

Abstract
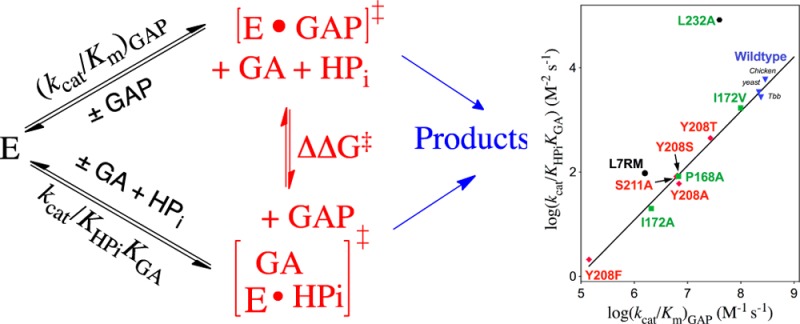
Values of (kcat/Km)GAP for triosephosphate isomerase-catalyzed reactions of (R)-glyceraldehyde 3-phosphate and kcat/KHPiKGA for reactions of the substrate pieces glycolaldehyde and HPO32– have been determined for wild-type and the following TIM mutants: I172V, I172A, L232A, and P168A (TIM from Trypanosoma brucei brucei); a 208-TGAG for 208-YGGS loop 7 replacement mutant (L7RM, TIM from chicken muscle); and, Y208T, Y208S, Y208A, Y208F and S211A (yeast TIM). A superb linear logarithmic correlation, with slope of 1.04 ± 0.03, is observed between the kinetic parameters for wild-type and most mutant enzymes, with positive deviations for L232A and L7RM. The unit slope shows that most mutations result in an identical change in the activation barriers for the catalyzed reactions of whole substrate and substrate pieces, so that the two transition states are stabilized by similar interactions with the protein catalyst. This is consistent with a role for dianions as active spectators, which hold TIM in a catalytically active caged form.
Triosephosphate isomerase (TIM) catalyzes the stereospecific and reversible conversion of dihydroxyacetone phosphate (DHAP) to (R)-glyceraldehyde 3-phosphate (GAP), by a proton transfer mechanism through enzyme-bound cis-enediolate intermediates (Scheme 1).1−4 The 12 kcal/mol stabilization of the transition state by interactions between TIM and the remote phosphodianion group of substrate accounts for ∼80% of total transition-state stabilization.5 These binding interactions not only anchor substrate to the enzyme active site but also play a role in activating TIM for deprotonation of bound carbon acid, as shown by the large effect of the deletion of a phosphodianion gripper loop on kcat for TIM-catalyzed isomerization of GAP6 and by the large activation by exogenous phosphite dianion (HPi) of TIM-catalyzed deprotonation of glycolaldehyde (GA)7 and isomerization of [1-13C]-glycolaldehyde ([1-13C]-GA) to [2-13C]-GA.8 This utilization of the binding energy of the nonreacting phosphodianion in enzyme activation, observed here and for other enzymatic reactions,7,9−13 is a critical difference between reactions catalyzed by enzymes14,15 and catalysis by small molecules.16
Scheme 1.
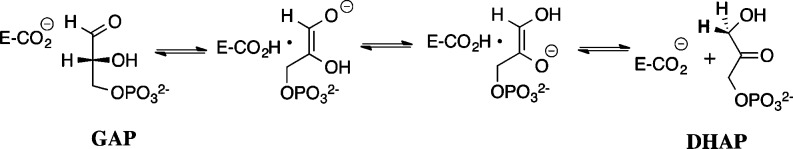
We report here a linear free-energy relationship, with slope of 1.0, between the kinetic parameters for the reactions of GAP and the pieces GA and HPi catalyzed by wild-type and structural mutants of TIM. This correlation shows that the transition states for the two reactions are stabilized by similar interactions with the protein catalyst and that the reactions proceed through similar transition states.
The I172 V,17 I172A,17 L232A,17,18 and P168A19,20 mutants of TIM from Trypanosoma brucei brucei (TbbTIM) and the 208-TGAG for 208-YGGS loop 7 replacement mutant (L7RM)19,21 of TIM from chicken muscle (cTIM) were examined in earlier work. The Y208T, Y208S, Y208A, Y208F, and S211A mutants of yeast TIM (yTIM) were prepared, purified, and characterized as described in the Supporting Information (SI). The positions of these amino acid residues are shown in Figure 1, for the complex between DHAP and TIM from yeast.22 The kinetic parameters determined for the Y208F and S211A enzyme-catalyzed isomerization of GAP are in good agreement with the published values of Sampson and Knowles.23
Figure 1.
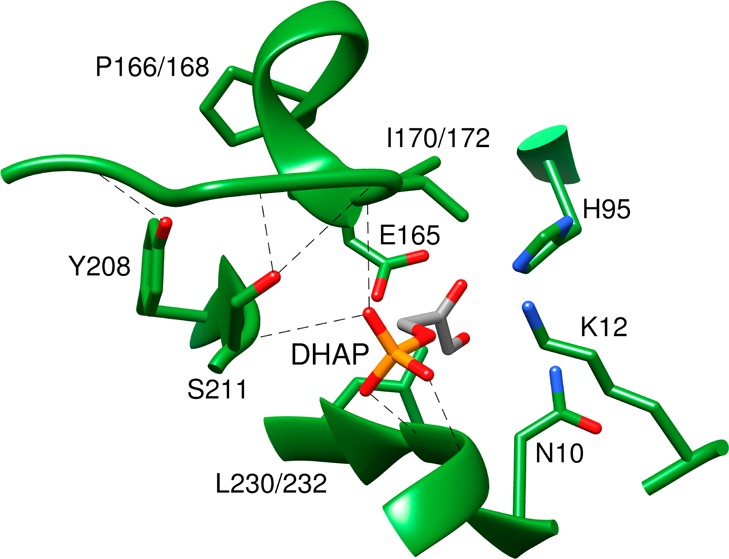
A model, from an X-ray crystal structure, of the complex between TIM from yeast and DHAP (PDB entry 1NEY) showing the amino acids mutated in this work. The side chains of H95, K12, and N10 play key roles in catalysis of the isomerization reaction.2,3Small differences in the numbering of the amino acids at TIM from the different sources used in these studies are noted, where appropriate: (cTIM or yTIM and TbbTIM): Pro 166 and 168, I170 and 172, L230 and 232.
The second-order rate constants (kcat/Km)obs for TIM-catalyzed reactions of [1-13C]-GA in D2O and the fractional yields, (fP)E, of products [2-13C]-GA, [2-13C, 2-2H]-GA, and [1-13C, 2-2H]-GA (Scheme 2) were determined by monitoring the disappearance of [1-13C]-GA and the formation of products by 1H NMR.8Tables S1–S6 report kinetic and product data for the reactions catalyzed by wild-type yTIM and by the Y208T, Y208S, Y208A, Y208F, and S211A mutant enzymes. A significant yield of [1-13C, 2,2-di-2H]-GA is sometimes observed from the TIM-catalyzed reactions of [1-13C]-GA,8,17,19 where the dideuterium-labeled product is formed by a nonspecific protein-catalyzed reaction.8,24,25 This is a minor product (5–10% yield) of the Y208T, Y208S Y208A, S211A mutant enzyme-catalyzed reactions of [1-13C]-GA in the presence of HPi and the major product (30 – 40% yield) of the reactions catalyzed by the severely crippled Y208F mutant. When the total yield of [2-13C]-GA, [2-13C, 2-2H]-GA and [1-13C, 2-2H]-GA [∑(fP)E, eq 1] is less than quantitative, the second-order rate constants (kcat/Km) for reactions at the enzyme active site (Scheme 2) were determined from the observed second-order rate constant (kcat/Km)obs and the sum of the yields of the three products [∑(fP)E], using eq 1.17,19
| 1 |
Scheme 2.

Figure 1 shows the active site for a complex between yTIM and DHAP.22 I172 and L232 from TbbTIM function in a hydrophobic clamp.26 The basicity of the side chain of E167, which reacts to deprotonate the carbon acid substrate, is enhanced by interactions with the side chain of I172.27 Steric interactions between the side chains of P168 and loop 7, induced by the ligand-gated conformational change,1,2 force the E167 (Tbb numbering) carboxylate toward the carbon acid substrate.20,28 This conformational change is enabled by formation of hydrogen bonds between the side-chain hydroxyls of Y208 and S211 from loop 7, respectively, with the backbone amide nitrogen of A176 and G173 from loop 6 and a hydrogen bond between the carbonyl oxygen of A169 and the γ-O of S211. The I172 V,17 I172A,17 L232A,17,18 and P168A19,20 mutations of TbbTIM, the 208-TGAG for 208-YGGS loop 7 replacement mutation (L7RM) of cTIM,19,21 and the Y208 and S211 mutations of yTIM each modify the enzyme structure in the region of the active site. Most of these structural mutations result in a decrease in the kinetic parameters for the TIM-catalyzed reactions of whole substrates and substrate pieces. This reflects the destabilization of the respective transition states for the mutant enzyme-catalyzed reactions, which result from subtle effects of these mutations on enzyme structure.
Figure 2 shows the dependence on [HPO32–] of kcat/Km for the reactions of [1-13C]-GA catalyzed by Y208 and S211 mutants of TIM. The third-order rate constants kcat/KGAKHPi (Scheme 3) reported in Table S7 were determined as the slopes of linear plots of kcat/Km against [HPO32–] for the reactions catalyzed by the Y208F, Y208A, and S211A mutants or as the slopes of the linear portions of the plots of data at low [HPO32–] for the reactions catalyzed by Y208S and Y208T mutants. Table S7 also reports the kinetic parameters kcat and (kcat/Km)GAP for wild-type and mutant TIM-catalyzed isomerization of GAP.
Figure 2.

Dependence of kcat/Km for the TIM-catalyzed turnover of the free carbonyl form of [1-13C]-GA in D2O on [HPO32–] at pD 7.0, 25 °C, and an ionic strength of 0.10 (NaCl). (A) Reactions catalyzed by the Y208T, Y208S, Y208A, and S211A mutants of yTIM. (B) Reactions catalyzed by the Y208F mutant.
Scheme 3.

The activation barrier for conversion of TIM and GAP to the transition state for enzyme-catalyzed isomerization of GAP [ΔG⧧GAP] is defined by the second-order rate constant (kcat/Km)GAP, while the barrier to formation of the transition state for the TIM-catalyzed reaction of the substrate pieces GA + HPi [ΔG⧧GA+HPi] is defined by the third-order rate constant kcat/KHPiKGA (Scheme 4). Figure 3 presents the linear logarithmic free energy correlation between the activation barriers for wild-type and mutant-TIM-catalyzed reactions of the whole substrate GAP and the substrate pieces GA + HPi. This correlation with slope of 1.04 ± 0.03 (95% confidence interval; 0.97–1.11) shows that most of these mutations, which alter the interactions of ligands with flexible loops 6 and 7 (Figure 1),29 result in the same destabilization of the transition states for the catalyzed reactions of the whole substrate and substrate pieces. We conclude that these transition states show strikingly similar interactions with TIM and that by this criteria are remarkably similar. The slope of 1.0 for Figure 3 reflects the constant difference in activation barriers for the reaction of whole substrate and the substrate in pieces: ΔΔG⧧ = 6.6 ± 0.3 kcal/mol (Scheme 4). This difference is the entropic advantage to the binding of the transition state for the reaction of the whole substrate compared with the transition state for reaction of the two pieces.30 This result is in good agreement with other estimates of the catalytic advantage obtained from covalent attachment of the reactants in a bimolecular reaction.31
Scheme 4.
Figure 3.
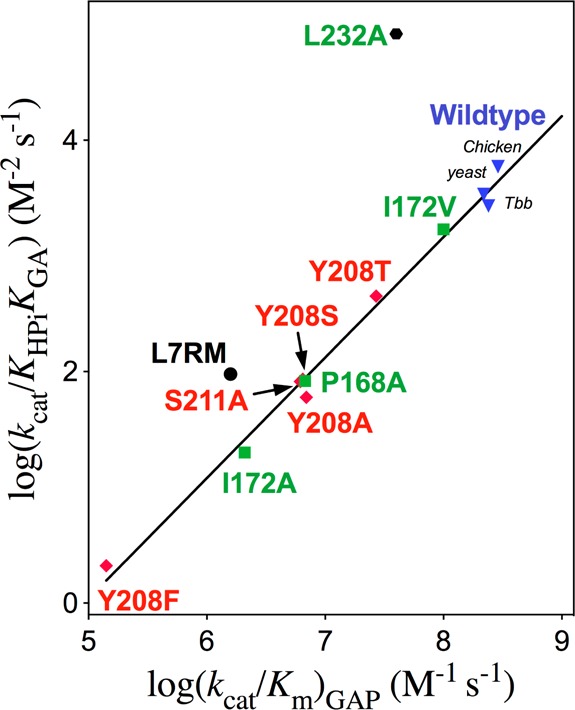
Linear free energy relationship, with slope 1.04 ± 0.03, between the second-order rate constant [log (kcat/Km)GAP] for wild-type and mutant TIM-catalyzed isomerization of GAP and the corresponding third-order rate constant [log (kcat/KHPiKGA)] for the enzyme-catalyzed reactions of the substrate pieces GA and HPi. Key: Green, TbbTIM; black, cTIM; red, yTIM.
The large positive deviation of the point for L232A mutant TbbTIM from the correlation in Figure 3 reflects the 25-fold larger value of kcat/KHPiKGA for the L232A mutant compared to wild-type TIM.17,18 We have proposed that the L232A mutation results in a 25-fold increase in the equilibrium constant Kc for the thermodynamically unfavorable conversion of TIM from the dominant inactive open form Eo to an active loop-closed form Ec, which is reflected by a ∼25-fold increase in concentration of the active enzyme (Scheme 5).8,17,18 We are uncertain of the explanation for the smaller positive deviation from this linear correlation of the kinetic parameters for the complex loop 7 replacement mutation of cTIM (Figure 3).
Scheme 5.

Wolfenden proposed that optimal enzymatic catalysis is sometimes obtained when the substrate is trapped in a protein cage, at an active site that provides for maximum enzyme-ligand contacts.32 This catalytic cage is created when substrate or transition-state analogs bind to TIM, by the closure of a flexible gripper loop over the ligand phosphodianion22,26,33−35 and presumably over the HPi activator. The linear correlation from Figure 3 provides evidence that dianions play a role as active spectators in creating the caged catalytic complex. The dianions are active and serve as a type of glue to hold TIM in a high-energy closed active form (Ec, Scheme 5), but in a different sense they are spectators, which provide no direct stabilization of the transition state for the unactivated reaction19 and which do not affect the transition-state structure.
The results of previous studies on TIM and the decarboxylation catalyzed by orotidine 5′-monophophate decarboxylase show that each enzyme is composed of a catalytic domain, which is competent to carry out the catalyzed reaction, and a dianion binding domain, where strong binding interactions with dianions are utilized to activate these enzymes for catalysis.36−38 The present results provide evidence that interactions between TIM and a spectator dianion lock the enzyme into an active conformation that is otherwise present at low concentrations (Scheme 5). Part of the dianion binding energy is used to drive desolvation of the carboxylate side-chain of Glu-165 (cTIM and yTIM) or Glu-167 (TbbTIM), which enhances the side-chain basicity toward deprotonation of carbon.27 The dianion binding interactions might also be utilized to organize/position the catalytic side chains at the enzymatic transition state, consistent with the notion that there is a high degree of “preorganization” of these side chains at the active sites of efficient enzyme catalysts.39−41
Acknowledgments
This work was supported by Grant GM39754 from the National Institutes of Health.
Supporting Information Available
Experimental procedures for the preparation and purification of Y208T, Y208S, Y208A, Y208F and S211A mutants of yTIM; procedures for determination of the product yields and kinetic parameters for wild-type and mutant yTIM-catalyzed reactions of [1-13C]-GA; Tables S1–S6, second-order rate constants and fractional product yields for the reaction of [1-13C]-GA catalyzed by wild-type yTIM (Table S1); and, by Y208T, Y208S, Y208A, Y208F and S211A mutants (Tables S2–S6, respectively); Table S7, kinetic parameters for wild-type and mutant yTIM-catalyzed reactions of GAP and of GA and HPi. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Richard J. P. Biochemistry 2012, 51, 2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga R. K.; Kapetaniou E. G.; Venkatesan R. Cell. Mol. Life Sci. 2010, 67, 3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles J. R. Nature 1991, 350, 121. [DOI] [PubMed] [Google Scholar]
- O’Donoghue A. C.; Amyes T. L.; Richard J. P. Biochemistry 2005, 44, 2622. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; O’Donoghue A. C.; Richard J. P. J. Am. Chem. Soc. 2001, 123, 11325. [DOI] [PubMed] [Google Scholar]
- Pompliano D. L.; Peyman A.; Knowles J. R. Biochemistry 1990, 29, 3186. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Biochemistry 2007, 46, 5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Amyes T. L.; Richard J. P. Biochemistry 2009, 48, 5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray W. J. Jr.; Long J. W.; Owens J. D. Biochemistry 1976, 15, 4006. [DOI] [PubMed] [Google Scholar]
- Kholodar S. A.; Murkin A. S. Biochemistry 2013, 52, 2302. [DOI] [PubMed] [Google Scholar]
- Goryanova B.; Amyes T. L.; Gerlt J. A.; Richard J. P. J. Am. Chem. Soc. 2011, 133, 6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W.-Y.; Amyes T. L.; Richard J. P. Biochemistry 2008, 47, 4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P.; Tait J. J. J. Am. Chem. Soc. 2005, 127, 15708. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Biochemistry 2013, 52, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. Adv. Enzymol. Relat. Areas Mol. Biol. 1975, 43, 219. [DOI] [PubMed] [Google Scholar]
- Morrow J. R.; Amyes T. L.; Richard J. P. Acc. Chem. Res. 2008, 41, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Koudelka A. P.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2012, 134, 10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2011, 133, 16428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Wierenga R. K.; Loria J. P.; Richard J. P. Biochemistry 2013, 52, 5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteleijn M. G.; Alahuhta M.; Groebel K.; El-Sayed I.; Augustyns K.; Lambeir A.-M.; Neubauer P.; Wierenga R. K. Biochemistry 2006, 45, 15483. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Berlow R. B.; Loria J. P. Biochemistry 2009, 48, 4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jogl G.; Rozovsky S.; McDermott A. E.; Tong L. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson N. S.; Knowles J. R. Biochemistry 1992, 31, 8482. [DOI] [PubMed] [Google Scholar]
- Go M. K.; Malabanan M. M.; Amyes T. L.; Richard J. P. Biochemistry 2010, 49, 7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Koudelka A.; Amyes T. L.; Richard J. P. Biochemistry 2010, 49, 5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kursula I.; Wierenga R. K. J. Biol. Chem. 2003, 278, 9544. [DOI] [PubMed] [Google Scholar]
- Malabanan M. M.; Nitsch-Velasquez L.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2013, 135, 5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnini S.; Groenhof G.; Wierenga R. K.; Juffer A. H. Proteins 2006, 64, 700. [DOI] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. Cur. Opin. Struct. Biol. 2010, 20, 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page M. I.; Jencks W. P. Proc. Nat. Acad. Sci. U.S.A. 1971, 68, 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfenden R. Mol. Cell. Biochem. 1974, 3, 207. [DOI] [PubMed] [Google Scholar]
- Alahuhta M.; Wierenga R. K. Proteins Struct., Funct., Bioinf. 2010, 78, 1878. [DOI] [PubMed] [Google Scholar]
- Davenport R. C.; Bash P. A.; Seaton B. A.; Karplus M.; Petsko G. A.; Ringe D. Biochemistry 1991, 30, 5821. [DOI] [PubMed] [Google Scholar]
- Lolis E.; Petsko G. A. Biochemistry 1990, 29, 6619. [DOI] [PubMed] [Google Scholar]
- Spong K.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2013, 135, 18343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Amyes T. L.; Gerlt J. A.; Richard J. P. Biochemistry 2013, 52, 7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Ming S. A.; Goldman L. M.; Wood B. M.; Desai B. J.; Gerlt J. A.; Richard J. P. Biochemistry 2012, 51, 4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A. J. Biol. Chem. 1998, 273, 27035. [DOI] [PubMed] [Google Scholar]
- Adamczyk A. J.; Cao J.; Kamerlin S. C. L.; Warshel A. Proc. Nat. Acad. Sci. U.S.A. 2011, 108, 14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerlin S. C. L.; Sharma P. K.; Chu Z. T.; Warshel A. Proc. Nat. Acad. Sci. U.S.A. 2010, 107, 4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.