkenazi S.; Wang L. H. V. Wires Nanomed. Nanobi. 2009, 1, 360. [DOI] [PubMed] [Google Scholar]
- Wilson K.; Homan K.; Emelianov S. Nat. Commun. 2012, 3, 618. [DOI] [PubMed] [Google Scholar]
- Schipper M. L.; Nakayama-Ratchford N.; Davis C. R.; Kam N. W. S.; Chu P.; Liu Z.; Sun X.; Dai H.; Gambhir S. S. Nat. Nano 2008, 3, 216. [DOI] [PubMed] [Google Scholar]
- Lu W.; Melancon M. P.; Xiong C.; Huang Q.; Elliott A.; Song S.; Zhang R.; Flores L. G. II; Gelovani J. G.; Wang L. V.; Ku G.; Stafford R. J.; Li C. Cancer Res. 2011, 71, 6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Broek B.; Devoogdt N.; D’Hollander A.; Gijs H.-L.; Jans K.; Lagae L.; Muyldermans S.; Maes G.; Borghs G. ACS Nano 2011, 5, 4319. [DOI] [PubMed] [Google Scholar]
- Lal S.; Link S.; Halas N. J. Nat. Photonics 2007, 1, 641. [Google Scholar]
- Halas N. J.; Lal S.; Chang W.-S.; Link S.; Nordlander P. Chem. Rev. 2011, 111, 3913. [DOI] [PubMed] [Google Scholar]
- Giannini V.; Fernández-Domínguez A. I.; Heck S. C.; Maier S. A. Chem. Rev. 2011, 111, 3888. [DOI] [PubMed] [Google Scholar]
- Mallidi S.; Larson T.; Tam J.; Joshi P. P.; Karpiouk A.; Sokolov K.; Emelianov S. Nano Lett. 2009, 9, 2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakr O. M.; Wunsch B. H.; Stellacci F. Chem. Mater. 2006, 18, 3297. [Google Scholar]
- Orendorff C. J.; Sau T. K.; Murphy C. J. Small 2006, 2, 636. [DOI] [PubMed] [Google Scholar]
- Nehl C. L.; Liao H. W.; Hafner J. H. Nano Lett. 2006, 6, 683. [DOI] [PubMed] [Google Scholar]
- Chen S. H.; Wang Z. L.; Ballato J.; Foulger S. H.; Carroll D. L. J. Am. Chem. Soc. 2003, 125, 16186. [DOI] [PubMed] [Google Scholar]
- Hao E.; Bailey R. C.; Schatz G. C.; Hupp J. T.; Li S. Y. Nano Lett. 2004, 4, 327. [Google Scholar]
- Kothapalli S.; Te-Jen M.; Vaithilingam S.; Oralkan O.; Khuri-Yakub B. T.; Sam Gambhir S. IEEE Trans. Biomed. Eng. 2012, 59, 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You E.-A.; Zhou W.; Suh J. Y.; Huntington M. D.; Odom T. W. ACS Nano 2012, 6, 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon G. D.; Choi S.-W.; Cai X.; Li W.; Cho E. C.; Jeong U.; Wang L. V.; Xia Y. J. Am. Chem. Soc. 2011, 133, 4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M. R.; Bondi J. F.; Schaak R. E. Nat. Chem. 2012, 4, 37. [DOI] [PubMed] [Google Scholar]
- Smith B. R.; Cheng Z.; De A.; Koh A. L.; Sinclair R.; Gambhir S. S. Nano Lett. 2008, 8, 2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Daimon H.; Onodera T.; Koda T.; Sun S. Angew. Chem., Int. Ed. 2008, 47, 3588. [DOI] [PubMed] [Google Scholar]
- Wang C.; Tian W.; Ding Y.; Ma Y. Q.; Wang Z. L.; Markovic N. M.; Stamenkovic V. R.; Daimon H.; Sun S. J. Am. Chem. Soc. 2010, 132, 6524. [DOI] [PubMed] [Google Scholar]
- Cho E. C.; Kim C.; Zhou F.; Cobley C. M.; Song K. H.; Chen J. Y.; Li Z. Y.; Wang L. H. V.; Xia Y. N. J. Phys. Chem. C 2009, 113, 9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkinen H. Nat. Chem. 2012, 4, 443. [DOI] [PubMed] [Google Scholar]
- Yang M.; Cheng K.; Qi S. B.; Liu H. G.; Jiang Y. X.; Jiang H.; Li J. B.; Chen K.; Zhang H. M.; Cheng Z. Biomaterials 2013, 34, 2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokari T.; Sztrum C. G.; Salant A.; Rabani E.; Banin U. Nat. Mater. 2005, 4, 855. [Google Scholar]
- Costi R.; Saunders A. E.; Banin U. Angew. Chem., Int. Ed. 2010, 49, 4878. [DOI] [PubMed] [Google Scholar]
- Jokerst J. V.; Cole A. J.; Van de Sompel D.; Gambhir S. S. ACS Nano 2012, 6, 10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C. H. J.; Zuckerman J. E.; Webster P.; Davis M. E. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakor A. S.; Luong R.; Paulmurugan R.; Lin F. I.; Kempen P.; Zavaleta C.; Chu P.; Massoud T. F.; Sinclair R.; Gambhir S. S. Sci. Transl. Med. 2011, 3, 79ra33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Z.; Ren G.; Liu H.; Jiang L.; Cheng Z. J. Biomed. Opt. 2010, 15, 036007. [DOI] [PubMed] [Google Scholar]
- Cheng Z.; De Jesus O. P.; Kramer D. J.; De A.; Webster J. M.; Gheysens O.; Levi J.; Namavari M.; Wang S.; Park J. M.; Zhang R.; Liu H.; Lee B.; Syud F. A.; Gambhir S. S. Mol. Imaging Biol. 2010, 12, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Z.; Ren G.; Liu H.; Jiang L.; Cheng Z. Bioconjugate Chem. 2010, 21, 947. [DOI] [PubMed] [Google Scholar]
- Knoess C.; Siegel S.; Smith A.; Newport D.; Richerzhagen R.; Winkeler A.; Jacobs A.; Goble R. N.; Graf R.; Wienhard K.; Heiss W. D. Eur. J. Nucl. Med. Mol. I 2003, 30, 737. [DOI] [PubMed] [Google Scholar]
- de la Zerda A.; Bodapati S.; Teed R.; May S. Y.; Tabakman S. M.; Liu Z.; Khuri-Yakub B. T.; Chen X.; Dai H.; Gambhir S. S. ACS Nano 2012, 6, 4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi J.; Kothapalli S. R.; Ma T.-J.; Hartman K.; Khuri-Yakub B. T.; Gambhir S. S. J. Am. Chem. Soc. 2010, 132, 11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M. F.; de la Zerda A.; Jokerst J. V.; Zavaleta C. L.; Kempen P. J.; Mittra E.; Pitter K.; Huang R.; Campos C.; Habte F.; Sinclair R.; Brennan C. W.; Mellinghoff I. K.; Holland E. C.; Gambhir S. S. Nat. Med. 2012, 18, 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.