

Abstract
His216 is a well-conserved residue in pyruvate carboxylases and, on the basis of structures of the enzyme, appears to have a role in the binding of MgATP, forming an interaction with the 3′-hydroxyl group of the ribose ring. Mutation of this residue to asparagine results in a 9-fold increase in the Km for MgATP in its steady-state cleavage in the absence of pyruvate and a 3-fold increase in the Km for MgADP in its steady-state phosphorylation by carbamoyl phosphate. However, from single-turnover experiments of MgATP cleavage, the Kd of the enzyme·MgATP complex is essentially the same in the wild-type enzyme and H216N. Direct stopped-flow measurements of nucleotide binding and release using the fluorescent analogue FTP support these observations. However, the first-order rate constant for MgATP cleavage in the single-turnover experiments in H216N is only 0.75% of that for the wild-type enzyme, and thus, the MgATP cleavage step is rate-limiting in the steady state for H216N but not for the wild-type enzyme. Close examination of the structure of the enzyme suggested that His216 may also interact with Glu218, which in turn interacts with Glu305 to form a proton relay system involved in the deprotonation of bicarbonate. Single-turnover MgATP cleavage experiments with mutations of these two residues resulted in kinetic parameters similar to those observed in H216N. We suggest that the primary role of His216 is to coordinate the binding of MgATP and the deprotonation of bicarbonate in the reaction to form the putative carboxyphosphate intermediate by participation in a proton relay system involving Glu218 and Glu305.
Pyruvate carboxylase (PC, EC 6.4.1.1.1) is a member of the family of biotin-dependent metabolic enzymes and supplies oxaloacetate for gluconeogenesis and the replenishment of tricarboxylic acid cycle intermediates that have been removed to synthesize other molecules such as amino acids and fatty acids.1−3 In vertebrates and many bacteria, the enzymic activity is tightly regulated by the allosteric activator acetyl CoA.4,5 As in the other biotin-dependent carboxylases, the initial step in the reaction catalyzed by pyruvate carboxylase involves the ATP-dependent carboxylation of the biotin prosthetic group in the biotin carboxylase (BC) domain active site. This reaction is thought to proceed via a carboxyphosphate intermediate produced by the transfer of a phosphoryl group from MgATP to bicarbonate3,6 (Figure 1, reaction 1). This intermediate is then thought to decarboxylate in the BC domain active site to produce carbon dioxide that carboxylates the biotin (Figure 1, reaction 2). The carboxybiotin thus formed then moves to the carboxyl transferase (CT) domain where it transfers its carboxy group to pyruvate to form oxaloacetate (Figure 1, reaction 3). From the structure of Rhizobium etli PC (RePC)7,8 and kinetic analyses of mutant forms of the enzyme, Zeczycki et al.9 proposed a mechanism for the formation of carboxyphosphate involving deprotonation of bicarbonate by Glu305, with transfer of a proton to Glu218 that also interacts with Lys245 (see Figure 2).
Figure 1.

Partial reactions catalyzed by pyruvate carboxylase that together form the overall pyruvate carboxylation reaction.
Figure 2.

Reaction scheme adapted from ref (9) showing the proposed involvement of Glu218, Glu305, and Lys245 in the nucleophilic attack of bicarbonate on MgATP, resulting in the formation of carboxyphosphate in the BC domain active site of RePC.
Another residue that is highly conserved in the active site of the BC domain is His216, which hydrogen bonds with the 3′-hydroxyl group of bound MgATP and the carboxyl group of Glu218 in RePC8 and with the carboxyl group of the equivalent residue in Staphylococcus aureus PC (Glu211).10 Thus, His216 provides a link between the bound nucleotide and the residues that participate in the formation of carboxyphosphate from bicarbonate and MgATP.
In this work, we explore the role of His216 in the binding of MgATP to the BC domain of RePC and the cleavage of ATP resulting in the formation of carboxyphosphate as an intermediate in the carboxylation of biotin.
Experimental Procedures
Materials
IPTG, malate dehydrogenase, and lactate dehydrogenase were obtained from Roche. HisPur cobalt IMAC resin was obtained from Thermo Scientific. All other materials were purchased from Sigma-Aldrich
Construction of the RePC Mutants
Mutagenesis was conducted on the 1.0 kb XhoI–SacII PC gene fragment corresponding to the allosteric domain using a Quickchange site-directed mutagenesis kit (Stratagene). Mutations were verified by DNA sequencing (Macrogen). The primers used to generate H216N, E218Q, and E305A mutants were H216N-F (5′-GAGCGCGCCCGCAACGTCGAAAGCC-3′; underlining indicates nucleotide substitution), H216N-R (5′-GGCTTTCGACGTTGCGGGCGCGCTG-3′), E218Q-F (5′-GCCCGCCACGTCCAAAGCCAGATCC-3′), E218Q-R (5′-GGATCTGGCTTTGGACGTGGCGGGC-3′), E305A-F (5′-CGCATCCAGGTCCAACACACGGTGAC-3′), and E305A-R (5′-GTCACCGTGTGTTGGACCTGGATGCG-3′), respectively. The corrected mutations were verified by DNA sequencing (Macrogen). The equivalent fragment of the wild-type RePC gene in the expression clone8,11 was then replaced with the mutagenized fragments.
Expression and Purification of RePC
The bacterium Escherichia coli BL21(DE3), containing the pCY216 plasmid,12 which encodes the E. coli BirA gene, was transformed with either the wild-type RePC or mutant RePC plasmid. The cultures were grown in 8 L of Luria-Bertani broth supplemented with 6.25 g/L arabinose, 10 mg/L biotin, 200 mg/L ampicillin, and 30 mg/L chloramphenicol at 37 °C until an OD600 of 1.0–1.2 was reached. The cultures were subsequently cooled on ice for 30 min, induced via addition of 0.1 mM IPTG, and incubated for approximately 36 h at 16 °C. The cells were harvested by centrifugation at 4039g and 4 °C for 15 min. The harvested cells were disrupted by incubation with 1 mg/mL lysozyme followed by mechanical disruption of the cells using a Bead-Beater (Biospec). Nucleic acids were removed from the lysate by protamine sulfate precipitation, and the total proteins were subsequently precipitated with 36% (w/v) saturated ammonium sulfate. The total proteins were then suspended in loading buffer [300 mM NaCl, 50 mM NaH2PO3, and 10 mM imidazole (pH 7.4)] prior to being loaded onto 20 mL of HisPur cobalt resin. RePC was selectively eluted from the resin using elution buffer [300 mM NaCl, 50 mM NaH2PO3, and 150 mM imidazole (pH 7.4)]. Purified PC was stored at −80 °C in storage buffer containing 30% (v/v) glycerol, 0.1 M Tris-HCl (pH 7.8), and 1 mM DTE.13
Determination of the Biotin Content of RePC
Aliquots of the enzyme were digested in triplicate with 0.2% (w/v) chymotrypsin (Sigma) in 0.2 M KH2PO4 (pH7.2) at 37 °C for 24 h followed by digestion with 0.45% (w/v) protease from Streptomyces griseus at 37 °C for 48 h. The biotin assay was performed as described by Rylatt et al.14 in triplicate. The enzyme concentrations mentioned are determined by the total amount of enzymic biotin in the purified wild-type and mutant RePC proteins.
Sedimentation Analysis of the Enzymic Quaternary Structure
Sedimentation velocity analytical centrifugation was performed with a Beckman Proteome Lab XL-A (Beckman-Coulter, Palo Alto, CA) ultracentrifuge using the absorbance optics system to visualize the protein at a wavelength of 280 nm. Two-sector cells were used, and data were acquired every 0.003 cm. Data were collected as 300 absorbance scans with a nominal time increment of 1 min at 30 °C at a speed of 40000 rpm. In all cases, enzyme samples were prepared at a concentration of 1.56, 0.5, or 0.4 μM in 0.1 M Tris-HCl (pH7.8) and 1 mM DTE with or without 5 mM MgCl2, 10 mM pyruvate, and 0.1 mM acetyl CoA. The computer-captured data were analyzed with SEDFIT.15 The partial specific volume of the enzyme was calculated from the amino acid composition using SEDNTERP (http://www.bbri.org/RASMB).15 The density of the Tris-HCl buffer (1.005 g/mL) was assumed to be the density of the enzyme solution.
Pyruvate Carboxylation Activity Assays
The initial rates of the enzymatic carboxylation of pyruvate were determined using a coupled spectrophotometric assay in which the oxaloacetate was converted to malate using malate dehydrogenase. The concomitant oxidation of NADH was measured by the change in absorbance at 340 nm.16 The enzymic activity was determined at 30 °C in a 1 mL reaction mixture containing 0.1 M Tris-HCl (pH 7.8), 6 mM MgCl2, 1 mM ATP, 20 mM NaHCO3, 10 mM pyruvate, 0.25 mM acetyl CoA, 0.22 mM NADH, and 5 units of malate dehydrogenase. kcat values were calculated by dividing the measured reaction velocity by the biotin concentration of the RePC used in the assay.
Oxamate-Stimulated Oxaloacetate Decarboxylation Assays
All assays were performed as described by Attwood and Cleland16 at 30 °C in 0.1 M Tris-HCl (pH 7.8) with 0.02 mM oxaloacetate, 1 mM oxamate, and 0.25 mM acetyl CoA. Assays were initiated by the addition of the wild type to a final concentration of 0.45 μM and H216N to a final concentration 0.64 μM; however, prior to the addition, a background rate of oxaloacetate decarboxylation was established, and this rate was subtracted from the rate in the presence of the enzyme.
Steady-State Bicarbonate-Dependent ATP Cleavage Activity Assays
The specific activities for the wild-type- and H216N-catalyzed, bicarbonate-dependent ATP cleavage were determined in triplicate using a coupled spectrophotometric assay in which pyruvate kinase and lactate dehydrogenase were used as coupling enzymes.17 The reactions were performed at 30 °C in a 1 mL reaction volume containing 0.1 M Tris-HCl (pH 7.8), 20 mM NaHCO3, 0.25 mM acetyl CoA, 10 mM phosphoenolpyruvate, 0.22 mM NADH, 5 units of pyruvate kinase, and 4 units of lactate dehydrogenase. MgATP concentrations were varied between 1 and 400 μM, and free Mg2+ concentrations were maintained at 5 mM by varying the concentration of added MgCl2.
Single-Turnover Bicarbonate-Dependent ATP Cleavage Activity Assays
The rate of ATP hydrolysis by wild-type RePC was measured using a rapid chemical quench-flow instrument (RQF-3, KinTek Corp., Austin, TX) at a constant temperature of 30 °C in 0.1 M Tris-HCl buffer (pH 7.8) containing 0.25 mM acetyl CoA, 5 mM MgCl2, and 20 mM NaHCO3. A 19 μL mixture of 1 μCi of an [α-32P]ATP solution (10 nM, final concentration) was mixed with 19 μL enzyme solution (with increasing reaction concentrations of the enzyme of 2, 3.5, 5, 7, 10, 20, and 40 μM). The reaction was quenched by 2 M HCl at various times after initiation (between 0.1 and 80 s) and the mixture expelled from the instrument; 1 μL of the quenched solution was spotted onto a PEI-cellulose TLC plate prespotted with 1 μL of a 20 mM ATP/ADP mixture and subsequently developed in an ATP/ADP separation solution containing 0.5 M LiCl and 1 M formic acid. The bands corresponding to ATP and ADP were visualized with a UV lamp and cut into pieces, and radiolabeled nucleotides were quantified by Cherenkov counting. From these measurements, the fractions of the original ATP converted to ADP were calculated at the different reaction times. To estimate the amount of ATP that spontaneously hydrolyzed in the reaction time, [α-32P]ATP was mixed with water instead of enzyme and analyzed, and any required corrections were made to the sample data. To estimate the end point of the ATP enzymatic hydrolysis, 19 μL of an enzyme solution was manually mixed with 19 μL of an [α-32P]ATP solution and the reaction was allowed to proceed for 5 min, after which the reaction was quenched by the addition of 280 μL of 2 M HCl. The fractions of the original ATP converted to ADP were plotted versus reaction times, and first-order exponential curves were fit by nonlinear least-squares regression to estimate the observed first-order rate constant for MgATP cleavage at each enzyme concentration. A secondary plot of these observed first-order rate constants (kobs) versus enzyme concentration was produced, and the data were fit to eq 1 using nonlinear least-squares regression to obtain estimates of the Kd for the binding of MgATP to the enzyme and the intrinsic rate constant (kcat) for MgATP cleavage.
| 1 |
The rates of ATP hydrolysis by H216N, E218Q, and E305N mutant RePC were measured by manual acid quench experiments. An enzyme solution was prepared containing 9 nM [α-32P]ATP (5 μCi), 0.25 mM acetyl CoA, 5 mM MgCl2, 20 mM NaHCO3 in 0.1 M Tris-HCl buffer (pH 7.8), and increasing concentrations of the mutant enzymes (2.2, 4.5, 8.9, 17.8, and 35.6 μM) in a total volume of 100 μL. Various times after the reaction had been started (0.5–20 min), 5 μL aliquots of the reaction mixture were withdrawn and added to 40 μL of 2 M HCl. The rate constants of MgATP hydrolysis at each concentration of the mutant enzymes, the Kd of MgATP binding, and the kcat of MgATP hydrolysis were determined as for the wild-type enzyme.
Phosphorylation of MgADP by Carbamoyl Phosphate
The rate of ADP phosphorylation by carbamoyl phosphate was determined for the wild type and H216N using a spectrophotometric assay in which hexokinase and glucose-6-phosphate dehydrogenase were used as coupling enzymes.18 Reactions were performed at 30 °C in a 1 mL reaction mixture containing 0.1 M Tris-HCl (pH 7.8), 5 mM MgCl2, 5 mM MgADP, 0.25 mM acetyl CoA, 0.5 mM glucose, 0.5 mM NADP, 5 units of hexokinase, and 4 units of glucose-6-phosphate dehydrogenase. Carbamoyl phosphate concentrations were varied between 0.5 and 40 mM.
Stopped-Flow Measurements of Mg·Formycin-A-5′-Triphosphate (MgFTP) Binding to RePC
The fluorescence measurement of the kinetics of Mg·formycin-A-5′-triphosphate (MgFTP) binding and displacement by MgATP were as described by Geeves et al.,19 except experiments were performed at 30 °C. A KinTek SF-2004 instrument (KinTek Corp.) was used in these measurements. Excitation was at 310 nm, and a 350 nm cut-on filter was used to monitor emission. Measurements were taken at 30 °C in 0.1 M Tris-HCl buffer (pH 7.8) containing 5 mM MgCl2 and 20 mM NaHCO3. In binding experiments, the solution of MgFTP (10 μM, final concentration) was rapidly mixed with the solution of either wild-type RePC or H216 (0.5 μM, final concentration), and the trace of fluorescence emission produced by the excitation of FTP with 310 nm light was recorded using a 350 nm cut-on filter. In displacement experiments, the solution of preformed complex MgFTP·WT or MgFTP·H216N (10 μM MgFTP and 0.5 μM wild type or H216N, final concentrations) was rapidly mixed with the solution to give a final MgATP concentration of 1 mM, and the kinetic trace of fluorescence quenching was recorded.
Results
Sedimentation Analysis of the Quaternary Structure of Wild-Type RePC and H216N
Table 1 shows that from the analytical ultracentrifugation analyses, the distribution of quaternary structures in H216N is very similar to that of the wild-type enzyme in the presence and absence of acetyl, pyruvate, and MgCl2. This indicates that the mutation of His216 has not destabilized the quaternary structure of the enzyme.
Table 1. Analyses of Multimeric Species Present in Wild-Type RePC (WT) and H216N in the Presence and Absence of Acetyl CoA, Pyruvate, and MgCl2 from Analytical Ultracentrifugation Studies.
| monomer |
dimer |
tetramer |
||||
|---|---|---|---|---|---|---|
| RePC enzyme | molecular mass (kDa) | % | molecular mass (kDa) | % | molecular mass (kDa) | % |
| WTa | 163 ± 26 | 14 | 287 ± 38 | 14 | 450 ± 32 | 72 |
| H216Na | 152 ± 41 | 12 | 304 ± 51 | 15 | 499 ± 82 | 73 |
| WTb | mixture of monomers and dimers | 14 | 472 ± 52 | 86 | ||
| H216Nb | mixture of monomers and dimers | 14 | 562 ± 57 | 86 | ||
In 0.1 M Tris-HCl (pH 7.8) and 1 mM DTE.
In 0.1 M Tris-HCl (pH 7.8), 0.1 mM acetyl CoA, 10 mM pyruvate, 5 mM MgCl2, and 1 mM DTE.
Steady-State Pyruvate Carboxylation and Oxamate-Stimulated Oxaloacetate Decarboxylation
The kcat values for wild-type RePC- and H216N-catalyzed pyruvate carboxylation are listed in Table 2. Thus, the reaction catalyzed by H216N occurs at only 0.45% of the rate of that catalyzed by wild-type RePC. The kcat values for oxamate-stimulated decarboxylation of oxaloacetate are listed in Table 2. Thus, the rate of the reaction catalyzed by H216N is 36% of that catalyzed by wild-type RePC.
Table 2. Kinetic Parameters for Steady-State Reactions, Including Pyruvate Carboxylation, Oxamate-Dependent Oxaloacetate Decarboxylation, Bicarbonate-Dependent MgATP Cleavage, and Carbamoyl Phosphate-Dependent MgADP Phosphorylation Catalyzed by RePC.
| wild-type RePC | H216N | |
|---|---|---|
| pyruvate carboxylation | ||
| kcat (s–1) | 11.0 ± 0.3 | 0.052 ± 0.007 |
| oxaloacetate decarboxylation | ||
| kcat (s–1) | 0.28 ± 0.01 | 0.10 ± 0.01 |
| MgATP cleavage | ||
| kcat (s–1) | 0.0617 ± 0.0002 | 0.0117 ± 0.0007 |
| Kma (μM) | 9.0 ± 0.9 | 82 ± 16 |
| kcat/Km (mM–1 s–1) | 6.86 | 0.14 |
| MgADP phosphorylation | ||
| kcat (s–1) | 1.65 ± 0.07 | 0.022 ± 0.003 |
| Kmb (μM) | 2.8 ± 0.4 | 8.1 ± 2.4 |
| kcat/Km (mM–1 s–1) | 589 | 2.7 |
Km for MgATP.
Km for carbamoyl phosphate.
Steady-State Bicarbonate-Stimulated MgATP Cleavage and MADP Phosphorylation by Carbamoyl Phosphate
From Table 2, H216N showed a 9-fold increase in Km for MgATP compared to that of wild-type RePC while having a kcat that was 19% of that for the wild-type enzyme, resulting in a decrease in kcat/Km of 98% compared to that of wild-type RePC. For MgADP phosphorylation, H216N showed a 2.9-fold increase in Km for carbamoyl phosphate compared to that of wild-type RePC, and the kcat was only 1.3% of that for the wild-type enzyme, resulting in a decrease in kcat/Km of 99.5% compared to that of wild-type RePC (see Table 2).
Single-Turnover Measurements of the Bicarbonate-Dependent MgATP Cleavage Reaction
Figure 3a shows the time courses of bicarbonate-dependent MgATP cleavage reactions at various concentrations of wild-type RePC. At most enzyme concentrations, the reactions were complete within 20 s, with essentially all of the MgATP converted to MgADP. The rates of approach of the reactions to completion are clearly dependent on enzyme concentration, and Figure 3c shows a plot of the observed first-order rate constants derived from fits of the data sets in Figure 3a to single-exponential processes (solid lines in Figure 3a) versus the concentration of wild-type RePC. Figure 3b shows similar time courses for reactions with mutant H216N, and in these cases, reactions were mainly complete within 1200 s, indicating much slower reactions were catalyzed by mutant H216N and those rates were also dependent on the concentration of H216N. Figure 3d shows a plot of the observed first-order rate constants derived from fits of the data sets in Figure 3b to single-exponential processes (solid lines in Figure 3b) versus the concentration of wild-type RePC. The solid line represents a fit of the data to eq 1. Similar experiments were also performed with mutants E218Q and E305A (data not shown). The values of Kd and kcat obtained from fits of the kobs data versus enzyme concentration to eq 1 for wild-type RePC and its mutants are listed in Table 3. The values of Kd were similar for both wild-type and mutant forms of RePC, indicating that none of the mutations had large effects on binding of MgATP to the enzyme. However, the mutations all had very marked effects on kcat, with mutant H216N having a kcat that was ∼1% of that of wild-type RePC and those of E218Q and E305A being ∼2%.
Figure 3.

Time courses of MgATP cleavage in single-turnover experiments at varying concentrations of (a) wild-type RePC [(◆) 1.9, (○) 3.8, (△) 7.6, (●) 9.5, (□) 22, and (▲) 40 μM] and (b) H216N [(◆) 2.2, (○) 4.5, (●) 8.9, (□) 17.8, and (▲) 35.6 μM]. Other reaction conditions are as described in Experimental Procedures. MgATP cleavage is expressed as the ratio of radioactivity present as ADP ([ADP]) to the original radioactivity present as ATP before the start of the reactions ([ATP]o). Solid lines represent nonlinear least-squares regression fits of the data set at each concentration of RePC to a single exponential. (c) Plot of observed first-order rate constants (kobs) derived from the fits of the data in panel a vs the concentration of RePC. The solid line represents a fit of the data to eq 1 (see Experimental Procedures). (d) Plot of observed first-order rate constants (kobs) derived from the fits of the data in panel b vs the concentration of H216N. The solid line represents a fit of the data to eq 1 (see Experimental Procedures).
Table 3. Analysis of the Kinetics of Single-Turnover MgATP Cleavage Reactions in Wild-Type and Mutant RePC.
| enzyme | Kd (μM) | kcat (s–1) | kcat (% of WT value) |
|---|---|---|---|
| WT | 10 ± 4 | 1.400 ± 0.004 | 100 |
| H216N | 12 ± 2 | 0.011 ± 0.001 | 0.8 |
| E218Q | 11 ± 3 | 0.032 ± 0.003 | 2.2 |
| E305A | 8 ± 5 | 0.032 ± 0.010 | 2.2 |
Direct Measurement of Nucleotide Binding to Wild-Type RePC and H216N by Stopped-Flow Fluorescence Spectroscopy
Panels a and b of Figure 4 show the time course of the increase in fluorescence associated with the binding of MgFTP to wild-type RePC and mutant H216N, respectively. Panels c and d of Figure 4 show the time course of the decrease in fluorescence as MgFTP dissociates from MgFTP·wild-type RePC and MgFTP·H216N complexes, respectively. The observed first-order rate constants derived from fits of these data to single exponentials are listed in Table 4. Both binding and dissociation rate constants are similar for wild-type RePC and mutant H216N, indicating that the H216N mutation has little effect on binding of the nucleotide to the enzyme and supporting the observations from the single-turnover MgATP cleavage experiments.
Figure 4.

Time courses of stopped-flow fluorescence experiments to measure either the kinetics of binding of MgFTP to (a) wild-type RePC and (b) mutant H216N or the kinetics of dissociation of (c) the wild-type RPC·MgFTP complex and (d) the H216N·MgFTP complex on displacement by MgATP. Reactions were performed at 30 °C. For panels a and b, 20 μM MgFTP was present in one syringe and 1 μM enzyme in the other. For panels c and d, 20 μM MgFTP and 1 μM wild-type RePC or H216N were present in one syringe and 2 mM MgATP was present in the other. Other reaction conditions were as described in Experimental Procedures.
Table 4. Kinetic Parameters for Binding of MgFTP to RePC and H216N and Its Dissociation from the Enzymes upon Displacement by MgATP, Determined from Fits of Single Exponentials to the Stopped-Flow Fluorescence Data in Figure 5.
| kbindingobs (s–1) | kdisplacementobs (s–1) | |
|---|---|---|
| wild-type RePC | 302 ± 75 | 241 ± 37 |
| H216N | 263 ± 27 | 254 ± 73 |
Discussion
The mutation of His216 to asparagine does not affect the stability of the quaternary structure of the enzyme. Hence, the effects of the mutation are due to direct effects on the reactions of the enzyme and are informative of the role of this residue in those reactions. His216 obviously has an important role or roles in catalysis as mutation of this residue reduces the rate of the overall pyruvate carboxylation reaction by >99%. As might be expected, the main effects of the mutation lie in the reactions that occur in the BC domain of the enzyme where MgATP cleavage and biotin carboxylation occur and not in the CT domain, where pyruvate carboxylation itself occurs. This is evidenced by the relatively weak effect of the mutation of His216 to asparagine on oxamate-stimulated oxaloacetate cleavage that occurs in the CT domain and the much larger effects on MgATP cleavage and MgADP phosphorylation that occur in the BC domain.
Steady-state bicarbonate-dependent MgATP cleavage is coupled with biotin carboxylation and primarily rate-limited in the absence of pyruvate by a combination of the rates of decarboxylation of carboxyphosphate and carboxybiotin.20,21 The decrease in kcat for bicarbonate-dependent MgATP cleavage observed in mutant H216N may be as a result of a change in the rate-limiting step, perhaps to that of the formation of carboxyphosphate. The increase in Km in H216N may be in part due to a decrease in the affinity of the enzyme for MgATP. From the structure of the enzyme,8 His216 appears to play a role in the binding of the nucleotide by interacting with its 3′-hydroxyl group, with N1 of the imidazole ring of His216 positioned 3.0 Å from the 3′-hydroxyl oxygen of the bound ATPγS (see Figure 6). The strong effect of the His216 mutation on MgADP phosphorylation in terms of the reduction in kcat indicates that His216 has a major role in the formation of carboxyphosphate and the cleavage of MgATP. This is because carbamoyl phosphate is an analogue of carboxyphosphate and the MgADP phosphorylation reaction is analogous to the reverse of reaction 1 in Figure 1.18
Figure 6.

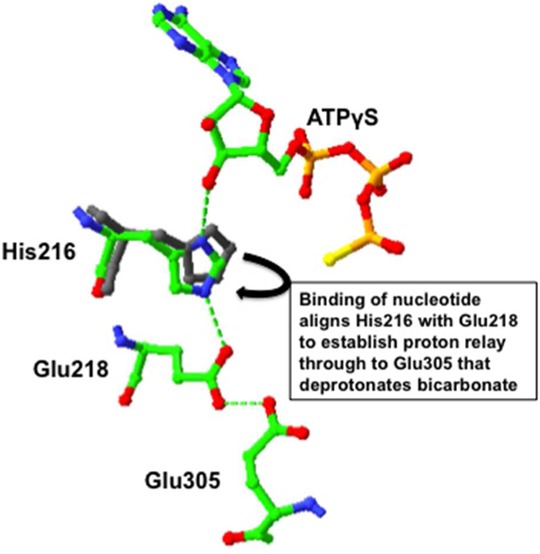
Reaction scheme showing the proposed proton relay involving His216, Glu218, and Glu305 in the BC domain active site leading to the deprotonation of bicarbonate and the transfer of a phosphoryl group from bound MgATP leading to the formation of carboxyphosphate (1). Carboxyphosphate decarboxylates, and the resultant PO43– deprotonates biotin while the carbon dioxide carboxylates it; carboxybiotin and Pi then leave the BC domain (2). Glu305 is deprotonated by the solvent leading to reversal of the proton relay (3). When MgADP dissociates, His216 moves away from Glu218. Note that bound Mg2+ ions are not illustrated for the sake of clarity.
To ascertain whether the major effect of the H216N mutation is on MgATP binding or the reaction in which the transfer of a phosphoryl group from MgATP to bicarbonate occurs, the kinetics of the single-turnover bicarbonate-dependent MgATP cleavage reaction were measured. This represents the first application of this kinetic approach to the study of pyruvate carboxylase and provides a direct way of determining the intrinsic rate constant of MgATP cleavage and the dissociation constant of the enzyme·MgATP complex. The results described above clearly show that the H216N mutation has little effect on the binding of MgATP to the enzyme, but a very strong effect on the catalytic step in the reaction compared to that of wild-type RePC. The lack of an effect of the H216N mutation on nucleotide binding is supported by the similarity of the kinetics of MgFTP binding and dissociation between wild-type and H216N RePC. This indicates that either the interaction between N1 of the imidazole of His216 and the 3′-hydroxyl group of MgATP (see Figure 5) provides little in the way of binding stabilization or the replacement of His216 with asparagine still allows this interaction to occur in much the same way as with histidine. This lack of an effect of the H216N mutation on MgATP binding suggests that its effects on the steady-state bicarbonate-dependent MgATP cleavage reaction are largely due to the inhibition of the MgATP cleavage step itself. In fact, the value of kcat determined from the single-turnover experiment is very similar to the kcat determined in the steady-state experiments for mutant H216N. This indicates that in the steady-state reaction, the phosphoryl transfer step between MgATP and bicarbonate in mutant H216N has become slow enough to be rate-limiting.
Figure 5.
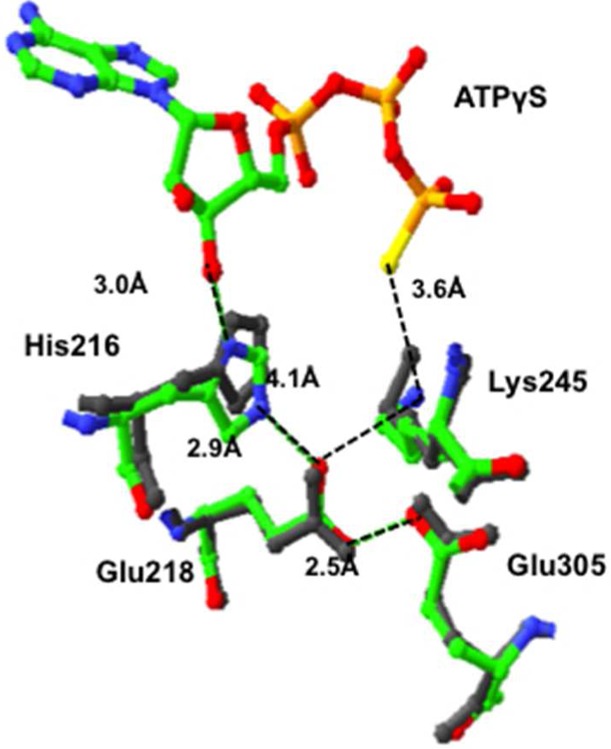
Molecular model showing the position of His216 relative to Glu218 and Glu305 in the BC domain of RePC in a subunit with ATPγS bound (colored residues, Protein Data Bank entry 2QF7) and in a subunit of RePC without nucleotide bound (gray residues, Protein Data Bank entry 3TW7). Distances between atoms are indicated by the black dotted lines and are in units of angstroms. Distances are measured between atoms in the structure of RePC with ATPγS bound (Protein Data Bank entry 2QF7).
Similar single-turnover MgATP cleavage experiments were also performed with mutants E218Q and E305A. The resulting kinetics are remarkably similar to those of the H216N mutant, with the mutations having little effect on MgATP binding but strong inhibitory effects on the phosphoryl transfer reaction itself. Because neither Glu218 nor Glu305 has direct contact with bound MgATP in the BC domain active site, the lack of an effect of mutation of these residues on MgATP binding is not unexpected. As shown in Figure 6, in the active site of the BC domain of RePC with MgATPγS bound, His216 is positioned to potentially form hydrogen bonds with both the 3′-hydroxyl group of the nucleotide and the carboxyl group of Glu218, which in turn could hydrogen bond with that of Glu305. In the BC domain with no bound nucleotide, His216 moves away from and out of alignment with Glu218, so that it no longer interacts so strongly with that residue. This suggests that the binding of the nucleotide correctly positions His216 to optimally interact with Glu218. The presence or absence of the acetyl CoA bound to the subunit does not affect the positioning of these residues, indicating that this is not a locus of activation of the enzyme by this allosteric activator. In the presence of a bound nucleotide, N3 of the imidazole ring of His216 is >1 Å closer to Glu218 than the side chain amino group nitrogen of Lys245, which is closer to the γ-thiophosphoryl group of the bound ATPγS and is indeed important for MgATP binding as shown by Zeczycki et al.9
If we accept the proposal of Zeczycki et al.9 that Glu305 deprotonates bicarbonate concomitant with the transfer of a phosphoryl group from MgATP and there is a proton relay system involving proton transfer from Glu305 to Glu218, how might His216 fit into this scheme? We can postulate that the binding of MgATP to the BC domain active site results in the repositioning of His216 through its interaction with the 3′-hydroxyl group (see Figure 5). This places N3 of the imidazole ring adjacent to the protonated carboxyl group of Glu218. If N3 is unprotonated, it could extract the proton from Glu218, triggering the series of proton transfers that ultimately results in the deprotonation of bicarbonate and the formation of carboxyphosphate. Once biotin has been carboxylated and leaves the BC domain active site, the proton relay would be reversed and dissociation of MgADP from the active site would result in the movement of His216 away from Glu218. The positioning of residues and proton transfer leading to phosphoryl transfer are coordinated by the binding of the nucleotide through its interaction with His216.
If this proton relay scheme is correct, the mutation of any of the three residues involved would be expected to have a similar effect on the catalysis of MgATP cleavage, which is exactly what was observed in the single-turnover MgATP cleavage experiments described above. Adina-Zada et al.13 measured pH profiles of bicarbonate-dependent MgATP cleavage catalyzed by a mutant form of PC from Bacillus thermodenitrificans that lacked biotin and thus decoupled MgATP cleavage from biotin carboxylation. A residue with a pK of 6.6 was observed in the pH profile for the reaction; this residue needed to be deprotonated for the reaction to occur and could correspond to His216.13 In the reverse reaction, His216 would need to be protonated so that the transfer of a proton to carbonate could occur as MgATP is formed from carboxyphosphate.
Apart from being well-conserved among pyruvate carboxylases, residues equivalent to His216, Glu218, and Glu305 in RePC are also well-conserved among other biotin-dependent carboxylases, including acetyl CoA carboxylase, propionyl CoA carboxylase, and methylcrotonyl CoA carboxylase. This suggests that the proposed mechanism of coordination of MgATP binding with catalysis and the proton relay mechanism is also likely to occur in these related enzymes. The biotin carboxylase subunit of acetyl CoA carboxylase also has an ATP grasp fold,22 as does carbamoyl phosphate synthetase,23 which shares some mechanistic similarities with the biotin-dependent carboxylases. However, sequence alignments show that in the carbamoyl phosphate synthetases, the residues equivalent to His216 and Glu305 in RePC are glutamate and serine, respectively (Glu215 and Ser307, respectively, in the E. coli enzyme). In the structure of E. coli carbamoyl phosphate synthetase, Glu215 interacts with both the 2′- and 3′-hydroxyls of the bound nucleotide but is not positioned to interact with Glu217 (the residue equivalent to Glu218 in RePC). Thus, the proposed proton relay mechanism may be restricted to the biotin-dependent carboxylases.
In this work, we have provided evidence that supports the proposal that three residues in the BC domain active site of RePC form a proton relay system to promote the phosphorylation of bicarbonate. We have also proposed that the binding of the nucleotide in the BC domain active site repositions His216 to establish this proton relay system; thus, His216 coordinates catalysis with substrate binding.
Glossary
Abbreviations
- acetyl CoA
acetyl coenzyme A
- PC
pyruvate carboxylase
- RePC
R. etli pyruvate carboxylase
- BC
biotin carboxylase
- CT
carboxyl transferase
- BCCP
biotin carboxyl carrier protein.
The authors declare no competing financial interest.
This work was supported by the National Institutes of Health Grant GM070455.
Funding Statement
National Institutes of Health, United States
References
- Jitrapakdee S.; Wallace J. C. (1999) Structure, function and regulation of pyruvate carboxylase. Biochem. J. 340(Part 1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jitrapakdee S.; Wallace J. C. (2003) The biotin enzyme family: Conserved structural motifs and domain rearrangements. Curr. Protein Pept. Sci. 4, 217–229. [DOI] [PubMed] [Google Scholar]
- Jitrapakdee S.; St Maurice M.; Rayment I.; Cleland W. W.; Wallace J. C.; Attwood P. V. (2008) Structure, mechanism and regulation of pyruvate carboxylase. Biochem. J. 413, 369–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adina-Zada A.; Zeczycki T. N.; Attwood P. V. (2012) Regulation of the structure and activity of pyruvate carboxylase by acetyl CoA. Arch. Biochem. Biophys. 519, 118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adina-Zada A.; Zeczycki T. N.; St Maurice M.; Jitrapakdee S.; Cleland W. W.; Attwood P. V. (2012) Allosteric regulation of the biotin-dependent enzyme pyruvate carboxylase by acetyl-CoA. Biochem. Soc. Trans. 40, 567–572. [DOI] [PubMed] [Google Scholar]
- Attwood P. V.; Wallace J. C. (2002) Chemical and catalytic mechanisms of carboxyl transfer reactions in biotin-dependent enzymes. Acc. Chem. Res. 35, 113–120. [DOI] [PubMed] [Google Scholar]
- Lietzan A. D.; Menefee A. L.; Zeczycki T. N.; Kumar S.; Attwood P. V.; Wallace J. C.; Cleland W. W.; St Maurice M. (2011) Interaction between the biotin carboxyl carrier domain and the biotin carboxylase domain in pyruvate carboxylase from Rhizobium etli. Biochemistry 50, 9708–9723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Maurice M.; Reinhardt L.; Surinya K. H.; Attwood P. V.; Wallace J. C.; Cleland W. W.; Rayment I. (2007) Domain architecture of pyruvate carboxylase, a biotin-dependent multifunctional enzyme. Science 317, 1076–1079. [DOI] [PubMed] [Google Scholar]
- Zeczycki T. N.; Menefee A. L.; Adina-Zada A.; Jitrapakdee S.; Surinya K. H.; Wallace J. C.; Attwood P. V.; St Maurice M.; Cleland W. W. (2011) Novel insights into the biotin carboxylase domain reactions of pyruvate carboxylase from Rhizobium etli. Biochemistry 50, 9724–9737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang S.; Tong L. (2008) Crystal structures of human and Staphylococcus aureus pyruvate carboxylase and molecular insights into the carboxyltransfer reaction. Nat. Struct. Mol. Biol. 15, 295–302. [DOI] [PubMed] [Google Scholar]
- Zeczycki T. N.; St Maurice M.; Jitrapakdee S.; Wallace J. C.; Attwood P. V.; Cleland W. W. (2009) Insight into the carboxyl transferase domain mechanism of pyruvate carboxylase from Rhizobium etli. Biochemistry 48, 4305–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman-Smith A.; Turner D. L.; Cronan J. E. Jr.; Morris T. W.; Wallace J. C. (1994) Expression, biotinylation and purification of a biotin-domain peptide from the biotin carboxy carrier protein of Escherichia coli acetyl-CoA carboxylase. Biochem. J. 302(Part 3), 881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adina-Zada A.; Jitrapakdee S.; Surinya K. H.; McIldowie M. J.; Piggott M. J.; Cleland W. W.; Wallace J. C.; Attwood P. V. (2008) Insights into the mechanism and regulation of pyruvate carboxylase by characterisation of a biotin-deficient mutant of the Bacillus thermodenitrificans enzyme. Int. J. Biochem. Cell Biol. 40, 1743–1752. [DOI] [PubMed] [Google Scholar]
- Rylatt D. B.; Keech D. B.; Wallace J. C. (1977) Pyruvate carboxylase: Isolation of the biotin-containing tryptic peptide and the determination of its primary sequency. Arch. Biochem. Biophys. 183, 113–122. [DOI] [PubMed] [Google Scholar]
- Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood P. V.; Cleland W. W. (1986) Decarboxylation of oxalacetate by pyruvate carboxylase. Biochemistry 25, 8191–8196. [DOI] [PubMed] [Google Scholar]
- Attwood P. V.; Graneri B. D. (1992) Bicarbonate-dependent ATP cleavage catalysed by pyruvate carboxylase in the absence of pyruvate. Biochem. J. 287(Part 3), 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood P. V.; Graneri B. D. (1991) Pyruvate carboxylase catalysis of phosphate transfer between carbamoyl phosphate and ADP. Biochem. J. 273(Part 2), 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeves M. A.; Branson J. P.; Attwood P. V. (1995) Kinetics of nucleotide binding to pyruvate carboxylase. Biochemistry 34, 11846–11854. [DOI] [PubMed] [Google Scholar]
- Branson J. P.; Nezic M.; Wallace J. C.; Attwood P. V. (2002) Kinetic characterization of yeast pyruvate carboxylase isozyme pyc1. Biochemistry 41, 4459–4466. [DOI] [PubMed] [Google Scholar]
- Legge G. B.; Branson J. P.; Attwood P. V. (1996) Effects of acetyl CoA on the pre-steady-state kinetics of the biotin carboxylation reaction of pyruvate carboxylase. Biochemistry 35, 3849–3856. [DOI] [PubMed] [Google Scholar]
- Thoden J. B.; Blanchard C. Z.; Holden H. M.; Waldrop G. L. (2000) Movement of the biotin carboxylase B-domain as a result of ATP binding. J. Biol. Chem. 275, 16183–16190. [DOI] [PubMed] [Google Scholar]
- Thoden J. B.; Raushel F. M.; Wesenberg G.; Holden H. M. (1999) The binding of inosine monophosphate to Escherichia coli carbamoyl phosphate synthetase. J. Biol. Chem. 274, 22502–22507. [DOI] [PubMed] [Google Scholar]