

Abstract
The assembly of hundreds of identical proteins into an icosahedral virus capsid is a remarkable feat of molecular engineering. How this occurs is poorly understood. Key intermediates have been anticipated at the end of the assembly reaction, but it has not been possible to detect them. In this work we have used charge detection mass spectrometry to identify trapped intermediates from late in the assembly of the hepatitis B virus T = 4 capsid, a complex of 120 protein dimers. Prominent intermediates are found with 104/105, 110/111, and 117/118 dimers. Cryo-EM observations indicate the intermediates are incomplete capsids and, hence, on the assembly pathway. On the basis of their stability and kinetic accessibility we have proposed plausible structures. The prominent trapped intermediate with 104 dimers is attributed to an icosahedron missing two neighboring facets, the 111-dimer species is assigned to an icosahedron missing a single facet, and the intermediate with 117 dimers is assigned to a capsid missing a ring of three dimers in the center of a facet.
Introduction
Virus capsids containing hundreds of subunits have evolved to assemble with high fidelity in a short period of time. In many cases, capsid formation is sufficiently robust that, given the right conditions, purified capsid proteins spontaneously assemble into icosahedral virus-like particles.1,2 Despite a large effort, the underlying principles of capsid assembly are far from fully understood.3−5 The geometric control shown in forming a virus capsid far exceeds our ability to control assembly in man-made chemical systems. Thus, understanding virus assembly at a molecular level will not only increase our knowledge of a process of great biological and medical importance, but it will also help to develop new self-assembly strategies for materials science.6−9
Capsid assembly reactions necessarily have a single starting point of bulk subunit and a single ending point of complete capsid.10,11 The number of possible intermediates grows combinatorially from both ends of the assembly landscape. However, the concentrations of almost all the intermediates are exceedingly low during assembly.12,13 A small number of key intermediates, which may lead to kinetic bottlenecks, are anticipated at both ends of the assembly reaction, due to nucleation at the beginning and capsid closure at the end. The early intermediates are expected to be small fragments of the capsid while the late ones are expected to be almost complete capsids that are missing just a few subunits. Recent experiments have begun to identify early intermediates;14−19 however, whether late-assembly bottlenecks exist has remained unknown because it has not been possible to detect and identify them.
The assembly of hepatitis B virus (HBV) is of particular interest because it is a devastating pathogen and because it is an attractive target for the development of new assembly-directed antiviral molecules.20,21 Around 360 million people suffer from chronic HBV infection, which kills around 600,000 people annually. HBV self-assembly is usually studied with the core protein assembly domain, a 149-residue protein, Cp149. The building block for the HBV capsid is the Cp149 dimer. Under the right conditions, the dimers spontaneously assemble to yield icosahedral capsids with 90 dimers (in a T = 3 structure) and 120 dimers (in T = 4).22,23 Icosahedral capsids are usually described using a triangulation number (T), which gives the number of monomers (60T) in the capsid.24 The 60T monomers are arranged into 12 pentamers and 10(T–1) hexamers. The interdimer association energy for HBV is only about −15 kJ/mol at physiological temperature and ionic strength, but this supports formation of globally stable capsids because Cp149 dimers are tetravalent.25−28 A stronger association energy, which can be contrived by high salt assembly,25 results in kinetic trapping of intermediates on the assembly pathway because the high frequency of nucleation events and minimal dissociation of partial capsids deplete free subunits.29 In this work we analyze the trapped intermediates by charge detection mass spectrometry (CDMS) and cryo-electron microscopy (cryo-EM). These two techniques are complementary: CDMS provides information about the number of dimers in the trapped intermediates, and class averages from cryo-EM provide information about their structures.
Over the past few years, mass spectrometry (MS) has emerged as a powerful tool to investigate the composition of biological assemblies30−35 including viruses. Stockley, Ashcroft, and co-workers have used MS to probe the early steps in the assembly of the MS2 bacteriophage capsid,14−16 and Heck and collaborators have investigated the early stages of HBV capsid assembly.18 In conventional MS the mass-to-charge ratio (m/z) spectrum is measured, and for large multiply charged ions the charge must be deduced from the charge state ensemble to determine the mass. This approach starts to become problematic for masses around 100 kDa. Though the m/z peaks are usually still resolved in this range, they broaden and shift due to salt adduction and complex formation. The m/z peaks often do not shift by the same amount, making it difficult to assign the charge state. In some cases, the charge state resolution can be improved by purification, or the mass can be determined because the analyte identity is known. However, as the mass and heterogeneity increase, the problems are compounded until it is no longer possible to determine the mass from the m/z spectrum.
Here we have used charge detection mass spectrometry (CDMS), a single-particle technique where the mass is directly determined from m/z and z measurements for each ion. With this approach, masses can be measured for heterogeneous mixtures of large ions that resist analysis by conventional MS. CDMS was first used to measure the masses of micrometer-sized metal particles in 1960.36 The application of CDMS to electrospray ions was described by Fuerstenau and Benner in 1995,37 and the use of CDMS to measure the masses of intact viruses was reported in 2001.38 While groundbreaking, the mass resolution achieved in these early studies was rather poor (the peak for rice yellow mottle virus with a mass of ∼6.5 MDa had a full width at half-maximum of ∼12 MDa). Consequently, the technique received little attention. In recent papers,39−41 we have described a number of key improvements to CDMS that have led to large gains in the mass resolution and the limit of detection. Without these improvements, the measurements reported here would not have been possible.
Methods
Charge Detection Mass Spectrometry
Mass spectra were measured using a home-built charge detection mass spectrometer that is described in detail elsewhere.39−41 Briefly, electrosprayed ions enter the instrument through a heated capillary. They are separated from the ambient gas flow by three differentially pumped regions incorporating an RF ion funnel, an RF hexapole, and an RF quadrupole. Ions are extracted from the quadrupole, accelerated through a 100 V potential, and focused into the entrance of a dual hemispherical energy analyzer. The energy-selected ions are then focused into a modified cone trap that contains the charge detection cylinder. Ions oscillate in the trap, passing back and forth through the charge detector, for 129 ms. A charge-sensitive preamplifier incorporating a liquid nitrogen-cooled FET detects the image charge. The amplified signal is digitized and transmitted to a computer where the signals are analyzed using a fast Fourier transform (FFT). The fundamental frequency from the FFT is used to derive the m/z of the ion and the magnitude of the FFT is used to obtain its charge (z). The masses determined for each ion are then binned to yield the mass spectrum. Results for ions trapped for less than the full 129 ms are discarded. Figure S1 in Supporting Information (SI) shows an example of the charge detection signal and its FFT. Though a rare event, some ions lose charge while trapped. The loss of charge usually results in a sudden change in the ion’s oscillation frequency. In these cases, the ions are rejected by the data analysis program.
The measured masses are systematically overestimated by around 1% due to the presence of adducts (the addition of small molecules and counterions to the virus capsid) and imperfect calibration of the m/z and z measurements. The relationship between the oscillation frequency of an ion in the trap and the m/z is determined from SIMION simulations and the charge is calibrated by introducing test charges, ranging from approximately 1500–10000 elementary charges (e), through a capacitor. Though the calibration is performed with high charges, we have checked that it is valid down to charges as low as 7 e by measuring the m/z spectrum of cytochrome c and ubiquitin with CDMS. In this m/z range (<1500 Da/e), we were able to resolve individual charge states. This allows us to compare the charge deduced from the m/z spectrum (i.e., m/z charge) to the image charge (i.e., measured charge). For the +7–18 charge states, the m/z charge and the measured charge were in agreement. These results will be published elsewhere. To correct for the presence of adducts and small inaccuracies in m/z and z, the measured masses were calibrated to the T = 3 and T = 4 peaks of HBV using the sequence mass for the Cp149 monomer. The presence of two internal standards helps ensure the accuracy of the mass scale.
The mass resolution is limited by the uncertainties in the m/z and z measurements. With the 129 ms trapping time used here, the absolute root-mean-square (rms) deviation of the measured charge from its true value is expected to be 1.2 e.41 The kinetic energy distribution of the ions entering the trap limits the m/z resolution. The hemispherical energy analyzer was operated under low resolution conditions in these experiments, where the relative rms deviation of the m/z is around 0.010. The relative error in m/z is constant for all measurements; the absolute error in charge is constant for a given trapping time. Combining the contributions from the charge and the m/z, the relative rms deviation of the mass for the experiments reported here is expected to be around 0.013–0.014. Because the ions are highly charged (>100 e) and they are trapped for a long time, the chance of a false positive is essentially zero.
Sucrose Gradient Centrifugation and Cryo-EM Sample Preparation
Assembly of 40 μM (1.3 mg/mL) Cp149 dimer in 50 mM HEPES pH 7.5, was induced by addition of NaCl to 1 M final concentration. The resulting solution was allowed to equilibrate for up to 48 h at room temperature. A 200 μL aliquot of the assembly reaction was loaded onto a continuous 10–40% (w/v) sucrose gradient, containing 300 mM NaCl in 50 mM HEPES and centrifuged for 5 h at 200000g. For the cryo-electron microscopy images presented here, the protein layer above the T = 4 particle layer, including intermediate and T = 3 particles, was removed from the gradient, dialyzed into 300 mM NH4OAc and concentrated to a final concentration of 3.7 mg/mL protein.
Electron Microscopy
For cryo-EM specimen preparation, 4 μL of purified HBV sample from the sucrose gradient was applied to a glow-discharged carbon-coated 300 Cu mesh grid. The sample was left on the carbon film for 25 s, blotted for 4 s, and vitrified by plunge-freezing into liquid ethane using an FEI Vitrobot. The frozen-hydrated samples were visualized using a JEOL-3200 FS electron microscope (EM) equipped with a Gatan 626 cryotransfer system. The EM was operated at 300 kV with an in-column zero-loss energy filter using a slit width of 20 eV to improve image contrast. Grids were viewed under low-dose condition (15–20 e–/Å2) at a nominal magnification of 40000× (corresponding to 3.26 Å/pixel). Images were recorded on a Gatan 4k × 4k CCD camera at a defocus range of 2.0 to 4.0 μm.
Image Processing
Particles were semi-manually selected using a 200 × 200 pixel box, centered using EMAN2,42 and then normalized and filtered using Xmipp.43,44 A pool of 5681 particles was subjected to 2-D multireference alignment and classification based on the iterative hierarchical clustering method using CL2D from Xmipp.45 Details about the clustering schemes are given below.
Results
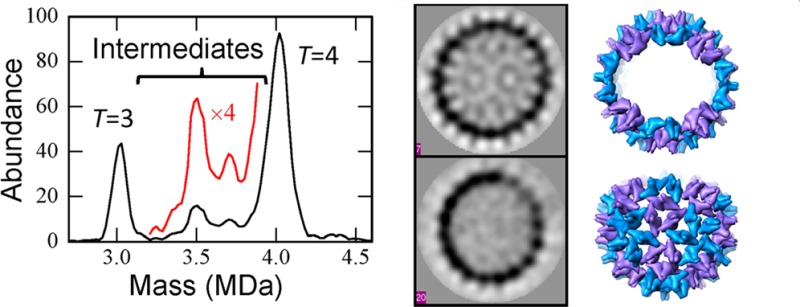
To trap intermediates, HBV capsids were assembled under aggressive conditions. Figure 1 shows mass distributions measured by CDMS for HBV capsids assembled for 24 h in 1 M NaCl and then dialyzed into 300 mM NH4OAc for 24 h. Samples were dialyzed into NH4OAc because this salt is compatible with electrospray. The spectrum in Figure 1 was measured around 6 h after dialysis. The prominent peaks near 3 and 4 MDa are due to the T = 3 and T = 4 capsids. The average number of charges on the T = 3 capsids is +129 (ranging from 122 to 136 e at fwhm) and the average number of charges on the T = 4 capsids is +150 (ranging from 143 to 157 e at fwhm). In addition to the T = 3 and T = 4 peaks, there are prominent features at around 3.5 and 3.7 MDa due to trapped intermediates. The mass spectrum was measured at regular intervals for a week, and no changes were observed, indicating that the observed intermediates are stable in 300 mM NH4OAc. Similar measurements were made under milder assembly conditions (0.3 M NaCl followed by dialysis into 100 mM NH4OAc). These mass spectra also show the peaks at 3.5 MDa and 3.7 MDa, though with significantly lower abundance. An example is shown in Figure S2 of SI. With the milder conditions, most of the trapped intermediates annealed over the course of a week, presumably by a process where some species dissociate to provide free subunits for the remainder to be completed. An example of a spectrum dominated by complete T = 3 and T = 4 capsids is shown in Figure S3 of SI.
Figure 1.
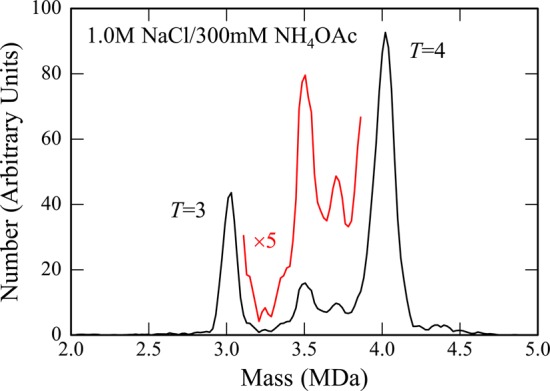
Mass spectrum measured by CDMS for HBV capsids assembled under aggressive conditions (1 M NaCl) and dialyzed into 300 mM NH4OAc. The mass spectrum is a histogram obtained by binning the measured masses for 5893 individual ions with 20 kDa bins. After binning, the histograms were smoothed with a five point Savitsky-Golay algorithm. The red line shows an expanded view of the region between the T = 3 and T = 4 peaks.
The mass resolution achieved in these measurements is insufficient to resolve an intermediate with a specific number of dimers from its immediate neighbors. The peak resulting from a single species is expected to be Gaussian with a width dictated by the experimental resolution (which is determined by the uncertainty in the charge and m/z measurements). To obtain better information about the size of the trapped intermediates, the measured mass distributions were analyzed by fitting them with Gaussian functions. A Gaussian function was centered and fixed at the mass of all possible intermediates with an integral number of dimers. The widths were governed by the experimental mass resolution. Initially, the intensities of all the intermediates were set to the same value, and then they were optimized by Monte Carlo sampling to obtain the best fit to the measured spectrum using a least-squares criterion. The fit to the spectrum in Figure 1 is shown in Figure 2. The points are the measured spectrum, and the red curve shows the best fit. The blue histogram in Figure 2 shows the optimized Gaussian intensities for each intermediate. Note that many of the possible intermediates have intensities that are near zero. This is a robust result that is independent of the initial intensities.
Figure 2.
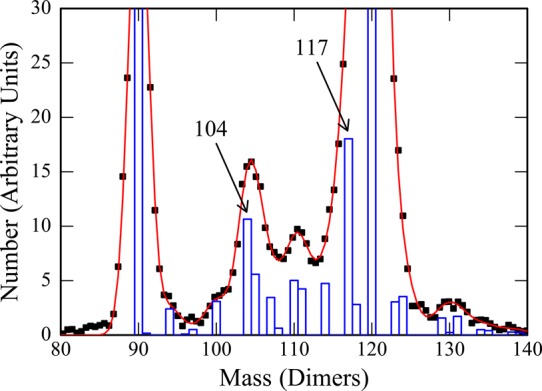
Analysis of the CDMS mass spectrum in Figure 1. The points are the measured mass spectrum and the red curve is a least-squares fit to the points using the model described in the text. The blue histogram shows the intensities of the intermediates that provide the best fit to the measured spectrum. The mass scale is in terms of the number of dimers.
It is possible to draw a number of conclusions from the fit shown in Figure 2. First, the peak at around 3.5 MDa is mainly due to trapped intermediates of 104 and 105 dimers. Second, the 3.7 MDa peak is attributed mainly to intermediates of 110 and 111 dimers. Third, the simulations shows that there is a low mass tail on the T = 4 peak, which corresponds mainly to an intermediate with 117 dimers with a smaller component at 118. Other spectra that we have analyzed lead to similar results, though in some of them the 118-dimer intensity is larger. In addition to the 104/105, 110/111, and 117/118 features discussed above, there are peaks in the histogram at 107 and 114 dimers. These are not associated with clear peaks in the mass distribution; thus, their assignments are not as reliable as the features discussed above. In some of the other spectra we have analyzed, the peak at 107 dimers shifts to 108; however, the 114 peak does not move.
In addition to the main features discussed above, there is a small signal ∼10 dimer units above the T = 4 peak that may result from off-pathway assembly into metastable, non-icosahedral structures beyond the T = 4 capsid. These species were not detected in the cryo-EM studies discussed below (their abundance is quite small, and they probably have low symmetry, and thus, their absence is not surprising).
HBV capsids were further investigated by cryo-EM, which allows visualization of particles undistorted by staining and drying artifacts. Images were analyzed for evidence of incomplete or irregular particles by hierarchical class averaging. To minimize confusion of authentic T = 4 particles with intermediates during EM analysis, the sample was harvested from a sucrose gradient fraction including the T = 3 band and extending to, but not including, the T = 4 band (red brace in Figure 3a). This way, all intermediate species leading up to the T = 4 particles are imaged, with a minimal number of obscuring T = 4 particles. In a typical micrograph (Figure 3b and Figure S4 of SI), along with numerous T = 3 and T = 4 particles, we observe apparently incomplete particles, arcs, and ellipsoids, as well as Gemini structures suggestive of two partial capsids.
Figure 3.

Characterization of the HBV capsids. (a) HBV capsids separated on a sucrose gradient showing the extracted fraction (red brace) used for cryo-EM analysis. (b) A representative cryo-EM micrograph of a frozen-hydrated HBV specimen. Black arrowhead, T = 3 capsid. White arrowhead, T = 4 capsid. Red arrowhead, capsid with defects. Scale bar, 50 nm. (c) Selected class averages show a T = 3 average, a T = 4 average, and three averages with defects, respectively. The red dashed lines indicate regions of weak density on the capsid. Scale bar = 10 nm. The averages in panel (c) correspond to classes 3, 7, 20, 8, and 1, respectively, in Figure S2 of SI.
To overcome the noise inherent in cryo-EM we subjected 5681 images (excluding arcs) to hierarchical class averaging. The results are shown in Figure 3c and Figure S5 of SI. We started with four classes (Figure S5a of SI) that were progressively split to provide greater definition of the characteristics of a given group of images. A hallmark of a good clustering structure is that, as classes are iteratively refined, the number of images switching between classes decreases. At 4, 8, and 16 classes, classification was very stable. At the final level of classification (28 classes, Figure S5b of SI), however, a large portion of images repeatedly switched classes during iteration, indicating that some classes were redundant and that few additional features would be observed by increasing the number of classes. Fifteen of the 28 final classes (3119 images) correspond to T = 3 particles based on their ∼32 nm diameter. Six of 28 classes (1293 images) appear to be intact T = 4 particles. Both the T = 3 and T = 4 averages have well-defined internal features. The remaining seven classes (1269 images) show evidence of significant defects. These averages all lack clearly defined internal density. They all show irregularities in the density of the protein ring outlining the averaged particle, suggesting a missing wedge of protein (see the red dashed lines in Figure 3c). They are slightly elliptical and inconsistent with an icosahedral particle (see Figure S5b of SI, numbers 0, 1, 15, 18, and 20). We did not attempt further analysis to obtain more detailed structural information. The broad distribution of species present in the mass range below T = 4 and the paucity of particles in any one class would make particle selection and reconstruction very challenging. In a separate experiment we selected a sucrose gradient fraction with less mass than the T = 3, but we did not find any incomplete T = 3 capsids. This is consistent with the CDMS results where only incomplete T = 4 capsids were found.
A control 2-D cryo-EM experiment verified that capsids with missing density were a function of assembly conditions and not an artifact of the classification scheme. HBV capsids were assembled for 24 h in a low salt buffer (0.15 M NaCl) to produce full capsids. They were then visualized using the procedures described above. Focusing on T = 4 capsids to identify incomplete particles, we selected 8064 images and used the classification scheme described above. The resulting class averages are consistent with intact T = 4 capsids viewed from different orientations (Figure S6 of SI).
Discussion
The CDMS and cryo-EM results presented here all indicate that high-molecular weight intermediates can be trapped by assembly in high salt conditions where Cp149-Cp149 association is strong; a strong association energy is predicted by theory to trap intermediates because the high frequency of nucleation events depletes free subunits that are required to complete a nascent particle.29 The prominent trapped intermediates observed in the CDMS spectra must persist because these species are resistant to dimer addition and loss. The most likely explanation for this behavior is that they have a lower free energy than their neighbors. Kinetic trapping is expected to trap intermediates on the assembly pathway. The cryo-EM measurements support this view. If we had observed aberrant structures in the cryo-EM class averages, they would probably be off the assembly pathway. However, we found incomplete T = 4 capsids, which indicates that they are on-pathway. Many paths and intermediates are possible; it is surprising we see so few intermediates persist.
The masses of the partial capsids along with the missing density in the cryo-EM images provide a basis for developing structural models for the prominent trapped intermediates. Theory suggests constraints consistent with the cryo-EM data and with persistent, on-path, metastable intermediates; their structures should be an incomplete capsid with one contiguous hole, edges of this hole should have dimers that are in high-affinity sites, and addition of dimers to this hole should be to low-affinity contacts.11 These rules provide the basis for a non-icosahedral particle that is more stable than complexes with one more or one less dimer. Examples of structures that satisfy the stability rules are shown in Figure 4. The model structures shown in the figure are based on coordinates for HBV T = 4 from ref (46) and are displayed with UCSF Chimera.47 A plausible 117-mer (Figure 4a) lacks a trimer of dimers, such as the three dimers central to a facet, leading to a roughly circular hole satisfying the requirements described above; the dimers at the edge of the hole are bound to three other dimers, and if another dimer is added, it can only bind to two dimers. A likely metastable 111-mer (Figure 4b) has a roughly triangular hole corresponding to the loss of a full facet. A possible 110-mer (Figure 4c) is missing the five dimers around a five-fold axis plus the five surrounding dimers resulting in a roughly circular hole. A metastable 104-mer complex can be modeled by extending the hole in the 111-mer to two icosahedral facets (Figure 4e). Viewed from the side, these models appear slightly elliptical and could easily result in cryo-EM projections that have relative weaknesses in density (e.g., Figure 4f).
Figure 4.

Models of possible structures for the trapped intermediates in HBV T = 4 assembly. T = 4 capsids missing (a) 3; (b) 9; (c) 10; (d) 12; and (e) 16 Cp149 dimers. (f) shows a side view of the T = 4 capsid missing 16 dimers, illustrating its slightly elliptical appearance. There are two classes of dimers in a HBV T = 4 capsid: blue dimers extend from pentameric to hexameric vertices, and purple dimers extend between hexameric vertices.22 In all of the proposed structures, the dimers surrounding the hole are in contact with at least three other dimers.
Structures that satisfy the stability rules outlined above can only be found for some of the possible intermediates, in particular, for the 104-mer and above; structures with all the dimers in high affinity sites can only be found for 104, 106, 108, 110, 111, 113, 115, and 117. Of these we unambiguously detect 104, 110, 111, and 117, which are notably symmetric. The unobserved 106, 113, and 115 metastable complexes are not symmetric. The only symmetric structure that we do not unambiguously detect is the 108-mer. A structure with 108 dimers that satisfies the stability rules is missing the six dimers around a two-fold axis, plus the surrounding six dimers. This structure (Figure 4d), is closely related to the 104-mer. While we do not see a strong peak for the 108-mer in Figure 3, there are peaks in the histogram at 107/108 which could be due to this species. The stability rules are based on counting the number of dimer contacts while keeping the capsid rigid. Rigorous optimization of the energies of possible intermediates may better explain why some are apparently preferred and others are not.
The most prominent trapped intermediates for the T = 4 capsid appear to be associated with missing icosahedral facets. The intermediates with 104, 111, and 117 dimers are consistent with one assembly path with multiple metastable pause points. However, the 110-mer, missing a pentamer of dimers and its surrounding five dimers, appears to be on a different path.
Finally, trapped intermediates were not observed for the T = 3 capsid in either the mass spectra or the cryo-EM measurements. The different geometries of the T = 3 and T = 4 capsids result in differences in the species that satisfy the stability rules. However, we have not been able to deduce an unequivocal explanation for the lack of T = 3 intermediates on the basis of the currently available literature.
Conclusions
Late intermediates for the assembly of the HBV T = 4 capsid are kinetically trapped by assembly under high salt conditions. Intermediates have been detected and identified by charge detection mass spectrometry. Prominent intermediates were found with 104/105, 110/111, and 117/118 dimers. Cryo-EM measurements indicate that the intermediates are incomplete T = 4 capsids. The detection of incomplete capsids rather than aberrant structures suggests that they are on-path intermediates. From their stability and kinetic accessibility we have proposed plausible structures. The prominent intermediate with 104 dimers is assigned to an icosahedron missing a two neighboring facets. The intermediate with 111 dimers is attributed to an icosahedron missing a single facet, and the 117-dimer species is assigned to a capsid missing a ring of three dimers in the center of a facet. Though assembly conditions used in our experiments are not physiological, we detected intermediates that are kinetically accessible, thermodynamically stable and that may help define the paths of HBV assembly.
Acknowledgments
We gratefully acknowledge the support of the NSF through Award Number 0832651 to M.F.J. and the NIH through Award Number R01-AI077688 to A.Z. and a subcontract to A.Z. from 5R01-GM100071 to Stephen Jacobson. E.E.P. and D.Z.K. were supported by Indiana University Quantitative and Chemical Biology Training Program Fellowships. Electron microscopy was performed in the cryo-TEM facility at IU.
Supporting Information Available
Six additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): The authors except A.Z. declare no competing financial interests. A.Z. is associated with a company that is developing anti-viral compounds.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bancroft J. B.; Hills G. J.; Markhari R. Virology 1967, 31, 354. [DOI] [PubMed] [Google Scholar]
- Zlotnick A.; Mukhopadhyay S. Trends Microbiol. 2011, 19, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luque A.; Reguera D.; Morozov A.; Rudnick J.; Bruinsma R. J. Chem. Phys. 2012, 136, 184507. [DOI] [PubMed] [Google Scholar]
- Roos W. H.; Bruinsma R.; Wuite G. J. L. Nat. Phys. 2010, 6, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan M. F.Modeling viral capsid assembly. Adv. Chem. Phys. 2014, 155 (in press). Available at http://arxiv.org/abs/1301.1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. E.; Speir J. A. J. Mol. Biol. 1997, 269, 665. [DOI] [PubMed] [Google Scholar]
- Mannige R. V.; Brooks C. L. PLoS ONE 2010, 5, e9423 10.1371/journal.pone.0009423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. T.; Meeuwissen S. A.; Noltea R. J. M.; van Hest J. C. M. Nanoscale 2010, 2, 844. [DOI] [PubMed] [Google Scholar]
- Dedeo M. T.; Finley D. T.; Francis M. B. Prog. Mol. Biol. Transl. Sci. 2011, 103, 353. [DOI] [PubMed] [Google Scholar]
- Endres D.; Zlotnick A. Biophys. J. 2002, 83, 1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisant P.; Neeman H.; Zlotnick A. Biophys. J. 2010, 99, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porterfield J. Z.; Zlotnick A. In Emerging Topics in Physical Virology; Stockley P. G., Twarock R., Eds.; Imperial College Press: London, 2010. [Google Scholar]
- Jack R. L.; Hagan M. F.; Chandler D. Phys. Rev. E. 2007, 76, 021119. [DOI] [PubMed] [Google Scholar]
- Stockley P. G.; Rolfsson O.; Thompson G. S.; Basnak G.; Francese S.; Stonehouse N. J.; Homans S. W.; Ashcroft A. E. J. Mol. Biol. 2007, 369, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton V. L.; Stockley P. G.; Stonehouse N. J.; Ashcroft A. E. Mass Spectrom. Rev. 2008, 27, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapman T. W.; Morton V. L.; Stonehouse N. J.; Stockley P. G.; Ashcroft A. E. Rapid Commun. Mass Spectrom. 2010, 24, 3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker G. K.; van Duijn E.; Crawford S. E.; Uetrecht C.; Baclayon M.; Roos W. H.; Wuite G. J.; Estes M. K.; Prasad B. V.; Heck A. J. R. Mol. Cell. Proteomics 2010, 9, 1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht C.; Barbu I. M.; Shoemaker G. K.; van Duijn E.; Heck A. J. R. Nat. Chem. 2011, 3, 126. [DOI] [PubMed] [Google Scholar]
- Tresset G.; Le Coeur C.; Bryche J.-F.; Tatou M.; Zeghal M.; Charpilienne A.; Poncet D.; Constantin D.; Bressanelli S. J. Am. Chem. Soc. 2013, 135, 15373. [DOI] [PubMed] [Google Scholar]
- Billioud G.; Pichoud C.; Puerstinger G.; Neyts J.; Zoulim F. Antiviral Res. 2011, 92, 271. [DOI] [PubMed] [Google Scholar]
- Katen S. P.; Chirapu S. R.; Finn M. G.; Zlotnick A. ACS Chem. Biol. 2010, 5, 1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther R. A.; Kiselev N. A.; Böttcher B.; Berriman J. A.; Borisova G. P.; Ose V.; Pumpens P. Cell 1994, 77, 943. [DOI] [PubMed] [Google Scholar]
- Wingfield P. T.; Stahl S. J.; Williams R. W.; Steven A. C. Biochem. 1995, 34, 4919. [DOI] [PubMed] [Google Scholar]
- Caspar D. L.; Klug A. Cold. Spring Harbor Symp. Quant. Biol. 1962, 27, 1. [DOI] [PubMed] [Google Scholar]
- Zlotnick A.; Cheng N.; Conway J. F.; Booy F. P.; Steven A. C.; Stahl S. J.; Wingfield P. T. Biochemistry 1996, 35, 7412. [DOI] [PubMed] [Google Scholar]
- Ceres P.; Zlotnick A. Biochemistry 2002, 41, 11525. [DOI] [PubMed] [Google Scholar]
- Katen S. P.; Zlotnick A. Methods Enzymol. 2009, 455, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnick A. Virology 2003, 315, 269. [DOI] [PubMed] [Google Scholar]
- Zlotnick A. J. Mol. Biol. 2007, 366, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo J. A. Mass Spectrom. Rev. 1997, 16, 1. [DOI] [PubMed] [Google Scholar]
- Ashcroft A. E. Nat. Prod. Rep. 2005, 22, 452. [DOI] [PubMed] [Google Scholar]
- Hernandez H.; Robinson C. V. Nat. Protoc. 2007, 2, 715. [DOI] [PubMed] [Google Scholar]
- Uetrecht C.; Versluis C.; Watts N. R.; Wingfield P. T.; Steven A. C.; Heck A. J. R. Angew. Chem., Int. Ed. 2008, 47, 6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht C.; Versluis C.; Watts N. R.; Roos W. H.; Wuite G. J. L.; Wingfield P. T.; Steven A. C.; Heck A. J. R. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A.; Reading E.; Hopper J. T. S.; Robinson C. V. Nat. Protoc. 2013, 8, 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton H.; Hendricks C. D.; Wuerker R. F. J. Appl. Phys. 1960, 31, 1243. [Google Scholar]
- Fuerstenau S. D.; Benner W. H. Rapid Commun. Mass Spectrom. 1995, 9, 1528. [DOI] [PubMed] [Google Scholar]
- Fuerstenau S. D.; Benner W. H.; Thomas J. J.; Brugidou C.; Bothner B.; Siuzdak G. Angew. Chem., Int. Ed. 2001, 40, 541. [PubMed] [Google Scholar]
- Contino N. C.; Jarrold M. F. Int. J. Mass Spectrom. 2013, 345–347, 153. [Google Scholar]
- Contino N. C.; Pierson E. E.; Keifer D. Z.; Jarrold M. F. J. Am. Soc. Mass. Spectrom. 2013, 24, 101. [DOI] [PubMed] [Google Scholar]
- Pierson E. E.; Keifer D. Z.; Contino N. C.; Jarrold M. F. Int. J. Mass Spectrom. 2013, 337, 50. [DOI] [PubMed] [Google Scholar]
- Tang G.; Peng L.; Baldwin P. R.; Mann D. S.; Jiang W.; Rees I.; Ludtke S. J. J. Struct. Biol. 2007, 157, 38. [DOI] [PubMed] [Google Scholar]
- Sorzano C. O.; Marabini R.; Velázquez-Muriel J.; Bilbao-Castro J. R.; Scheres S. H.; Carazo J. M.; Pascual-Montano A. J. Struct. Biol. 2004, 148, 194. [DOI] [PubMed] [Google Scholar]
- Sorzano C. O. S.; de la Fragab L. G.; Clackdoylec R.; Carazoa J. M. Ultramicroscopy 2004, 101, 129. [DOI] [PubMed] [Google Scholar]
- Sorzano C. O.; Bilbao-Castro J. R.; Shkolnisky Y.; Alcorlo M.; Melero R.; Caffarena-Fernández G.; Li M.; Xu G.; Marabini R.; Carazo J. M. J. Struct. Biol. 2010, 171, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne C.; Finn M. G.; Zlotnick A. J. Virol. 2006, 80, 11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. J. Comput. Chem. 2004, 25, 1605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.