

Abstract
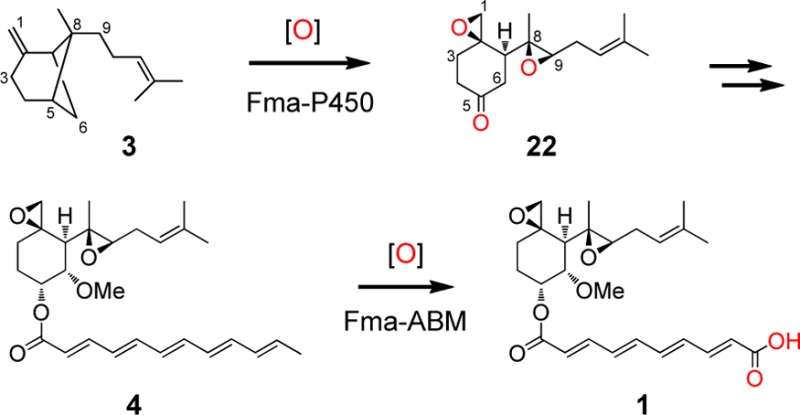
Fumagillin (1), a meroterpenoid from Aspergillus fumigatus, is known for its antiangiogenic activity due to binding to human methionine aminopeptidase 2. 1 has a highly oxygenated structure containing a penta-substituted cyclohexane that is generated by oxidative cleavage of the bicyclic sesquiterpene β-trans-bergamotene. The chemical nature, order, and biochemical mechanism of all the oxygenative tailoring reactions has remained enigmatic despite the identification of the biosynthetic gene cluster and the use of targeted-gene deletion experiments. Here, we report the identification and characterization of three oxygenases from the fumagillin biosynthetic pathway, including a multifunctional cytochrome P450 monooxygenase, a hydroxylating nonheme-iron-dependent dioxygenase, and an ABM family monooxygenase for oxidative cleavage of the polyketide moiety. Most significantly, the P450 monooxygenase is shown to catalyze successive hydroxylation, bicyclic ring-opening, and two epoxidations that generate the sesquiterpenoid core skeleton of 1. We also characterized a truncated polyketide synthase with a ketoreductase function that controls the configuration at C-5 of hydroxylated intermediates.
Introduction
Cytochrome P450 enzymes (P450s) are heme-containing monooxygenases that catalyze a variety of oxidative transformations in primary and secondary metabolism.1 P450 enzymes are widely distributed in natural product biosynthetic pathways from bacteria, fungi, and plants. These enzymes play a key role in the generation of structural complexity, including hydroxylation by activation of C–H bonds,2 epoxidation,2b C–C bond cleavage,3 and functional group rearrangement/migration.4 In addition, P450s from secondary metabolism have also been shown to catalyze less common reactions, including recent examples of heterocycle formation1b and cationic terpene cyclization.5 An increasing number of oxidative enzymes have been shown to catalyze multistep oxidation at distinct carbon atoms of substrates, resulting in drastic structural transformations (Figure 1).5,6 For example, during lovastatin biosynthesis, LovA catalyzes consecutive oxidations at opposite sides of the decalin ring to yield the precursor monacolin J,7 while TamI catalyzes successive hydroxylation and epoxidation reactions to afford the bicyclic ketal system in tirandamycin.6d Considering the central roles of such P450s in biosynthetic pathways, as well as the potential applications of these enzymes as C–H activation biocatalysts, discovery of novel multifunctional P450s from nature is of both fundamental mechanistic significance and practical importance.
Figure 1.
Examples of natural products of which multifunctional P450s play important roles in the biosynthesis. Functional groups introduced by P450s are shown in red.6a,6d,7,8
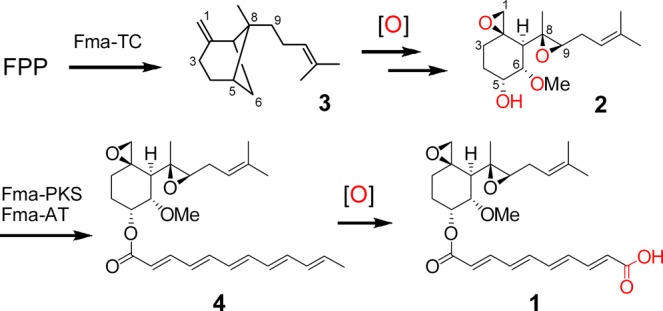
Fumagillin (1) from Aspergillus fumigatus is a meroterpenoid that has numerous biological activities. Compound 1 and its derivatives have been intensely studied for their potential use in the treatment of amebiasis,9 microsporidiosis,10 rheumatoid arthritis,11 and for their antiangiogenic properties by the irreversible inhibition of human type 2 methionine aminopeptidase (MetAP2).12,13 Structurally, 1 consists of a highly oxygenated, rearranged sesquiterpene esterified to a polyketide-derived tetraenoic diacid. The terpenoid portion, fumagillol (2) is derived by cleavage and multiple oxidation of the bicyclic sesquiterpene hydrocarbon intermediate β-trans-bergamotene (3)14 (Figure 2). The transformation of 3 to 2 involves a dramatic skeletal rearrangement in which the strained cyclobutane bridge is opened to yield the 1,8-bisepoxide-containing cyclohexanediol with six contiguous stereocenters. Therefore, given the unusual C–C cleavage and the requisite pair of C–H activation reactions starting from 3, it appeared likely that a P450 might be centrally involved in the biosynthesis of 2 and 1.
Figure 2.
Multiple oxidative-tailoring steps in fumagillin (1) biosynthesis.
We recently discovered the biosynthetic gene cluster for 1 in A. fumigatus.15 This fma cluster contains the first reported membrane bound class I terpene cyclase (Fma-TC) that catalyzes the formation of 3 from farnesyl pyrophosphate (FPP). The fma cluster also encodes a polyketide synthase (Fma-PKS) that synthesizes a dodecapentaenoate precursor that is trans-esterified to 2 by the acyltransferase Fma-AT to yield the intermediate prefumagillin 4.15 It was proposed that the polyene portion of 4 would be oxidatively cleaved to yield 1. Following our discovery, Wiemann et al. reported that the fma gene cluster is embedded within a supercluster on chromosome 8 of A. fumigatus that also encodes the biosynthetic pathways for fumitremorgin and pseurotin.16 The fma gene cluster (Figure 3A) contains four oxygenases that are most likely responsible for the oxidative tailoring steps that transform 3 to 1, including Af470 (Antibiotic Biosynthesis Monoxygenase superfamily monooxygenase), Af480 (nonheme iron-dependent dioxygenase), Af510 (cytochrome P450 monooxygenase) and Af440 (flavin-binding monooxygenase/methyltransferase). An additional redox enzyme in the fma gene cluster is the partial PKS (Af490), in which only the dehydratase (DH) and ketoreductase (KR) domains are present. The mechanistic role of each of these enzymes, especially that of the P450 (encoded by Af510), which is hypothesized to play a central role in the conversion of 3 to 2, has remained unknown.
Figure 3.
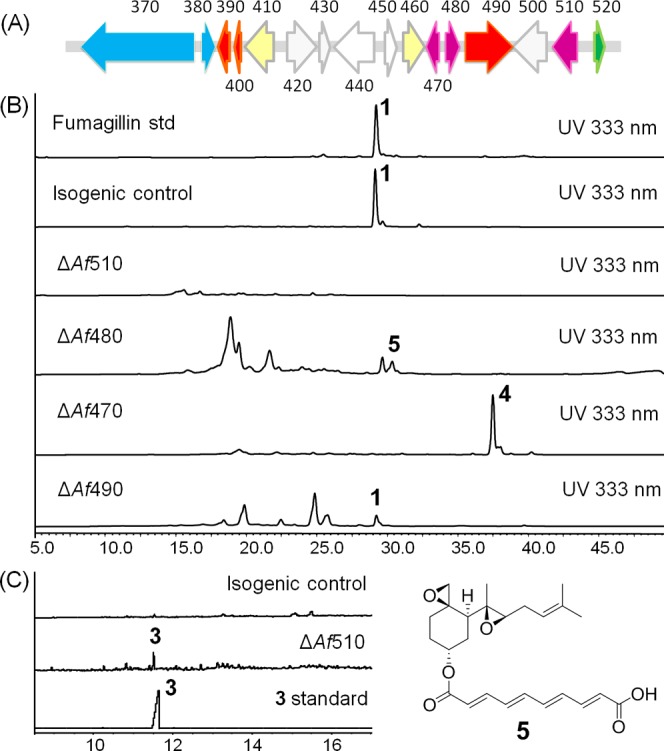
Genetic verification of Af510, Af480, Af470, and Af490. (A) The fma gene cluster. (B) HPLC analysis of metabolites extracted from isogenic control and ΔAf510, Af480, and Af470 strain showing loss of 1 and accumulation of 5 from ΔAf480 and 4 from ΔAf470 strain, respectively. (C) GC-FID analysis of the ΔAf510 strain showing the accumulation of 3.
Here we describe the complete characterization of the biosynthetic pathway of fumagillin (1). Using a combination of genetic knockout, chemical complementation, heterologous reconstitution in Saccharomyces cerevisiae, and in vitro biochemical assays, we have identified the role of each of the required pathway enzymes and biosynthetic intermediates. We show that Af510 indeed encodes a multifunctional P450 with a spectacular range of catalytic prowess, including hydroxylation of 3, oxidative cleavage of the bridging cyclobutane ring, and two epoxidation reactions. In addition to the on-pathway intermediates, we also demonstrate the range of off-pathway compounds that can be obtained using P450 starting from 3. Another notable finding includes the functional assignment of Af490 as a stereoselective ketoreductase in the biosynthesis of 2.
Results
Gene Inactivation Studies and Identification of Intermediates
We first set out to determine the roles and timing of the several redox enzymes in the fma gene cluster. We individually deleted five genes, Af510, Af490, Af480, Af470, and Af440 in A. fumigatus CEA17 akuBKU80 strain (pyrG89, ΔakuBKU80), which is deficient in nonhomologous end joining (Supporting Information).17 In comparison to the isogenic control strain, the metabolic profile of ΔAf510, ΔAf480 and ΔAf470 showed complete abolishment of the formation of 1 (Figure 3B), while ΔAf440 retained production of 1. The ΔAf490 strain showed decreased titers of 1 (86% decreasing in the culture of 4 days using CYA medium). The nonessential role of Af440 is in accordance with that reported by Wiemann et al., in which Af440 (which corresponds to psoF) was shown to be involved in pseurotin biosynthesis.16 However in contrast to the Wiemann report, in the analysis of the ΔAf470 profile, we found the accumulation of 4 with m/z 471 [M+Na]+ (Figure 3B), therefore suggesting a role for Af470 in the oxidative cleavage of the terminal alkene of the dodecapentaenoate side chain into the carboxylic acid present in 1. On the other hand, deletion of Af480 led to the accumulation of a polyene-containing compound 5, with maximum UV absorbance at 333 nm and m/z 451 [M+Na]+ (Figure 3B). Compound 5 was isolated from a 6-day liquid culture of A. fumigatus ΔAf480 mutant strain and structurally characterized. On the basis of the 1H, 13C and 2D NMR analyses, 5 was elucidated as 6-demethoxyfumagillin, in contrast to an aldehyde shunt product reported by Wiemann et al.16 The structure of 5 revealed that the nonheme iron-dependent oxygenase encoded by Af480 catalyzes hydroxylation of C6 in the biosynthesis of 1.
The only remaining unassigned oxygenase in the gene cluster was therefore the P450 gene, Af510, which shows the strongest sequence homology to the multifunctional P450 OrdA18 in aflatoxin biosynthesis (47% protein identity). Wiemann et al. reported that deletion of Af510 in A. fumigatus led to no detectable accumulation of any intermediates. While the same phenotype was observed here using LC–MS assay conditions, a more careful extraction with hexane and analysis of the hydrocarbon extract by GC-FID revealed the presence of β-trans-bergamotene (3) (Figure 3C). In the isogenic control, no accumulation of 3 was observed. This result suggested that the initial oxidation of 3 is catalyzed by the enzyme encoded by Af510.
Verification of Activities of Enzymes Encoded by Af470 and Af480
The enzyme encoded by Af470 (Fma-ABM) was initially revealed to harbor a DUF4188 conserved domain of unknown function by NCBI Conserved Domain Search. A further homology modeling search with Phyre219 showed, however, that Fma-ABM appears to be related to the cofactor-independent ABM superfamily monoxygenases with a ferrodoxin-like fold.20 To further test the role of Fma-ABM, we cloned the intron-less Af470 (for reconstruction of the gene from mRNA, see Supporting Information) into a yeast 2 μm expression vector which was transformed into S. cerevisiae strain BJ5464-NpgA. Upon supplementing 4 to the yeast culture expressing Af470, the biotransformation into 1 was detected (Figure 4A). On the basis of protein structure prediction (Supporting Information, Figure S7A), Fma-ABM was predicted to be a membrane-bound protein. Upon purification of the microsomal fractions of the yeast strain expressing Fma-ABM and incubation with 4, the same conversion to 1 was observed (Figure 4B). These results confirmed the role of Fma-ABM in the oxidative cleavage of 4 to 1.
Figure 4.
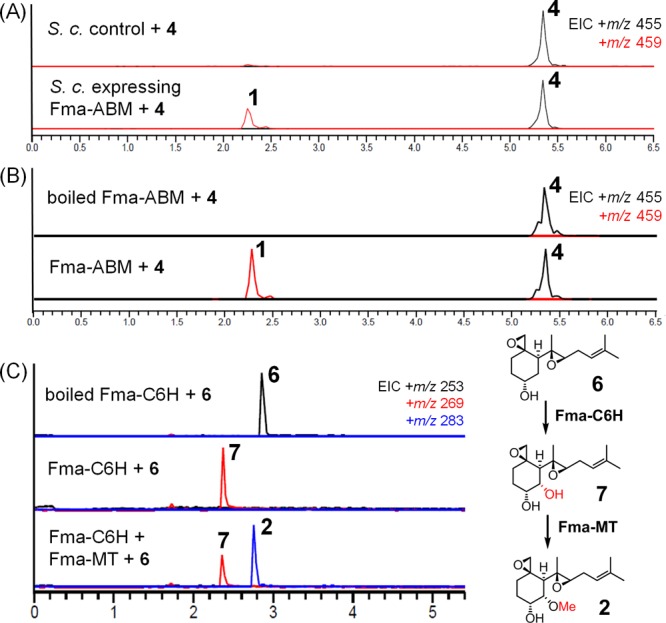
Verification of the function of Fma-ABM, Fma-C6H, and Fma-MT. (A) LC–MS detection of 1 upon expression of Fma-ABM in S. cerevisiae BJ5464-NpgA with supplement of 4. S. cerevisiae control is untransformed BJ5464-NpgA. (B) LC–MS analysis of in vitro assays of yeast microsomes containing Fma-ABM toward 4. (C) LC–MS analysis of in vitro assays of Fma-C6H with 6; and combined Fma-C6H and Fma-MT with 6, respectively.
To confirm the role of the enzyme encoded by Af480 (Fma-C6H), the 6xHis-tagged enzyme was solubly expressed from E. coli BL21 (DE3) and purified to homogeneity (Supporting Information, Figure S5). Bioinformatics analysis showed that Fma-C6H shares a similar sequence with the members of the phytanoyl-CoA dioxygenase (PhyH) superfamily, thereby requiring Fe (II) and α-ketoglutarate (α-KG) for its activity. Incubation of recombinant Fma-C6H with α-ketoglutarate, sodium ascorbate, and substrate 5, however, did not lead to the formation of any hydroxylated product. We therefore reasoned that Fma-C6H may function upstream in the pathway prior to attachment of the polyene portion. To obtain such a substrate, the hydrolysis of 5 under basic condition was performed to afford 6-demethoxyfumagillol (6) (Supporting Information). When recombinant Fma-C6H and 6 were incubated under the same condition as above, complete conversion of 6 to 6-hydroxylfumagillol (7) was obtained (Figure 4C). Furthermore, coincubation of Fma-C6H with the recombinant methyltransferase (Fma-MT) encoded by Af390–400 in the presence of the necessary cofactors (α-ketoglutarate, sodium ascorbate, and SAM) and 6 resulted in the formation of the expected 2 (Figure 4C). Collectively, these studies confirmed the function of Af480 and revealed that 6 (or a related compound) may be a key intermediate in the biosynthetic pathway of 1. To test this hypothesis, feeding of 6 to A. fumigatus blocked in bergamotene formation (deletion of Fma-TC, ΔAf520) restored biosynthesis of 1 as well as the production of 5 (see compilation of chemical complementation traces in Figure 7). With this information in hand, we then turned to investigation of the possible routes of oxidative modification of 3 into 6.
Figure 7.

LC–MS analysis of metabolites produced as a result of chemical complementation experiments. Compounds are supplemented at ∼40 μg/mL to (A) ΔAf520; (B) ΔAf510; (C) ΔAf480. (D) Structures of 21–23.
Yeast Reconstitution of Fma-P450 Activity and Characterization of Products
The transformation of 3 into 6 requires at least three oxidation steps to introduce the one C5 hydroxyl group and the two epoxide functionalities. In addition, the cleavage of the C–C bond between C5 and C8 must also take place. Several proposals for this mechanistically unresolved transformation have been advanced, including desaturation of the C5–C6 bond followed by concomitant cleavage of the C5–C8 bond.21 In light of the functional assignment of Af440, Af470, and Af480 described above, it appeared likely that the P450 encoded by Af510 (Fma-P450) is a multifunctional P450 that is responsible for most, if not all, of the oxidative steps between 3 and an advanced intermediate such as 6.
We previously showed that expression of Fma-TC alone in S. cerevisiae can lead to accumulation of β-trans-bergamotene (3).15 To investigate the activity of Fma-P450 toward 3, we cloned the intron-less Af510 into a yeast 2 μm expression vector for microsomal expression. To equip Fma-P450 with the optimal redox partner, we also cloned the A. fumigatus cytochrome P450 oxidoreductase (AfCPR) for coexpression.22 All three genes (encoding Fma-TC, Fma-P450, and AfCPR) were placed under the ADH2 promoter and transformed into BJ5464-NpgA. After four days of culturing followed by extraction with hexanes/EtOAc (1:1), we observed a series of sesquiterpene compounds (6, 8–19) that are derived from 3 (Figure5A). Of these compounds, 10, 11, 14, and 17 were the major products (0.6–1.1 mg/L); 8 and 12 were minor products (0.3–0.5 mg/L); and the remaining (6, 9, 13, 15, 16, 18, and 19) were present at less amounts (<0.3 mg/L). The same set of metabolites was also observed when 3 was directly supplied to the yeast culture expressing only Fma-P450 and AfCPR (Figure 5B). In the absence of 3 or Fma-P450, none of these 13 compounds was observable in the extract. To elucidate the structures of these products, large scale (12 L) fermentation of the triply transformed yeast strain was performed, followed by isolation and characterization of each compound (Figure 5D and Supporting Information).
Figure 5.
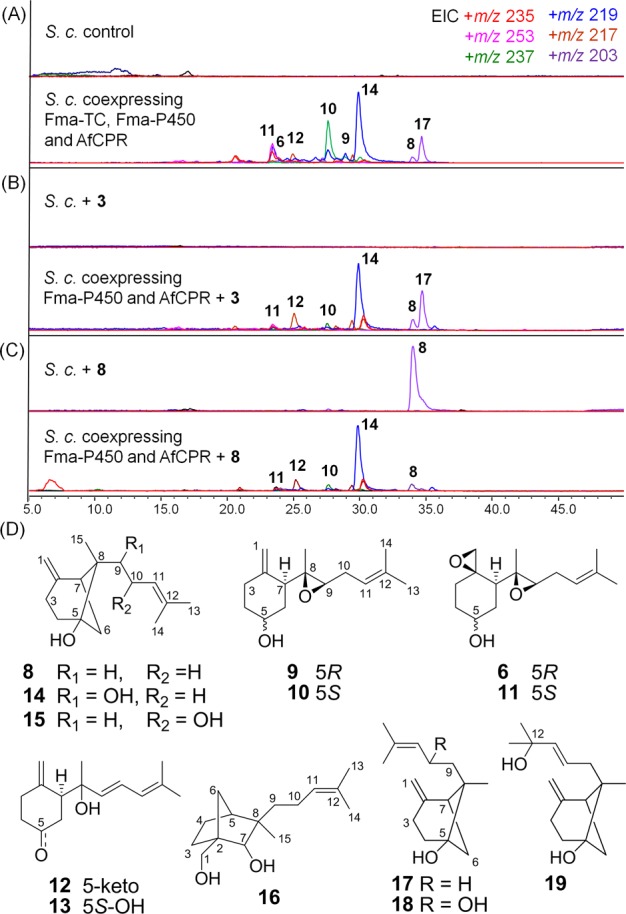
Products of Fma-P450 when reconstituted in S. cerevisiae. (A) LC–MS metabolic profiles of coexpression of Fma-TC (encoded by Af520), Fma-P450 (encoded by Af510), and AfCPR. Compounds 13, 15, 16, 18, and 19 are present at trace quantities and are not shown. (B) LC–MS analysis of coexpression of Fma-P450 and AfCPR in S. cerevisiae BJ5464-NpgA with supplement of 3; (C) same as trace B but with supplement of 8. (D) Elucidated structures of compounds 8–19.
Highly Oxidized Products
Compounds 6 and 9 were identified as 6-demethoxyfumagillol23 and cordycol,24 respectively, based on MS and NMR comparison to standards. 9 is the monoepoxide relative of 6 and may therefore be an immediate precursor of 6. Formation of these compounds does indicate that Fma-P450 alone is sufficient to transform 3 into 6. However, both compounds are found in very minute quantities (Figure 5A). Surprisingly, the 5-epimeric forms of 6 and 9, which are 5-epi-demethoxyfumagillol (11) and 5-epi-cordycol (10), respectively, were present as major products. The opposite stereochemistry of the C5 hydroxyl groups was readily established through the coupling patterns of H-5 in 1H NMR (6 and 9: br.s; 10 and 11: br.t, J = 9.4 and 10.5 Hz, respectively). To identify which of these compounds are on-pathway intermediates in the biosynthesis of 1, each compound was supplied to the ΔAf520 blocked mutant at ∼40 μg/mL. As expected, only 6 and 9 restored production of 1, even though each strain also produced the fumagillin analogues 5 and 20, respectively (Figure 7A and Supporting Information, Figure S11), through the direct esterification with the polyketide side chain (more on this below). In contrast, neither 10 nor 11 restored biosynthesis of 1 (Figure 7A and Supporting Information, Figure S11), thereby confirming that they are shunt products in S. cerevisiae, and no further epimerization of the C5 hydroxyl can take place in A. fumigatus. This result also provides insight into the intrinsic stereoselectivity of the Fma-AT which prefers the natural R-OH group at C5.
Less Oxidized Products
Compound 8 contains one additional oxygen atom compared to 3, and was found to be 5R-hydroxyl-β-trans-bergamotene (Supporting Information, Table S5/Figures S21–S26). 8 is therefore the product of direct oxidation of C5 of 3 by Fma-P450. Compounds 14 and 15 are dihydroxylated versions of 3. NMR characterization revealed the structures of 14 and 15 to be 5,9-dihydroxyl-β-trans-bergamotene and 5,10-dihydroxyl-trans-bergamotene, respectively (Supporting Information, Tables S11–S12/Figures S48–S57). All three compounds retained the trans-bergamotene scaffold found in 3, and therefore represent possible early intermediates in the pathway prior to the C5–C8 bond cleavage step. Interestingly, only the feeding of 8 to the ΔAf520 blocked mutant restored production of 1, while addition of either 14 or 15 failed to do so (Figure 7A and Supporting Information, Figure S11). Therefore 14 and 15 are shunt products of Fma-P450 activity in yeast. In particular, the high levels of 14 in the yeast culture extract hints that C9 is the site of the second P450 oxidation and may be subject to rapid hydroxylation (see discussion below). To prove the multifunctional nature of Fma-P450, 8 was added to the ΔAf510 mutant (Figure 7B). No restoration of 1 was observed, as would be expected for any additional role of Fma-P450 in the pathway. Finally, addition of purified 8 to the yeast culture expressing Fma-P450 and AfCPR similarly led to the formation of more oxidized products such as 11 and 14 (Figure 5C).
Ketone-Containing Product 12
Among the products isolated, 5-keto-isocordycol (12) contains a ketone functionality at C5 and an allylic alcohol resulting from prototopic ring-opening/rearrangement of the C8–C9 epoxide (Supporting Information, Table S9/Figure S40–S41). Although this compound is only present in minor quantities, the unexpected cyclohexanone shunt product suggests that the C5 hydroxyl (in 8, for example) may serve as the source of electrons in the oxidative cleavage of the cyclobutane ring, thereby transforming the bicyclic trans-bergamotene scaffold into an 2,5,7-trisubstituted cyclohexanone. The corresponding C5-reduced derivative, epi-isocordycol (13), was also found in the extract with the 5S-hydroxyl (Supporting Information, Table S10/Figures S42–S47). Both 12 and 13 will be revisited in the following sections.
Other Shunt Products
The other four compounds 16–19 were all found to be structurally related, but clearly off-pathway products. Compound 16 is particularly interesting with its trisubstituted sesquifenchyl core (Supporting Information, Table S13/Figures S58–S62). The 5-hydroxy-β-cis-bergamotene (17), 5,10-dihydroxyl-β-cis-bergamotene (18), and 19 all have the unexpected β-cis-bergamotene scaffold and are presumably formed by isomerization of reactive intermediates derived from 8 (Supporting Information, Tables S14–S16/Figures S63–S80). 17 is a major product from the yeast expression of Fma-P450, while 18 and 19 are further oxidized products of 17. The feeding of 17 to the ΔAf520 blocked mutant did not restore production of 1 (Figure 7A). Possible mechanisms of formation of 16–19 are shown in Figure 10 and will be discussed below.
Figure 10.

Proposed mechanism of Fma-P450. Radical mechanism from A to 21 was shown in Supporting Information, Figure S12.
Biochemical Assay of Fma-P450 and Identification of C5-Ketones as Biosynthetic Intermediates
The array of products recovered from S. cerevisiae clearly showcased the multifunctional capability of Fma-P450 (hydroxylation, epoxidation, rearrangement) in the oxidation of β-trans-bergamotene (3). The large number of shunt products, however, may be a result of the weak expression of fungal membrane-bound P450 in yeast. As a result of the low concentration of the P450, reactive intermediates may be readily intercepted or hydrolyzed to yield off pathway products such as 14 and 17. Therefore, to analyze the function of Fma-P450 under more controlled conditions, we prepared microsomal fractions from the yeast strain that overexpressed Fma-P450 and AfCPR for in vitro assay.
When 3 was incubated with 10 mg/mL of microsomal fractions and NADPH overnight, we detected 11 as essentially the single product in the extract (Figure 6A). Other major compounds observed in vivo, such as 10, 14, and 17, etc. are only present at very low levels when searched for using selective ion monitoring. Similarly, when the assay is repeated using 8 as the substrate, near complete conversion of 8 to 11 was observed with nearly no side products (Supporting Information, Figure S8). To analyze one of the individual steps catalyzed by the multifunctional P450, we assayed the epoxidation of 9 to 6 using the same microsomal extract. As shown in Figure 6B, we could observe incomplete conversion of 9 to 6 only in the presence of Fma-P450, thereby confirming the ability of this enzyme to catalyze the specific formation of epoxide.
Figure 6.
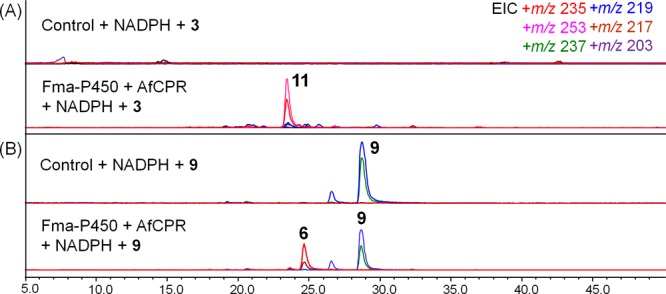
LC–MS analysis of in vitro assays of yeast microsomes containing Af510 and AfCPR toward (A) 3 and (B) 9.
Collectively, these in vivo and vitro results confirmed the multifunctional role of Fma-P450, including C5 hydroxylation, coupled or sequential 4e-oxidative cleavage, and rearrangement to yield epoxycyclohexanone, and the tandem epoxidation. At the same time, it is also evident that Fma-P450 alone is insufficient to generate the correct stereoisomer of 5R-demethoxyfumagillol 6, instead the 5S diastereomer 11 being formed in yeast or using yeast microsomes. Therefore, given that a ketone intermediate 12 could be isolated, the transformation of 3 most likely involves a C5 ketone intermediate that is stereoselectively reduced into the 5R isomer.
Several ketoreductases (KRs) such as the 3-KR in sterol biosynthesis and 3-KR for long-chain fatty acid synthase have been reported in S. cerevisiae,25 hence it is possible that an endogenous yeast KR could catalyze the reduction of the C-5 ketone to form the 5S-hydroxy moiety observed in compounds 10, 11, and 13. To test this hypothesis, we prepared both soluble and microsomal extracts of the untransformed host BJ5464-NpgA. We first tested if the isolated ketone 12 could be selectively reduced to 13. As shown in Supporting Information, Figure S9, complete reduction of 12 to 13 was observed. The R-isomer of 13 may be present at a very low concentrations as suggested by ion-monitoring, but the amount was insufficient for purification and characterization. To further test whether this endogenous yeast KR activity is responsible for formation of the other 5S-containing products shown in Figure 5D, we chemically prepared the three ketone substrates, 5-keto-cordycol (21), 5-keto-demethoxyfumagillol (22), and 5-keto-fumagillol (23) (Figure 7D, spectroscopic data in Supporting Information, Tables S17–S19 and Figures S81–S86). These compounds were synthesized from 10, 11, and 2 by PCC oxidation, respectively. When either 21 or 22 was added to the control yeast extract (soluble or microsomal), we observed the corresponding ketoreduction to 10 or 11 (Supporting Information, Figure S10), respectively, thereby validating the involvement of endogenous yeast KRs in the formation of compounds with the 5S hydroxyl functional group.
With the ketones 21–23 in hand, we performed chemical complementation experiments by feeding each of these compounds individually to the ΔAf520-blocked mutant. In each case, complete restoration of biosynthesis of 1 was observed (Figure 7A). Therefore, the C5-ketone moiety in each of the compounds can be processed correctly. Furthermore, chemical complementation of ΔAf510 mutant by 22 restored fumagillin production, indicating that Fma-P450 is not involved in the biosynthetic pathway beyond 22 and is not essential for reduction of the C5 ketone. Lastly, complementation of 23 to the ΔAf480 mutant efficiently restored fumagillin production, hinting that reduction of C5 may occur as the last step in the formation of 2.
Identification of Fma-KR as the Stereospecific 5-Ketoreductase
We hypothesized that a KR encoded in the Fma gene cluster might be responsible for the stereospecific 5R reduction of the ketone during the biosynthesis of 1. Although no standalone KR is found in the Fma cluster shown in Figure 3A, Af490 encodes a partial PKS in which only the DH-KR domains are present. Upon initial discovery of the fma cluster, the pseudo/partial PKS was assumed to be an inactive enzyme that had no likely role in the biosynthetic pathway. Although deletion of Af490 did not completely abolish the production of 1, we did notice an 86% decrease in the titer of fumagillin (1) (Figure 3B). The incomplete abolishment of the biosynthesis of 1 may be due to the presence of endogenous KRs in A. fumigatus that have overlapping functions to that encoded by the pathway enzyme Af490 (Fma-KR). To investigate the role of Fma-KR, the 110 kDa protein was therefore expressed as a 6xHis tag fusion and purified from S. cerevisiae (Supporting Information, Figure S6). Upon individual incubation of each of the ketone derivatives 21, 22, or 23 with recombinant Fma-KR, we observed complete conversion to the corresponding 5R-hydroxy-containing compounds, 9, 6, or 2, respectively (Figure 8A–C).
Figure 8.
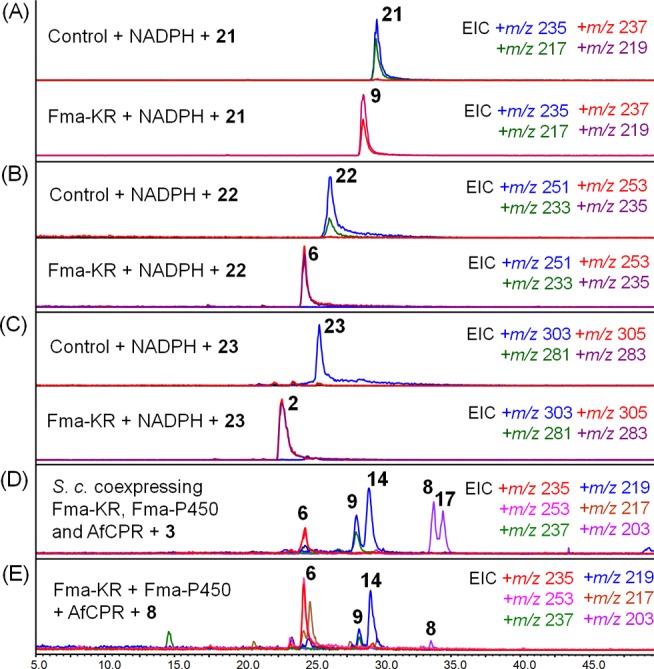
Fma-KR as the R-specific ketoreductase. (A–C) LC–MS analysis of in vitro assays of Fma-KR with 21–23. (D) LC–MS analysis of coexpression of Fma-KR, Fma-P450, and AfCPR in S. cerevisiae BJ5464-NpgA with supplement of 3. (E) LC–MS analysis of in vitro assay of Fma-KR, and yeast microsomes containing Af510 and AfCPR plus 8.
Lastly, to examine the product profile of Fma-P450 in the presence of Fma-KR, we repeated the biotransformation of 3 in S. cerevisiae by coexpression of Fma-P450, Fma-KR, and AfCPR. As shown in Figure 8D, the product profile is altered considerably to reflect the presence of the dedicated C5 reductase. The 5R-hydroxy isomers 6 and 9 are now clearly major products of the assay, while the 5S-hydroxy diastereomers 10 and 11 have essentially disappeared. The same results were obtained in the in vitro assay when Fma-P450-containing yeast microsomes were coincubated with purified Fma-KR. Whereas 11 was formed as the single product in the previous assay shown in Figure 6A, inclusion Fma-KR led to the production of the natural diastereomer 6 (Figure 8E).21b,26
The Complete Fumagillin Biosynthetic Pathway
On the basis of the new enzymes characterized and the new compounds isolated and identified in this work, we can now deduce a complete biosynthetic pathway for fumagillin (1), as shown in Figure 9. The pathway begins with the conversion of FPP to β-trans-bergamotene (3) by a membrane-bound Fma-TC.16 The initial oxidation of 3 by Fma-P450 involves C–H hydroxylation at the bridgehead C5 position to yield 8. Subsequently, a four electron oxidation initiated at C-9 coupled to cleavage of the cyclobutane C5–C8 bond of the bicyclo[3.1.1] core yields the on-pathway intermediate epoxyketone 21. An additional epoxidation reaction also catalyzed by Fma-P450 then furnishes the characteristic bisepoxide ketone 22. A possible mechanism for the Fma-P450-catalyzed conversion of 3 to 22 is shown in Figure 10 and will be elaborated further in the Discussion section.
Figure 9.

The fumagillin (1) biosynthetic pathway.
The diepoxyketone 22 is then subjected to successive C-6 hydroxylation and O-methylation by Fma-C6H and Fma-MT, respectively, to yield 23, which is then stereoselectively reduced by Fma-KR to 5R-hydroxy-seco-sesquiterpene 2. Acylation catalyzed by the Fma-AT with the dodecapentaenoyl group on Fma-PKS to yield 4 has been previously demonstrated.15 Finally, oxidative cleavage of 4 by the oxygenase (Fma-ABM) encoded by Af470 arrives at 1.
We have also shown that Fma-KR can stereospecifically reduce each of the three potential ketone intermediates 21–23 into the corresponding 5R-hydroxyl compounds, thereby implying that this ketoreduction step may occur at different stages during the biosynthesis of 2. In the proposed pathway, however, we have assigned the enzyme to catalyze reduction of the most advanced intermediate 23. We have shown that compounds 6 and 9, which can be produced by reduction of 22 and 21, respectively, can each be acylated efficiently by Fma-AT to yield the analogues 5 and 20. For example, feeding 6 to the ΔAf520 strain resulted in the production of both 5 and 1 (Figure 7A), with a relative ratio around 4:1. This indicated that C-6 hydroxylation by Fma-C6H may be slower than the C-5 acylation catalyzed by Fma-AT. The formation of 5 and 20 also demonstrates that Fma-AT has significant tolerance toward the substitution pattern of the terpene substrate, and can intercept these 5R-reduced products in vivo. In contrast, we have established that Fma-C6H is highly specific toward the unacylated intermediate 6. Therefore, 5 and 20 cannot be oxidized to 1. However, no trace of 5 or 20 is recovered from A. fumigatus, thereby suggesting that neither 6 nor 9 is an intermediate in vivo. Hence, we proposed that reduction of the C5 ketone takes place after the 6-methoxyl function has been installed in 23, thereby preventing premature C5 acylation of the terpenoid by Fma-AT.
Discussion
In this study, we have identified all the enzymes in the fumagillin pathway that transform the bicyclo[3.1.1] sesquiterpene 3 into the richly decorated pharmacophore 2, which acts by binding to type 1 and 2 MetAP.27 The highly oxidized cyclohexane portion of 2, which is also present in the metabolite ovalicin from Pseudeurotium ovalis,28 has been the subject of intensive synthetic efforts (see review in Yamagushi et al. and references therein).29 Considering the number of oxidative steps required to modify 3 into 2, it is remarkable that only two oxygenases are responsible. Most impressively, Fma-P450 encoded by Af510 alone catalyzes the eight electron oxidation of 3 into the intermediate 22 (Figure 9). The same set of reactions must therefore also be involved in the conversion of 3 into ovalicin, which is structurally identical to 23 except for the additional hydroxylation at C7 (Figure 8). The biosynthesis of ovalicin has previously been studied using labeled samples of mevalonate and of 3.14,30 From these investigations, several mechanisms and intermediates were ruled out, including the involvement 8,9-didehydrobergamotene that had been suggested by Birch (Figure 9).21a The pathway deduced from our genetic and biochemical experiments is fully consistent with these earlier findings.
The multifunctional Fma-P450 is the central player in the consecutive series of chemically and mechanistically distinct catalytic processes. This enzyme initiates the oxidative transformation on 3 by performing C-5 hydroxylation, followed by oxidative ring-opening coupled to epoxidation and a second tandem epoxidation to generate the key diepoxyketone intermediate 22 that serves as the substrate for later hydroxylation, methylation, ketoreduction, and acylation catalyzed by other pathway enzymes from the fma cluster. When Fma-P450 was reconstituted in S. cerevesaie, we observed numerous oxidized products derived from the bioconversion of 3. On the basis of the structures of these compounds, we can propose a detailed mechanism for Fma-P450. Either radical or cationic rearrangement mechanisms can be proposed for this enzyme and are shown in Figure 10 and Supporting Information, Figure S12. Although most of the P450-catalyzed C–H bond activation reaction has been shown to involve caged radical intermediates rather than carbocations,2b,4b,31 several recently discovered P450-catalyzed reactions almost certainly proceed through carbocationic mechanisms.5,32 It is possible that the various products produced by Af510 result from a partitioning between radical pair and cationic intermediates.
The first C–O bond installation occurs by C–H bond cleavage and hydroxylation at the C-5 of hydrocarbon 3 to form 5-hydroxybergamotene 8. Next, the hydrogen atom at C-9 is abstracted by the key ferryl-oxo intermediate (FeIV=O, porphyrin π cation radical) to generate the hydroxycyclobutylcarbinyl radical, A. This reactive intermediate can either undergo an oxygen rebound to generate 14 or be converted to a hydroxycyclobutylcarbinyl cation (B) after one-electron oxidation by the oxidative ion (IV)-hydroxo intermediate.33 Both reactions result in a ferric heme center and resume the catalytic cycle of Fma-P450 in the presence of AfCPR. Carbocation B can be subjected to attack by the “supernucleophile” iron(III)-peroxo intermediate generated from the following P450 cycle.1a The later reaction produces intermediate C and induces the subsequent ring-opening rearrangement through C5–C8 bond cleavage to form the 8,9-epoxide 21 (Figure 10), followed by a 1,2-epoxidation to generate 22. Nucleophilic addition from water or ferric hydroxo (FeIII–OH) complex to B also generates 14. This shunt pathway can readily take place, as indicated by the high level of 14 observed in yeast (Figure 5A). Formation of the shunt β-cis-bergamotene derivatives 17–19 can result from ring-opening/closure of the C-9 radical intermediate (D) to generate the isomeric radical (or cation) cis-5-hydroxybergamotene intermediate. Compounds 17–19 can then readily be obtained through direct nucleophilic attack of water or intervening 1,2-hydride shift. Lastly, if Fma-P450 epoxidizes the 1,2-exomethylene double bond of 3 prior to C-5 hydroxylation, the resulting epoxide intermediate could undergo well precedented cationic rearrangement of its bicyclo[3.1.1] skeleton to form the tetra-substituted sesquifenchol derivative 16.34 Overall, the oxidative cleavage of 8 to 21 is particularly intriguing, and would parallel the proposed conversion of loganin to secologanin,35 as well as that involved in the formation of furanocoumarin.36
The Af490 (Fma-KR) gene has previously been annotated as a PKS-like enzyme that harbors conserved motifs characteristic of the PKS-DH (smart00826) superfamily and PKS-KR (smart00822) domains as revealed by a NCBI Conserved Domain-Search. The discovery that the truncated PKS gene Af490 with only DH-KR domains encodes a functional enzyme that catalyzes stereospecific 5-ketoreduction of cyclohexanone 23 to 2 is intriguing given that PKS KR domain typically acts on β-ketoacyl thioester substrates attached on the acyl carrier proteins. Interestingly, a BLAST search for similar Fma-KR-like enzymes indicates that in A. fumigatus the hybrid PKS/NRPS (PsoA)6b in pseurotin biosynthesis (and the corresponding orthologues in A. clavatus and M. anisopliae)17 are the closest match with up to 56% protein identity (65% nucleotide identity) within the KR domain region. Hence, it appears that the Fma-KR may be an evolutionary product originating from classic gene duplication, divergence, and differential loss, but with the mechanisms acting on the functional domain level rather than on the entire gene. Although both A. clavatus and M. anisopliae have a close orthologue of psoA, the Fma-KR gene appears to be absent, suggesting the duplication has only occurred in A. fumigatus. Curiously, the pseurotin biosynthetic gene cluster6b is intertwined with the fma gene cluster and was shown recently to be coregulated by a single transcriptional factor (FapR) embedded in this fma-pso supercluster.16 The possibility that Af490 originated from a duplicated partial psoA gene but neofunctionalized to participate in the fma pathway further adds to the complex evolutionary history of this supercluster. Even more interesting, although we verified the role of Fma-KR in vitro and in S. cerevisiae, deletion of this gene in A. fumigatus did not lead to complete abolition of fumagillin (1) biosynthesis. It remains highly possible that the activity of the KR domain of PsoA can partially complement the deletion of Af490, therefore maintaining the ketoreductase activity, albeit at lower efficiency as indicated by the lowered titer of 1 in the ΔAf490 strain. Alternatively, other endogenous KRs in A. fumigatus may also possess this activity and catalyze the requisite reduction.
Fma-ABM encoded by Af470 has been shown to be the oxygenase responsible for the C10′-C11′ cleavage of the dodecapentaenoate of 4 and further oxidation of the aldehyde intermediate to the decatetraenedioic ester 1. Oxidative cleavage of olefins by a single oxygenase has been implicated in the biosynthesis of carotenoids,37 as well as in the formation of microbial secondary metabolites such as rifamycin.38 The carotenoid oxygenases are nonheme iron-dependent dioxygenases, while the cleavaging enzyme in the rifamycin pathway is a P450 monooxygenase. Interestingly, Fma-ABM belongs to a potentially new class of oxygenases that catalyze such reactions. It contains a DUF4188 conserved domain of still unknown function but homology modeling using Phyre2 suggests that the main portion of Fma-ABM is structurally related to aldoxime dehydratase (Supporting Information, Figure S7B) which is a heme-containing enzyme from Bacillus sp.,39 as well as the cofactor-free ABM superfamily of monooxygenases, which is typified by ActVA-Orf6 in actinorhodin biosynthesis and SnoaB in nogalonic acid biosynthesis.20 It is uncertain whether Fma-ABM requires a heme as a prosthetic group as it is much smaller than aldoxime hydratase (275 versus 373 amino acids). Further biochemical investigation of Fma-ABM enzyme is warranted as a BLAST search reveals that Fma-ABM homologues are ubiquitous in fungi.
Conclusions
We have uncovered the roles of three oxygenases, Fma-P450, Fma-C6H, and Fma-ABM, as well as an O-methyltransferase, Fma-MT, involved in the biosynthesis of fumagillin (1). Among these enzymes, Fma-P450, a P450 monooxygenase, catalyzes a multistep transformation that includes a simple hydroxylation followed by ring-opening of the bicyclic substrate coupled to generation of an epoxide, followed by a second epoxidation that results in the conversion of β-trans-bergamotene (3) to a highly oxygenated structure, 5-keto-demethoxyfumagillol (22). In the course of these studies we identified nine compounds as off-pathway products that shed light on the mechanism of action and catalytic potential of Fma-P450. The catalytic versatility of Fma-P450 therefore significantly augments the already impressive chemical virtuosity of this important class of enzymes.
We have also characterized the function of Fma-KR as the ketoreductase that controls the configuration at C-5 of hydroxylated intermediates that then undergo further acylation catalyzed by Fma-AT. This study has elucidated all the tailoring reactions in the biosynthesis of 1 and will provide opportunities for derivatization of 1 using enzymatic approaches.
Materials and Methods
Strains and Culture Conditions
A. fumigatus strain used in this study was A. fumigatus CEA17 akuBKU80 strain (pyrG89, ΔakuBKU80), which is deficient in nonhomologous end joining17 and was maintained on Czapek-Dox agar or glucose minimal agar (GMM).40 Details on AFUA_8G00390 and AFUA_8G00470–510 deletion mutants are shown in the Supporting Information. E. coli TOP10 (Invitrogen) and XL1-Blue (Stratagene) were used for DNA manipulation, and BL21 (DE3) was used for protein expression. Saccharomyces cerevisiae strain BJ5464-NpgA (MATα ura3–52 his3- Δ200 leu2- Δ1 trp1 pep4::HIS3prb1 Δ1.6R can1 GAL) was used as the yeast expression host.
General Techniques for DNA Manipulation
A. fumigatus genomic DNA was prepared using CTAB isolation buffer as described elsewhere.41 Polymerase chain reactions were performed using Phusion DNA Polymerase (New England Biolabs) or Platinum Pfx DNA polymerase (Invitrogen). DNA restriction enzymes were used as recommended by the manufacturer (New England Biolabs). RNA extraction was performed using a RiboPure Yeast Kit (Ambion), and ImProm-II Reverse Transcription System for RT-PCR (Invitrogen) was used to synthesize complementary DNA (cDNA) from total RNA. PCR products were subcloned to a pCR-Blunt vector (Invitrogen) and confirmed by DNA sequencing. Primers used to amplify the genes were synthesized by Integrated DNA Technologies and are listed in Supporting Information, Table S2. In vivo yeast recombination cloning was performed by transforming the S. cerevisiae BJ5464-NpgA with DNA fragments with >35 bp overlaps and includes a 2 μm plasmid backbone (derived from YEplac195 or YEplac112) using an S.c. EasyComp Transformation kit (Invitrogen).
Chemical Analysis
For production of 1 and other metabolites, A. fumigatus and mutant strains were cultured in CYA medium (Czapek-Dox agar supplemented with 5 g/L yeast extract). Total RNA for RT-PCR was extracted from A. fumigatus grown on Czapek-Dox liquid medium with 5 g/L yeast extract (CYB) after 4 days of cultivation. For chemical complementation of ΔAf520, ΔAf510, and ΔAf480 mutants, purified compounds are supplemented to the CYA medium at 0.2 mg/mL individually. LC–MS analyses of conversion of 7 to 8 by Af480 and 7 to 2 by Af480 and Af390–400 were performed on Prevail 3 μm, 2.1 × 100 mm2 C18 reversed-phase column (Alltech) and separated on a 5–95% (v/v) CH3CN linear gradient in H2O supplemented with 0.05% (v/v) formic acid at a flow rate of 125 μL/min. All LC–MS analyses except the aforementioned were performed on a Shimadzu 2010 EV LC–MS (Phenomenex Luna, 5 μm, 2.0 × 100 mm2, C18 column) using positive and negative mode electrospray ionization with a linear gradient of 5–95% MeCN-H2O in 30 min followed by 95% MeCN for 15 min with a flow rate of 0.1 mL/min. 1H, 13C, and 2D NMR spectra were obtained on Bruker AV500 spectrometer with a 5 mm dual cryoprobe at the UCLA Molecular Instrumentation Center.
Overexpression and Purification of His6-tagged Fma-C6H and Fma-MT
Af390 and Af400 were obtained as one transcript by RT-PCR verifying that Af400 was misannotated (for revised annotation, see Supporting Information). The DNA fragemnt of Af390–400 and Af480 from cDNA were inserted into pET21 (Novagen) digested with NdeI and XhoI to yield pKW20174 and pKW20172, respectively. Primers used for the amplification and cloning are listed in Supporting Information, Table S2. Recombinant enzymes were expressed with C-terminal His6-tagged in E. coli BL21 (DE3) and purified by nickel affinity chromatography. The cells were cultured at 37 °C, 250 rpm in 500 mL of LB medium with 35 μg/mL carbenicillin. Isopropylthio-β-d-galactoside (IPTG, 0.1 mM) to induce protein expression was added at OD600 between 0.4 to 0.6 and the cells were further cultured for 12–16 h at 16 °C. The cells were then harvested by centrifugation (3500 rpm, 15 min, 4 °C), resuspended in ∼25 mL of lysis buffer (100 mM Tris-HCL, pH 7.4, 0.1 M NaCl, 20 mM imidazole), followed by lysed by sonication on ice. Cell debris was removed by centrifugation (15 000 rpm, 30 min, 4 °C). The His6-tagged proteins were purified by using Ni-NTA agarose (Qiagen) according to manufacturer’s instructions. Purified enzyme was concentrated and exchanged into buffer A (50 mM Tris-HCl, pH 7.9, 2 mM EDTA, 2 mM DTT) + 10% glycerol with the centriprep filters (Amicon) and stored at −80 °C for enzyme assays.
Assay for Fma-C6H and Fma-MT Activity
For in vitro synthesis of 2 and 7, 10 μM Fma-C6H480 was incubated with 20 μM substrate 6, 1 mM sodium ascorbate, 1 mM α-ketoglutarate, and 100 mM NaCl in 100 mM Tri-HCl (pH 7.4). For in vitro synthesis of 2, the assay was performed using the same condition as above with additional 1 mM SAM and 10 μM Fma-MT. The reaction was incubated at 1 h and extracted twice with ethyl acetate. The organic phases were dried and dissolved in 20 μL of MeOH and subjected for analysis by LC–MS as described in Chemical Analysis.
Biotransformation in S.c
For biotransformation in S.c. of Fma-P450 and AfCPR, S. cerevisiae strain BJ5464-NpgA harboring both Fma-P450 and AfCPR plasmid were inoculated to 4 mL of Yeast Synthetic Drop-Out medium without uracil and leucine. The cells were grown for 72 h with constant shaking at 28 °C. A 15 μL aliquot of the seed culture was inoculated with 2 mL of YPD (10 g yeast extract, 20 g peptone, and 950 mL of Milli-Q water) supplemented with 1% dextrose. 3, 8, and 9 (0.5 mg in 10 μL of DMSO) was added to the culture after 48 h at 28 °C with shaking and the cells were cultivated for another 24 h. The cultures were extracted by hexanes-ethyl acetate (1:1) twice, the organic layers were concentrated in vacuo and redissolved in 100 μL of MeOH. A 10 μL aliquot of samples was further analyzed by LC–MS with the method described in Chemical Analysis. For biotransformation in S.c. of Af470, the culture of S. cerevisiae strain BJ5464-NpgA harboring Af470 was prepared by a similar method above and 4 (0.1 mg in 10 μL of DMSO) was added to the culture after 48 h.
Microsome Assay for Fma-P450 Activity
Details in preparation of Fma-P450 and AfCPR-containing microsomes for in vitro assay are shown in the Supporting Information. For in vitro microsomes assay, 10 mg/mL (wet weight) microsomal fractions containing Af510 and AfCPR, 1 mM substrates, 2 mM NADPH, and NADPH regeneration system (BD) solution A (5 μL) and B (1 μL), and 100 mM PBS, pH 7.4 were incubated in a 100 μL reaction. The reaction was incubated at room temperature for overnight and extracted with 100 μL of hexanes-ethyl acetate (1:1) twice. The organic phase was dried and redissolved in 20 μL of MeOH for analysis by LC–MS. The amount of protein in 10 mg/mL microsomes was calculated to be 180 μg/mL based on a modified Bradford assay against a BSA standard curve (protein samples were predenatured in 0.1 M NaOH).
Expression and Purification of Fma-KR from S. cerevisiae
S. cerevisiae BJ5464-NpgA was transformed with pHCfmaKR. For 1 L culture, the cells were grown at YPD (10 g/L yeast extract, 20 g/L peptone) supplemented with 1% dextrose and incubated at 28 °C with shaking for 72 h. The cells were harvested by centrifugation (3750 rpm at 4 °C for 10 min), and the cell pellet was resuspended in 20 mL of lysis buffer (50 mM NaH2PO4, 150 mM NaCl, 10 mM imidazole, pH 8.0) and lysed by sonication on ice in one minute intervals until homogeneous. To remove cellular debris, the homogeneous mixture was centrifuged at 17000 rpm for 1 h at 4 °C. Ni-NTA agarose resin was added to the supernatant (2 mL) and the solution was stirred at 4 °C overnight. Soluble Fma-KR was purified by gravity-flow column chromatography with increasing concentrations of imidazole in Buffer A (50 mM Tris-HCl, 500 mM NaCl, 20 mM–250 mM imidazole, pH 7.9). Purified protein was concentrated and buffer was exchanged into Buffer B (50 mM Tris-HCl, 2 mM EDTA, 100 mM NaCl, pH 8.0) using an Amicon Ultra-15 Centrifugal Filter Unit and stored in 10% glycerol. The purified Fma-KR was analyzed by SDS-PAGE (Supporting Information, Figure S6) and their concentration was calculated to be 8.7 mg/L, using the Bradford assay with BSA as a standard.
In Vitro Assays of Fma-KR
For in vitro synthesis of 6, 9, and 2, 10 μM Fma-KR was incubated with 1 mM substrates 21–23, respectively, 2 mM NADPH in 100 mM PBS, pH 7.4 in a total 100 μL reaction. The reaction was incubated at room temperature overnight and extracted with 100 μL of hexanes–ethyl acetate (1:1) twice. The organic phase was dried and dissolved in 20 μL of MeOH for analysis on LC–MS.
Preparation of 21–23
The method of oxidation by PCC followed that of Asami et al.42 A suspension of PCC (1 mg, 0.0046 mmol) and powdered molecular sieves 4A (3.0 mg) was added to a solution of 2 (2.3 mg) in 0.5 mL of CH2Cl2. The mixture was stirred in an ice–water bath and for 2.5 h at room temperature. Florisil and CH2Cl2 were added to the mixture, and the suspension was filtered through a combination of Celite and Florisil. The filtrate was concentrated in vacuo. The residue was purified by Silica gel plate (Merck, TLC Silica gel 60 F254, glass plates) with 25% acetone-hexanes developed by two times to give 23 (1.1 mg). A similar method was used as above to obtain 21 (0.8 mg) and 22 (1.0 mg) from 10 (2.2 mg) and 11 (3.0 mg), respectively.
Acknowledgments
H–C.L. is supported by National Science Council of Taiwan (102-2917-I-564-008). This work was supported by grants from the US National Institutes of Health to Y.T. (Grant Nos. 1R01GM085128 and 1DP1GM106413) and to D.E.C. (Grant No. 5R01GM030301), and from JSPS through the Funding Program for NEXT (Grant No. LS103) (K.W.) and by Nagase Science and Technology Foundation Japan (K.W). A.M.C and S.D. work was supported by Northern Illinois University. NMR instrumentation was supported by the NSF equipment Grant CHE-1048804. We thank Prof. Yongquan Li at Zhejiang University for a standard of 9. We thank Ralph A. Cacho, Dr. Youcai Hu, and Prof. Neil Garg for helpful discussions.
Supporting Information Available
Experimental details and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Meunier B.; de Visser S. P.; Shaik S. Chem. Rev. 2004, 104, 3947. [DOI] [PubMed] [Google Scholar]; b Mizutani M.; Sato F. Arch. Biochem. Biophys. 2011, 507, 194. [DOI] [PubMed] [Google Scholar]
- a Podust L. M.; Sherman D. H. Nat. Prod. Rep. 2012, 29, 1251. [DOI] [PMC free article] [PubMed] [Google Scholar]; b de Montellano P. R. O. Chem. Rev. 2010, 110, 932.19769330 [Google Scholar]
- Cryle M. J.; De Voss J. J. Chem. Commun. 2004, 86. [DOI] [PubMed] [Google Scholar]
- a Zhu D.; Seo M.-J.; Ikeda H.; Cane D. E. J. Am. Chem. Soc. 2011, 133, 2128. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jiang Y. Y.; He X.; de Montellano P. R. O. Biochemistry 2006, 45, 533.16401082 [Google Scholar]
- Chooi Y.-H.; Hong Y. J.; Cacho R. A.; Tantillo D. J.; Tang Y. J. Am. Chem. Soc. 2013, 135, 16805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tokai T.; Koshino H.; Takahashi-Ando N.; Sato M.; Fujimura M.; Kimura M. Biochem. Biophys. Res. Commun. 2007, 353, 412. [DOI] [PubMed] [Google Scholar]; b Maiya S.; Grundmann A.; Li X.; Li S.-M.; Turner G. ChemBioChem 2007, 8, 1736. [DOI] [PubMed] [Google Scholar]; c Anzai Y.; Li S.; Chaulagain M. R.; Kinoshita K.; Kato F.; Montgomery J.; Sherman D. H. Chem. Biol. 2008, 15, 950. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Carlson J. C.; Li S.; Gunatilleke S. S.; Anzai Y.; Burr D. A.; Podust L. M.; Sherman D. H. Nat. Chem. 2011, 3, 628. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Matsuda Y.; Awakawa T.; Wakimoto T.; Abe I. J. Am. Chem. Soc. 2013, 135, 10962. [DOI] [PubMed] [Google Scholar]
- Barriuso J.; Nguyen D. T.; Li J. W. H.; Roberts J. N.; MacNevin G.; Chaytor J. L.; Marcus S. L.; Vederas J. C.; Ro D.-K. J. Am. Chem. Soc. 2011, 133, 8078. [DOI] [PubMed] [Google Scholar]
- a Li S.; Tietz D. R.; Rutaganira F. U.; Kells P. M.; Anzai Y.; Kato F.; Pochapsky T. C.; Sherman D. H.; Podust L. M. J. Biol. Chem. 2012, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zocher G.; Richter M. E. A.; Mueller U.; Hertweck C. J. Am. Chem. Soc. 2011, 133, 2292. [DOI] [PubMed] [Google Scholar]
- Killough J. H.; Magill G. B.; Smith R. C. Science 1952, 115, 71. [DOI] [PubMed] [Google Scholar]
- Molina J. M.; Tourneur M.; Sarfati C.; Chevret S.; de Gouvello A.; Gobert J. G.; Balkan S.; Derouin F. N. Engl. J. Med. 2002, 346, 1963. [DOI] [PubMed] [Google Scholar]
- Bernier S. G.; Lazarus D. D.; Clark E.; Doyle B.; Labenski M. T.; Thompson C. D.; Westlin W. F.; Hannig G. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin N.; Meng L. H.; Wang M. Q. W.; Wen J. J.; Bornmann W. G.; Crews C. M. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger E. A.; Figg W. D. Expert Opin. Invest. Drugs 2000, 9, 1383. [DOI] [PubMed] [Google Scholar]
- Cane D. E.; Mcilwaine D. B. Tetrahedron Lett. 1987, 28, 6545. [Google Scholar]
- Lin H.-C.; Chooi Y.-H.; Dhingra S.; Xu W.; Calvo A. M.; Tang Y. J. Am. Chem. Soc. 2013, 135, 4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann P.; Guo C.-J.; Palmer J. M.; Sekonyela R.; Wang C. C. C.; Keller N. P. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Ferreira M. E.; Kress M. R.; Savoldi M.; Goldman M. H.; Hartl A.; Heinekamp T.; Brakhage A. A.; Goldman G. H. Eukaryot. Cell 2006, 5, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe K.; Chihaya N.; Hatabayashi H.; Kito M.; Hoshino S.; Zeng H.; Cai J.; Nakajima H. Fungal Genet. Biol. 2012, 49, 744. [DOI] [PubMed] [Google Scholar]
- Kelley L. A.; Sternberg M. J. E. Nat. Protoc. 2009, 4, 363. [DOI] [PubMed] [Google Scholar]
- Grocholski T.; Koskiniemi H.; Lindqvist Y.; Mantsala P.; Niemi J.; Schneider G. Biochemistry 2010, 49, 934. [DOI] [PubMed] [Google Scholar]
- a Birch A. J.; Hussain S. F. J. Chem. Soc. [Perkin 1] 1969, 11, 1473. [DOI] [PubMed] [Google Scholar]; b Cane D. E.; Buchwald S. L. J. Am. Chem. Soc. 1977, 99, 6132. [DOI] [PubMed] [Google Scholar]
- Munro A. W.; Girvan H. M.; Mason A. E.; Dunford A. J.; McLean K. J. Trends Biochem. Sci. 2013, 38, 140. [DOI] [PubMed] [Google Scholar]
- Kim D.; Min J.; Ahn S. K.; Lee H. W.; Choi N. S.; Moon S. K. Chem. Pharm. Bull. 2004, 52, 447. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Zhao Z.; Feng Q.; Xu Q.; Lu L.; Liu J.-K.; Zhang L.; Wu B.; Li Y.-Q. Helv. Chim. Acta 2013, 96, 76. [Google Scholar]
- Taramino S.; Teske B.; Oliaro-Bosso S.; Bard M.; Balliano G. Biochim. Biophys. Acta 2010, 1801, 1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane D. E.; Levin R. H. J. Am. Chem. Soc. 1976, 98, 1183. [DOI] [PubMed] [Google Scholar]
- Addlagatta A.; Matthews B. W. Protein Sci. 2006, 15, 1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cane D. E.; Mcilwaine D. B.; Oliver J. S. J. Am. Chem. Soc. 1990, 112, 1285. [Google Scholar]; b Cane D. E.; Mcilwaine D. B.; Harrison P. H. M. J. Am. Chem. Soc. 1989, 111, 1152. [Google Scholar]
- Yamaguchi J.; Hayashi Y. Chem.—Eur. J 2010, 16, 3884. [DOI] [PubMed] [Google Scholar]
- Cane D. E.; Levin R. H. J. Am. Chem. Soc. 1976, 98, 1183. [DOI] [PubMed] [Google Scholar]
- Newcomb M.; Letadicbiadatti F. H.; Chestney D. L.; Roberts E. S.; Hollenberg P. F. J. Am. Chem. Soc. 1995, 117, 12085. [Google Scholar]
- Zhu D. Q.; Seo M. J.; Ikeda H.; Cane D. E. J. Am. Chem. Soc. 2011, 133, 2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Auclair K.; Hu Z. B.; Little D. M.; de Montellano P. R. O.; Groves J. T. J. Am. Chem. Soc. 2002, 124, 6020. [DOI] [PubMed] [Google Scholar]; b Wen T. B.; Zhou Z. Y.; Jia G. C. Angew. Chem., Int. Ed. 2001, 40, 1951. [PubMed] [Google Scholar]
- Kim T. H.; Ito H.; Hatano T.; Takayasu J.; Tokuda H.; Nishino H.; Machiguchi T.; Yoshida T. Tetrahedron 2006, 62, 6981. [Google Scholar]
- Yamamoto H.; Katano N.; Ooi A.; Inoue K. Phytochemistry 2000, 53, 7. [DOI] [PubMed] [Google Scholar]
- Boland W.; Gabler A.; Gilbert M.; Feng Z. F. Tetrahedron 1998, 54, 14725. [Google Scholar]
- a Leuenberger M. G.; Engeloch-Jarret C.; Woggon W. D. Angew. Chem., Int. Ed. 2001, 40, 2614. [DOI] [PubMed] [Google Scholar]; b Schmidt H.; Kurtzer R.; Eisenreich W.; Schwab W. J. Biol. Chem. 2006, 281, 9845. [DOI] [PubMed] [Google Scholar]
- Xu J.; Wan E.; Kim C. J.; Floss H. G.; Mahmud T. Microbiology 2005, 151, 2515. [DOI] [PubMed] [Google Scholar]
- Sawai H.; Sugimoto H.; Kato Y.; Asano Y.; Shiro Y.; Aono S. J. Biol. Chem. 2009, 284, 32089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafer E. Adv. Genet. 1977, 19, 33. [DOI] [PubMed] [Google Scholar]
- Sambrook J.; Russell D. W.. Molecular Cloning: a Laboratory Manual; CSHL Press; Long Island, New York, 2001; Vol. 1. [Google Scholar]
- Asami Y.; Kakeya H.; Onose R.; Chang Y. H.; Toi M.; Osada H. Tetrahedron 2004, 60, 7085. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.