

Abstract
Despite recent advances in genome engineering made possible by the emergence of site-specific endonucleases, there remains a need for tools capable of specifically delivering genetic payloads into the human genome. Hybrid recombinases based on activated catalytic domains derived from the resolvase/invertase family of serine recombinases fused to Cys2-His2 zinc-finger or TAL effector DNA-binding domains are a class of reagents capable of achieving this. The utility of these enzymes, however, has been constrained by their low overall targeting specificity, largely due to the formation of side-product homodimers capable of inducing off-target modifications. Here, we combine rational design and directed evolution to re-engineer the serine recombinase dimerization interface and generate a recombinase architecture that reduces formation of these undesirable homodimers by >500-fold. We show that these enhanced recombinases demonstrate substantially improved targeting specificity in mammalian cells and achieve rates of site-specific integration similar to those previously reported for site-specific nucleases. Additionally, we show that enhanced recombinases exhibit low toxicity and promote the delivery of the human coagulation factor IX and α-galactosidase genes into endogenous genomic loci with high specificity. These results provide a general means for improving hybrid recombinase specificity by protein engineering and illustrate the potential of these enzymes for basic research and therapeutic applications.
Introduction
Targeted genetic engineering is driving progress in new areas of basic biological research, biotechnology, and gene therapy. Site-specific endonucleases, including zinc-finger nucleases (ZFNs),1,2 meganucleases,3,4 TAL effector nucleases (TALENs),5,6 and CRISPR/Cas systems,7,8 have dramatically enhanced the speed and efficiency with which researchers can introduce targeted genetic modifications into cells and organisms.9 Although site-specific nucleases are versatile and promote a broad range of genetic alterations, they rely on cellular DNA repair mechanisms, such as error-prone non-homologous end joining (NHEJ) and homology-directed repair (HDR), to induce custom alterations. The lack of availability of DNA repair pathways within certain cell types, however, may reduce the utility of this technology. In particular, poor induction of HDR via nuclease-induced DNA double-strand breaks (DSBs) or nicks has been shown to be a major limiting factor for achieving high rates of site-specific integration.10 Additionally, off-target DSBs induced by site-specific nucleases11,12 are difficult to comprehensively characterize in the absence of an accompanying donor template13,14 and can be potentially toxic to cells and organisms. Thus, there remains a continued need for the development of new tools capable of achieving highly precise targeted modifications with minimal toxicity.
Site-specific recombinases (SSRs, e.g., Cre, Flp, phiC31, and Bxb1) are a potentially powerful alternative to site-specific nucleases for targeted genetic engineering. SSRs are highly specialized enzymes that promote high-fidelity DNA rearrangements (e.g., integration, excision, or inversion) between defined segments of DNA.15 The strict target specificities demonstrated by many SSR systems, however, have limited their adoption in disciplines that require tools with highly flexible recognition capabilities. To overcome this, various protein engineering strategies have been used to alter SSR target specificity.16 While these approaches permit the design of SSR variants with new properties,17−19 they nevertheless typically lead to the emergence of relaxed specificity,20,21 an undesirable byproduct that limits the utility and safety of these enzymes.
Hybrid recombinases composed of catalytic domains derived from the resolvase/invertase family of serine recombinases (e.g., Gin, Hin, Tn3, and γδ)22 fused to custom-designed Cys2-His2 zinc-finger23,24 or TAL effector DNA-binding domains25 represent a unique solution to this problem (Figure 1a). In particular, zinc-finger recombinases (ZFRs) are a flexible class of chimeric proteins capable of introducing targeted modifications into mammalian cells.26,27 ZFRs promote site-specific recombination between DNA targets that consist of two inverted zinc-finger binding sites flanking a central 20-bp core sequence recognized by the recombinase catalytic domain (Figure 1a). Unlike targeted nucleases and conventional SSR systems, ZFR specificity is the cooperative product of modular site-specific DNA recognition and sequence-dependent catalysis. As such, new ZFRs with diverse targeting capabilities can be generated in a “plug-and-play” manner.26,28−31 In support of this, we have demonstrated that tailored ZFR variants can be rapidly assembled from a library of pre-selected Gin recombinase catalytic domains28,29 (referred to here as Gin α, β, γ, δ, ε, and ζ) and zinc-finger modules.32−36 This customization strategy allows for the design of synthetic recombinases that have the capacity to recognize a broad range of user-defined DNA targets and direct site-specific integration into endogenous genomic loci.29
Figure 1.

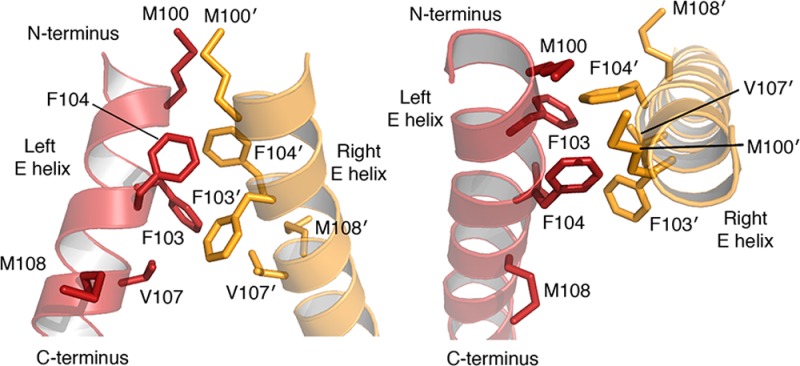
Structure of a zinc-finger recombinase (ZFR) and its dimer interface. (a) Top: ZFR monomers (“left”, red; “right”, yellow) consist of an activated serine recombinase catalytic domain fused to a Cys2-His2 zinc-finger DNA-binding domain. Zinc-finger proteins (ZFPs) can be replaced with TAL effector DNA-binding domains. Model shows the structure of an engineered ZFR, generated from the crystal structures of the γδ resolvase46 and Aart zinc-finger protein70 (PDB IDs: 1GDT and 2I13, respectively). Bottom: Cartoon of a ZFR dimer bound to DNA. Abbreviations are as follows: N indicates A, T, C, or G; R indicates G or A; Y indicates C or T; W indicates A or T; ZFBS indicates zinc-finger binding site. (b) Interactions at the Gin recombinase dimer interface from two vantage points.47 “Left” E helix colored red, “right” E helix colored yellow. Key residues shown as sticks (PDB ID: 3UJ3).
Despite their ability to specifically recognize DNA segments up to 56 bp in length, we previously observed that custom-designed ZFRs targeted integration with low specificity.29 One factor contributing to this is that the protein–protein interactions that govern ZFR-mediated recombination are not selective for the heterodimeric ZFR species. Indeed, expression of any two ZFR monomers required for genomic targeting inevitably leads to the formation of two side-product ZFR homodimers capable of inducing off-target genomic modifications. Similar phenomena have been observed with ZFNs and TALENs, which rely on dimerization of the FokI cleavage domain for DSB induction. To overcome this, numerous studies have utilized dimer interface redesign to generate enzyme variants with improved specificity.37−39 Most notably, structure-guided40,41 and selection-based42,43 approaches have yielded obligate heterodimeric variants of the FokI cleavage domain capable of enhancing ZFN and TALEN cleavage specificity. In addition, mutagenesis of the Cre recombinase dimer interface has led to the isolation of mutants with improved recombination specificity,44 presumably due to destabilization of Cre dimer binding cooperativity. Here, we employ rational design and directed evolution to redesign the serine recombinase dimerization interface and generate a new hybrid recombinase architecture that prevents formation of side-product recombinase homodimers by >500-fold. We show that ZFRs composed of these enhanced catalytic domains demonstrate substantially improved targeting specificity and efficiency, and enable the site-specific delivery of therapeutic genes into the human genome with low toxicity.
Results
Strategy for Dimer Interface Redesign
In order to redesign the Gin recombinase dimer interface and engineer ZFRs that preferentially heterodimerize, we sought to identify the specific amino acid residues that govern recombinase dimerization. To accomplish this, we examined the crystal structures of the γδ resolvase dimer45,46 and the activated, tetrameric configurations of the Gin47 and Sin48 recombinases. We focused our search on residues within the E helix—a key mediator of dimer–dimer interactions between recombinase subunits—and identified five residues that likely associate with one another via hydrophobic interactions: Met 100, Phe 103, Phe 104, Val 107, and Met 108 (all numbers hereafter according to the Gin recombinase; Figure 1b). In accordance with these structural observations, previous studies had revealed that introduction of Cys residues at positions 100, 103, and 107 leads to spontaneous cross-linking of two recombinase monomers.49,50 On the basis of these data, we hypothesized that substitution of these residues with complementary charged amino acids would (i) disfavor association of homodimers by charge and steric repulsion and (ii) promote heterodimer formation through favorable electrostatic contacts.
To evaluate the effect that charged substitutions within the dimer interface have on recombination, we created a collection of recombinase mutants based on the Gin α and ζ catalytic domains29 that contained either Arg (Gin α) or Asp (Gin ζ) substitutions at positions 100, 103, and 107 and evaluated their ability to recombine DNA as homodimers (i.e., Arg-Arg or Asp-Asp) and heterodimers (i.e., Arg-Asp). We determined recombination by split gene reassembly, a previously described method that links recombinase activity to antibiotic resistance.51 Notably, Gin homodimers that contained substitutions at position 103 showed a >10,000-fold reduction in recombination compared to wild-type enzymes (Figure S1). The corresponding heterodimer pair demonstrated a >100-fold increase in recombination compared to the inactivated recombinase mutants; however, no heterodimeric pair recombined DNA as efficiently as the wild-type enzyme (Figure S1). Furthermore, combining charge substitutions did not enhance the efficiency of heterodimer-mediated recombination, presumably due to suboptimal protein–protein interactions between recombinase monomers (Figure S1).
Selection for an Improved Recombinase Dimer Interface
In order to enhance ZFR heterodimer-mediated recombination, we employed directed evolution to select new dimer interface residues that more effectively facilitate heterodimerization. We randomized position 103 and the residues surrounding this region (i.e., positions 100, 104, 107, and 108) within the Gin ζ catalytic domain (Figure 1b) and held the complementary Gin α F103R monomer constant, as preliminary analysis indicated that Arg at position 103 was ∼2-fold more effective at preventing homodimerization than Asp (Figure S1). We selected recombinase variants by split gene reassembly using cells that already harbored the Gin α F103R mutant expression plasmid (Figure 2a). To ensure the formation of the intended heterodimeric species and reduce the possibility of homodimer-mediated survival, we fused the Gin ζ catalytic domain library and the Gin α F103R monomer to zinc-finger DNA-binding domains with orthogonal specificities. After only four rounds of selection, the activity of the mutant ZFR population increased by >500-fold in comparison to the parental Gin ζ F103D mutant (Figure 2b). We sequenced individual recombinase variants from the fourth round of selection and observed a striking degree of sequence similarity at positions 103 and 107, and significant diversity at positions 104 and 108 (Figure 2c). Intriguingly, we found that only ∼5% of selected clones contained a negatively charged residue at any position targeted for randomization. In particular, ∼93% of selected clones contained the native Phe residue at position 103. A nearly identical library that contained a fixed Asp substitution at Gin ζ position 103 yielded no enrichment following multiple rounds of selection in the presence of Gin α F103R (data not shown). These results suggest that Phe 103 could be contributing to critical protein–protein interactions that govern recombinase dimerization.
Figure 2.

Re-engineering the Gin recombinase dimer interface (a) Schematic representation of the split gene reassembly system used to evaluate heterodimer-mediated recombination. Expression of active recombinase variants leads to restoration of the β-lactamase coding sequence and host cell resistance to carbenicillin, an ampicillin analogue. Black triangles indicate cleavage site within the DNA target. TS indicates target site. Base positions 3 and 2 of the “left” half-site are indicated. (b) Selection of Gin ζ mutants that recombine DNA when paired with Gin α F103R. Asterisk indicates the selection step in which incubation time was decreased from 16 to 4 h. (c) Mutation frequencies (%) at positions targeted for randomization in the Gin ζ catalytic domain. Twenty-eight variants were sequenced after four rounds of selection. (d) Recombination by selected Gin ζ mutants on a symmetrical DNA target upon forced homodimerization (red) or on an asymmetrical target when paired with Gin α F103R (orange). Residues selected at each dimer position are indicated including the native Phe 103. (e) Recombination by various pairs of ZFRs that contain the YKWT/R dimer interface, with wild-type control. (f) Recombination specificity of the YKWT/R dimer interface compared to wild-type. DNA targets contained substitutions at base positions 3 and 2. Error bars indicate standard deviation (n = 3).
Virtually all selected Gin ζ recombinase monomers demonstrated high-activity (>25% recombination) in the presence of the complementary Gin α F103R monomer (Figure 2d). Despite the absence of negative selection pressure, we found that the majority of the selected variants also showed a reduction in recombination upon forced homodimerization on a symmetric DNA target (Figure 2d). One selected mutant (Gin ζ M100Y, F104K, V107W, and M108T; hereafter referred to as YKWT) demonstrated a 2000-fold enhancement in recombination on an asymmetric DNA target when paired with Gin α F103R compared to when used as a homodimer on a symmetric target (Figure 2e). This obligate heterodimer also recombined DNA ∼2-fold more efficiently than the counterpart ZFR composed of the wild-type dimer interface (Figure 2e). In order to determine whether the redesigned dimer interface negatively impacted ZFR catalytic specificity, we next evaluated obligate heterodimer-mediated recombination on DNA targets containing mutations within the 20-bp core site recognized by the Gin catalytic domain. Substitutions were introduced at core positions 3 and 2 (Figure 2a), as variations at these sites are highly tolerated by evolved serine recombinases with relaxed target specificity.29 In comparison to ZFRs that contained the wild-type dimer interface, ZFRs composed of the obligate heterodimeric architecture displayed a marked decrease in recombination on a non-cognate target harboring “CC” substitutions at positions 3 and 2 (Figure 2f). Taken together, these data indicate that the serine recombinase dimer interface can be effectively redesigned to favor heterodimerization, and that ZFRs composed of these enhanced catalytic domains display improved recombination efficiency and specificity in bacterial cells.
ZFR Heterodimers Recombine DNA in Mammalian Cells with Improved Specificity
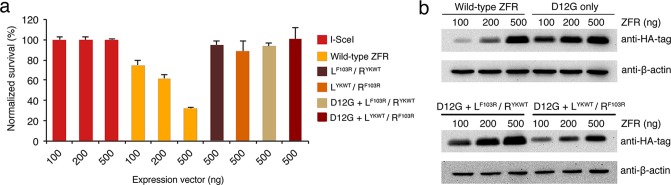
We next investigated whether the redesigned Gin recombinase dimer interface could improve ZFR specificity in mammalian cells. To test this, we introduced the YKWT and F103R substitutions into both the “left” (L) and “right” (R) monomers of a ZFR pair designed to target a 44-bp sequence from a non-protein coding region of human chromosome 4 (Figure 3a).29 Importantly, this ZFR pair provides an opportunity to directly assess the effectiveness of the redesigned dimer interface, as the “left–left” homodimer side product of this ZFR pair previously exhibited substantial recombination activity on the full-length ZFR target site in mammalian cells. We measured recombination using a transient reporter assay that correlates ZFR-mediated recombination with reduced luciferase expression in mammalian cells25,29 (Figure 3a). We co-transfected human embryonic kidney (HEK) 293T cells with a luciferase reporter plasmid containing the full-length ZFR target site and expression vectors for either the L or R ZFR monomers. We then directly compared the fold reduction in luciferase expression to that of 293T cells co-transfected with both L and R ZFR monomers and reporter plasmid. Impressively, we found that ZFR heterodimer pairs that contained the redesigned dimer interface demonstrated substantially improved specificity in comparison to the native ZFRs, reducing off-target homodimer-mediated recombination by >200-fold in both possible configurations (LYKWT/RF103R and LF103R/RYKWT, Figure 3b). However, these obligate heterodimeric pairs recombined DNA ∼2- to 5-fold less efficiently than the standard ZFRs (Figure 3c). Western blot analysis confirmed that the reduction in activity was not due to reduced levels of protein expression (Figure S2).
Figure 3.

Enhanced ZFRs recombine DNA with improved specificity in mammalian cells. (a) Schematic representation of the luciferase reporter system. ZFR-mediated recombination leads to excision of the SV40 promoter and reduced luciferase expression in mammalian cells. TS indicates target site. Black triangles indicate cleavage sites within DNA target. (b) Relative contribution to recombination from “left–right” heterodimers and “left–left” and “right–right” side-product homodimers among various ZFR pairs. The contribution of each homodimer to recombination was calculated by measuring the fold-reduction in luciferase expression in 293T cells transfected with either L- or R-only ZFR monomers, and dividing by the value obtained from cells transfected with both L and R ZFR monomers. (c) Recombination efficiency of wild-type and enhanced ZFR heterodimers with and without the D12G substitution. Recombination was normalized to the FLPe-FRT system. Renilla luciferase expression was used to normalize for transfection efficiency and cell number. Error bars indicate standard deviation (n = 3). (d) Crystal structure of the γδ resolvase (gray surface) in complex with DNA (orange sticks). Regions important for recombinase activity and specificity are highlighted and labeled.
To further improve the recombination efficiency of the obligate ZFR heterodimers, we searched our archive of evolved Gin recombinase catalytic domains24,51 and identified four mutations that were frequently observed among hyperactivated variants: D12G, N14S, K50E, and M70V. Analysis of the crystal structure of an activated mutant of the γδ resolvase catalytic domain indicates that these residues lie near the active site serine and may enhance catalysis by optimally positioning DNA for cleavage and strand exchange (Figure 3d; only D12G is shown).47 We introduced each mutation individually into both the L and R monomers of the obligate heterodimeric ZFR architecture and evaluated their impact on site-specific recombination. Two of the four substitutions (D12G and N14S) enhanced the catalytic activity of the obligate heterodimers (Figure S3). In particular, inclusion of D12G led to an increase in recombination efficiency that exceeded the standard ZFR heterodimer and was similar to FLPe,52 an evolved, highly efficient site-specific recombinase routinely used for cell-line engineering (Figure 3c). Comparison of the relative non-specific contribution of each ZFR homodimer to recombination revealed that the D12G/YKWT and D12G/F103R substitutions (hereafter referred to as enhanced ZFRs; eZFRs) retained the ability to fully prevent recombination by illegitimate homodimers (Figure 3b).
We next examined the portability of the eZFR architecture by introducing each of the enhancing mutations into three different ZFR pairs designed to target unique 44-bp sequences present on human chromosomes 1, 4, and X. Importantly, these ZFR pairs are composed of distinct combinations of Gin recombinase catalytic domains, each with evolved recognition specificities.28,29 As such, this analysis served to evaluate the compatibility of the redesigned dimer interface with our collection of re-engineered Gin catalytic domains. In comparison to wild-type ZFRs, the eZFR pairs targeting human chromosomes X and 4 demonstrated increased recombination efficiency on their intended DNA targets, while the eZFR pair designed to target chromosome 1 showed reduced activity (Figure S4). However, when analyzed on a panel of non-cognate target sites in the context of the luciferase reporter assay, these eZFRs showed improved recombination specificity on the majority of substrates evaluated (28 out of 32) (Figure S5). Taken together, these results demonstrate that dimer interface redesign improves the recombination specificity of custom-designed ZFRs in mammalian cells but that context-dependent interactions between the recombinase dimer interface and target site might influence recombination efficiency.
Dimer Interface Redesign Improves ZFR Integration Specificity
As the primary aim of this work was the improvement of ZFR specificity in the context of targeted genome engineering, we next sought to evaluate whether the eZFR framework improved the specificity of targeted integration in mammalian cells. To this end, we co-transfected HEK293T cells with enhanced and standard ZFR heterodimers designed to target the previously mentioned 44-bp sequence present on human chromosome 4 together with a 4.5-kb donor plasmid containing the cognate ZFR target site and a puromycin-resistance gene (Figure 4a). Importantly, the unmodified “left–left” and “right–right” side-product homodimers for this ZFR pair have been observed to catalyze integration at the selected genomic target. Thus, this ZFR pair allows for readout of the effectiveness of the eZFR architecture for preventing homodimerization29 (Figure 4b). We evaluated ZFR and eZFR-mediated integration by PCR, amplifying the 5′ and 3′ junctions between the donor plasmid and the chromosomal target 72 h after transfection. As anticipated, both eZFR configurations (LYKWT/RF103R and LF103R/RYKWT) catalyzed integration at the intended genomic locus (Figure 4b). In contrast to the standard ZFRs, no site-specific integration was observed after transfection with individual L or R eZFR monomers (Figure 4b). We determined the rate of genome-wide integration of the eZFR heterodimers by puromycin-selection and found that each configuration displayed improved targeting efficiency, with the LF103R/RYKWT configuration yielding a genome-wide integration rate near 0.8% (Figure 4b). These efficiencies are similar to those reported for phiC31-mediated site-specific integration in HEK293 cells.53−55 We next investigated the specificity of eZFR-mediated integration by PCR analysis of individually expanded puromycin-resistant clones. In total, 10 of 16 (63%) and 4 of 16 (25%) clones were positive for targeted integration by eZFRs containing the LF103R/RYKWT and LYKWT/RF103R heterodimeric configurations, respectively (Figure 4c). Compared to the standard ZFR architecture (2 of 17 clones; 11%), the LF103R/RYKWT eZFR heterodimers demonstrated a significant increase in targeted integration (χ2 = 9.23, p < 0.03), while the LYKWT/RF103R eZFR configuration did not (χ2 = 0.99, p > 0.8). DNA sequencing confirmed site-specific integration at the intended genomic locus.
Figure 4.

Site-specific integration into the human genome by enhanced ZFRs. (a) Schematic representation of sequence and location of the ZFR target site on human chromosome 4. Black triangles indicate cleavage sites within DNA target. Red line denotes the approximate position of the ZFR target site. (b) Bulk PCR analysis of HEK293 cells transfected with an empty donor plasmid containing only a puromycin-resistance gene and various ZFR pairs designed to target human chromosome 4. Integration was evaluated in the forward and reverse orientations. GAPDH indicates PCR control. DO indicates donor only (no eZFRs). Genome-wide integration rates indicated beneath each lane. (c) Clonal PCR analysis of puromycin-resistant cells transfected with empty donor and eZFRs in both orientations.
We next evaluated the toxicity of the eZFRs by measuring their impact on cell viability.56 Surprisingly, we observed that the standard ZFRs targeting human chromosome 4 induced high toxicity, leading to a ∼60% decrease in cell viability after 5 days at the highest concentrations tested (Figure 5a). In contrast, eZFRs showed no apparent toxicity and demonstrated a viability profile similar to the rare-cutting and non-toxic I-SceI homing endonuclease (Figure 5a). Furthermore, Western blot analysis revealed no difference in expression between ZFR and eZFR variants (Figures 5b and S2), indicating that the improved safety profiles of the eZFRs are not attributable to reduced expression levels. Together, these findings demonstrate that eZFRs promote targeted integration with improved specificity and demonstrate substantially lower toxicity than ZFRs composed of the wild-type dimer interface.
Figure 5.
Reduced cellular toxicity by enhanced ZFRs. (a) Cell viability of HEK293 cells transfected with increasing amounts of expression vector of standard or eZFRs. Toxicity was normalized to 293 cells transfected with the I-SceI endonuclease. Error bars indicate standard deviation (n = 3). (b) Western blot of lysate from HEK293 cells transfected with increasing amounts of expression vector of standard ZFRs or eZFRs. Samples were taken 48 h after transfection and probed with horseradish peroxidase-conjugated anti-HA and anti-β-actin (loading control) antibodies.
ZFR-Mediated Integration of the Human Factor IX and α-Galactosidase Genes
A potential application of ZFR technology is the site-specific integration of therapeutic genes into the human genome. To explore the feasibility of this goal using eZFRs, we constructed a 6.25-kb donor plasmid containing (i) the ZFR target site from human chromosome 4, (ii) a puromycin-resistance gene, and (iii) the cDNA for one of two disease-associated genes: the human coagulation factor IX (FIX) gene, whose deficiency leads to hemophilia B, and the human α-galactosidase (GLA) gene, which is necessary for lipid metabolism and whose mutation results in the metabolic disorder known as Fabry’s disease (Figure 4a). The phiC31 integrase has previously been used to deliver the human FIX gene into animal models, but these studies were based on random integration into pseudo-recognition sites.57 Co-transfection of HEK293 cells with donor plasmid and eZFRs with the LF103R/RYKWT dimeric configuration led to efficient integration of each therapeutic factor into the intended target site on chromosome 4, and puromycin selection revealed a genome-wide eZFR-mediated integration rate of ∼0.3 and ∼0.4% for the FIX- and GLA-harboring donor plasmids, respectively (Figure 6a). We evaluated eZFR-mediated integration specificity by PCR analysis of individual puromycin-resistant clones and found that 9 of 12 (75%) and 8 of 10 (80%) clones contained FIX and GLA cDNA, respectively, at the intended genomic target site (Figure 6b), which was verified by DNA sequencing. Notably, the specificity of transgene insertion achieved by these eZFRs is similar to those reported for ZFNs,58 TALENs,59 and CRISPR/Cas8 systems, indicating that eZFRs are effective tools for site-specific integration into the human genome. Lastly, toward characterizing the full integration landscape of the eZFRs, we computationally identified four potential off-target sites that contain up to three mismatches compared to the intended genomic target, and evaluated off-target integration from genomic DNA isolated from puromycin-resistant cells that were negative for targeted integration. We observed no transgene insertions at any of the four pseudo-integration sites (Figure S6). These findings indicate that more comprehensive genome-wide approaches are required to determine the full scope of eZFR-mediated off-target modifications.
Figure 6.

Targeted integration of the human coagulation factor IX and α-galactosidase genes by enhanced ZFRs. (a) Bulk PCR analysis of HEK293 cells transfected with eZFRs targeting human chromosome 4 and donor plasmids harboring either the human coagulation factor IX (FIX) or α-galactosidase genes (GLA). Integration was evaluated in the forward and reverse orientations. GAPDH indicates PCR control. DO indicates donor only (no eZFRs). Genome-wide integration rates indicated beneath each lane. (b) Clonal analysis of puromycin-resistant cells transfected with eZFRs and donor plasmids containing the FIX or GLA genes.
Discussion
Advances in targeted genetic engineering are driving progress in many fields, including biotechnology and gene therapy. While site-specific nucleases have facilitated many of these achievements, their capacity for inducing off-target mutations and reliance on DNA repair mechanisms could limit their effectiveness. In particular, the establishment of a new class of tools capable of specifically and safely delivering large payloads into the human genome would be broadly useful across diverse fields, including basic research, gene therapy and synthetic biology. Hybrid recombinases based on the serine resolvase/invertase family of enzymes are a class of reagents capable of delivering genetic payloads into the human genome with potentially few side effects. However, the specificity of these enzymes has proven low, primarily due to the formation of side-product homodimers capable of catalyzing off-target modifications. In this study, we have combined rational design and directed evolution to redesign the serine recombinase dimer interface to prevent formation of these deleterious homodimers, leading to the generation of a new class of hybrid recombinases that preferentially heterodimerize and catalyze site-specific integration into endogenous genomic loci with high specificity. This work expands upon our previous studies that focused on establishing a collection of re-engineered site-specific recombinases capable of targeting a broad range of genomic target sites.29 These results, and in particular our finding that eZFRs specifically introduce the human coagulation factor IX and α-galactosidase genes into the human genome with minimal toxicity, support the continued development of this technology for potential therapeutic applications. However, further studies are required to evaluate the activity and flexibility of these enzymes in primary cells and their potential to modify genomic “safe-harbor” regions with large multi-gene payloads. Future efforts will also focus on establishing optimal delivery methods by evaluating ZFR compatibility with integration-deficient lentiviral vectors60 or adeno-associated virus.61
In comparison to published results in similar cell lines, the eZFRs containing the LF103R/RYKWT dimeric configuration directed site-specific integration with specificities comparable to ZFNs,58 TALENs59 and CRISPR/Cas98 systems; however, the efficiency of eZFR-mediated integration remained lower than those typically observed with site-specific nuclease technologies. One reason for this is that ZFR-mediated recombination is reversible, and as such, insertion events may be excised shortly after integration. The design of integration-competent/excision-defective ZFR variants thus represents one potential solution for enhancing ZFR-mediated integration efficiency. As proof-of principle of this concept, Craig and co-workers recently reported the isolation of excision-competent/integration-defective variants of the piggyBac transposase.62
To our surprise, ZFRs composed of the wild-type dimer interface induced high levels of cellular toxicity. Because ZFR-mediated recombination necessitates the formation of covalent protein–DNA linkages that may activate the NHEJ repair pathway, we suspect that the high levels of cell death induced by the wild-type ZFR pair could be attributed to excessive amounts of cleavage at pseudo-recombination sites by ZFR homodimers or ZFR-mediated rearrangements spurred by the presence of excess homodimers. Thus, the dramatic reduction in toxicity observed with eZFRs can likely be attributed to the ability of the re-engineered dimer interface to prevent recombination by side-product homodimers. The improved efficiency and specificity demonstrated by eZFRs, as well as their ability to promote site-specific integration in the absence of DSBs, suggests that these tools might also be used to modify model organisms refractory to current genome engineering methods. Moreover, this new dimer interface is extensible and should be directly portable to a broad range of hybrid recombinases, including those based on TAL effector DNA-binding domains,5,6,25 and perhaps CRISPR/Cas technology.7,8,63 Although it remains unknown whether the previously described TAL effector architecture supports our expanded collection of Gin recombinase catalytic domains,29 the dimer interface substitutions described here should nonetheless improve the specificity of any TAL effector recombinase heterodimer that consists solely of the wild-type Gin catalytic domain. These enhanced recombinases may also find utility in synthetic biology64 by enabling implementation of complex computational tasks using orthogonal custom recombinases.65−67 Finally, the recombinase dimer interface mutations described in this work may facilitate new advances in the understanding of site-specific recombination.15 In particular, studies focused on the residues targeted in this work may shed new light on the mechanisms that govern the conformational changes during target site cleavage, strand exchange, and religation.68 In summary, our findings provide a general means for improving the targeting efficiency, specificity, and safety of customizable recombinases and illustrate the potential of these enzymes for diverse genome engineering applications, including therapeutic gene transfer.
Materials and Methods
Plasmids
The split gene reassembly vector (pBLA) was derived from pBluescriptII SK (−) (Stratagene) and modified to contain a chloramphenicol resistance gene and an interrupted TEM-1 β-lactamase gene under the control of a lac promoter.51 ZFR target sites were introduced into pBLA as previously described.51 Briefly, a GFPuv stuffer was PCR amplified with the primers GFP-ZFR-ζ-H1-α-P2-XbaI-Fwd and GFP-ZFR-ζ-H1-α-P2-HindIII-Rev and cloned into the SpeI and HindIII restriction sites of pBLA to generate the pBLA-ZFR substrate used for selections. Luciferase reporter plasmids were generated as previously described.29 Briefly, the Simian vacuolating virus 40 (SV40) promoter was PCR amplified from pGL3-Prm (Promega) with the primers SV40-ZFR-BglII-Fwd and SV40-ZFR-HindIII-Rev. PCR products were digested with BglII and HindIII and ligated into the same restriction sites of pGL3-Prm to generate the luciferase reporter vectors pGL3-ZFR-1, 2, 3, ..., 9. ZFR donor plasmids (pDonor; previously pBABE-Puromycin) were constructed as previously described26,29 with the following exceptions: cDNA for the human coagulation factor IX (FIX) and α-galactosidase (GLA) genes (Genecopoeia) were PCR amplified with the primers PstI-CMV-Donor-Fwd and BamH1-ZFR-Donor-Rev. PCR products were digested with PstI and BamH1 and ligated into the same restriction sites of pDonor. Correct construction of each plasmid was verified by sequence analysis (Tables S1 and S2). All primer sequences are provided in Table S3.
Selections and Recombination Assays
To construct the ZFR library, residues 1–115 of the Gin ζ catalytic domain28,29 were PCR amplified from pBLA-Gin-ζ-H1 with the primers pUC18-Prim-2 and Gin-Dimer-Lib-Rev. Mutations were introduced at positions 100, 103, 104, 107, and 108 with the degenerate codon DNK (D: A, T, or G; N: A, T, C, or G; and K: G or T), which encodes all amino acids except Pro, His and Gln. Residues 115 through 144 of the Gin ζ catalytic domain and the H1 zinc-finger protein24 were PCR amplified from pBLA-Gin-ζ-H1with the primers Gin-Dimer-Fwd and pUC18-Prim-1 and fused to the Gin ζ library by overlap PCR with the primers pUC18-Prim-1 and -2. The theoretical size of the ZFR library was ∼8 × 106. Fusion PCR products were digested with SacI and XbaI and ligated into the same restriction sites of pBLA. Ligations were ethanol precipitated and transformed by electroporation into E. coli TOP10F′ (Invitrogen) cells, which were modified to harbor the expression vector pPROLar-Gin-α-F103R-P2 (Clontech Laboratories, Inc.). Library size was determined to be ∼2 × 106. After 1 h recovery in Super Optimal Broth with Catabolite suppression (SOC) medium, cells were incubated with 100 mL of Super Broth (SB) with 30 μg mL–1 of chloramphenicol and cultured at 37 °C with shaking. At 16 h, 30 mL of cells was harvested by centrifugation and plasmid DNA was isolated by Mini-prep (Invitrogen); 3 μg of plasmid DNA was then used to transform E. coli TOP10F′. After 1 h recovery in 5 mL SOC, a portion of cells was plated on solid Lysogeny Broth (LB) with 30 μg mL–1 of chloramphenicol or 30 μg mL–1 of chloramphenicol and 100 μg mL–1 of carbenicillin, an ampicillin analogue. Recombination was determined as the number of colonies on chloramphenicol/carbenicillin plates, divided by the number of colonies on chloramphenicol-only plates. Colony number was determined by automated counting using the GelDoc XR Imaging System (Bio-Rad). The remaining recovery culture was incubated with 100 mL of SB medium with 30 μg mL–1 of chloramphenicol and 100 μg mL–1 of carbenicillin. At 16 h, cells were harvested, and plasmid DNA was purified by Maxi-prep (Invitrogen). Selected ZFRs were isolated by SacI and XbaI digestion and ligated into unmodified pBLA for further selection. After each round of selection, sequence analysis (Eton Biosciences) was performed on individual carbenicillin-resistant clones. Recombination assays with individually selected ZFRs was performed as described above.
ZFR Construction
For ZFR construction, the Gin α, β, γ, δ, ε, and ζ catalytic domains were PCR amplified from the previously described templates pBLA-Gin-α, β, γ, δ, ε, or ζ29 as two fragments with the primers Gin-HBS-D12G-Koz and Gin-YKWT-Rev or Gin-F103R-Rev and Gin-YKWT-Fwd or Gin-F103R-Fwd and Gin-AgeI-Rev. PCR products were fused by overlap PCR with the primers Gin-HBS-D12G-Koz and Gin-AgeI-Rev and cloned into the HindIII and AgeI restriction sites of pBH to generate the new SuperZiF-compatible69 sub-cloning vectors: pBH-D12G-Gin-α-, β-, γ-, δ-, ε-, or ζ-YKWT or F103R (YKWT denotes the mutations M100Y, F104K, V107W, and M108T). Previously constructed zinc-finger domains29 were ligated into the AgeI and SpeI restriction sites of the appropriate pBH-Gin sub-cloning vector to generate pBH-eZFR-L-or-R-1, 2, 3, 4, or 5 (eZFR: enhanced ZFR; L: left eZFR; R: right eZFR). Each eZFR gene was released from pBH by SfiI digestion and ligated into pcDNA 3.1 (Invitrogen) to generate pcDNA-eZFR-L- or-R-1, 2, 3, 4, or 5.
Luciferase Assays
Human embryonic kidney (HEK) 293 and 293T cells (American Type Culture Collection, ATCC) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS; Gibco) and 1% (v/v) antibiotic-antimycotic (Anti-Anti; Gibco). HEK293T cells were seeded onto 96-well plates at a density of 4 × 104 cells/well and established in a humidified 5% CO2 atmosphere at 37 °C. At 24 h after seeding, cells were transfected with 25–50 ng of pcDNA-eZFR-L-1 through 6, 25–50 ng of pcDNA-eZFR-R-1 through 6, 25 ng of pGL3-ZFR, and 1 ng of pRL-CMV (Promega) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. For cells transfected with only one ZFR monomer, empty pcDNA was substituted to maintain equal mass across transfections. At 48 h after transfection, cells were lysed with Passive Lysis Buffer (Promega), and luciferase expression was determined with the Dual-Luciferase Reporter Assay System (Promega) using a Veritas Microplate Luminometer (Turner Biosystems).
Integration Assays
HEK293 cells were seeded onto 24-well plates at a density of 1 × 105 cells/well and maintained in serum-containing media in a humidified 5% CO2 atmosphere at 37 °C. At 24 h after seeding, cells were transfected with 80 ng of pDonor, 10 ng of pcDNA-eZFR-L-1, 10 ng of pcDNA-eZFR-R-1, and 1 ng of pCMV-EGFP (Clontech) using Lipofectamine 2000 according to the manufacturer’s instructions. We note that eZFRs-L- and-R-1 target human chromosome 4. At 24 h after transfection, transfection efficiency was determined by flow cytometry analysis of EGFP expression (FACScan Dual Laser Cytometer; BD Biosciences; FACSDiva software). At 72 h after transfection, cells were harvested, and genomic DNA was isolated using Quick Extract DNA Extraction Solution (Epicentre). ZFR targets and GAPDH were PCR amplified from bulk genomic DNA by nested PCR with the following primer combinations: GAPDH-External-Fwd and GAPDH-External-Rev, GAPDH-Internal-Fwd and GAPDH-Internal-Rev (control); ZFR-1-External-Rev and CMV-External, ZFR-1-Internal-Rev and CMV-Internal (forward integration); ZFR-1-External-Fwd and CMV-External, ZFR-1-Internal-Fwd and CMV-Internal (reverse integration). For pDonor vectors that harbored the human FIX and GLA genes, the following primers were used for internal PCR: ZFR-1-Internal-Rev and FIX-Internal (FIX forward integration); ZFR-1-Internal-Fwd and FIX-Internal (FIX reverse integration); ZFR-1-Internal-Rev and GLA-Internal (GLA forward integration); ZFR-1-Internal-Fwd and GLA-Internal (GLA reverse integration). All primer sequences are provided in Table S1. For colony counting assays, at 72 h post-transfection, cells were split into 6-well plates at a density of 1 × 104 cells/well and maintained in serum-containing media with or without 2 μg mL–1 of puromycin. At 14–18 days, cells were stained with 0.2% crystal violet staining solution, and genome-wide integration rates were determined by counting the number of colonies formed in puromycin-containing media divided by the number of colonies formed in the absence of puromycin. Colony counting was determined by automated counting using the GelDoc XR System (Bio-Rad). For clonal analysis, at 72 after post-transfection, 1 × 104 cells were split onto a 100-mm dish and maintained in serum-containing media with 2 μg mL–1 of puromycin. Individual colonies were isolated with 10- × 10-mm open-ended cloning cylinders (Millipore) with sterile silicone grease (Millipore) and expanded in 96-well plates in the presence of 2 μg mL–1 of puromycin. Genomic DNA was isolated and used as the template for PCR as described above. Sequence analysis (Eton Biosciences) was performed across the 5′ and 3′ junctions for each amplicon.
Western Blots
At 48 h post-transfection, HEK293 cells were harvested and lysed with Laemmli buffer. ZFR expression was analyzed by SDS-PAGE with a Novex 4–20% Tris-Glycine Gel (Invitrogen). Samples were transferred onto a 0.2 μm nitrocellulose membrane and incubated for 2 h in Transfer Buffer (25 mM Tris-Base, 0.2 M glycine, 20% methanol, pH 8.5). Membranes were washed with 1× TBS (50 mM Tris-HCl, 150 mM NaCl, 0.05% Tween 20, pH 7.5) and visualized by automated chemiluminescence visualization using the Gel Doc XR Imaging System. ZFR was detected by horseradish peroxidase conjugated anti-HA antibody (Roche). β-Actin was used as an internal loading control and was detected with peroxidase-conjugated anti-β-actin antibody (Sigma).
Cell Viability Assays
HEK293 cells were seeded onto 24-well plates at a density of 1 × 105 cells/well. At 24 h after seeding, cells were transfected with 25–500 ng of pcDNA-ZFR-L-1, 25–500 ng of pcDNA-ZFR-R-1, 80 ng of pDonor, and 10 ng of pCMV-EGFP. At 30 h post-transfection, cells were collected, and EGFP fluorescence was measured by flow cytometry (FACScan Dual Laser Cytometer; BD Biosciences; FACSDiva software). For each sample, 50,000 live events were collected, and data were analyzed using FlowJo (Tree Star, Inc.). At 5 days post-transfection, cells were again collected, and EGFP fluorescence was measured via flow cytometry as before. ZFR-mediated toxicity was calculated by dividing the number of total viable cells (i.e., EGFP-positive cells) measured at 5 days post-transfection by the number of EGFP-positive cells at 30 h post-transfection.
Acknowledgments
We thank F. Ekman for providing technical assistance. Molecular graphics were generated using PyMol (http://pymol.org). This work was supported by the U.S. National Institutes for Health (DP1CA174426) and The Skaggs Institute for Chemical Biology. T.G. was supported by a U.S. National Institute of General Medicine Sciences fellowship (T32GM080209).
Supporting Information Available
Impact of charged substitutions within the dimer interface; expression of wild-type and obligate ZFR heterodimers; impact of hyperactivating mutations on recombination; portability of the enhanced recombinase architecture; specificity of enhanced recombinases; analysis of off-target effects; amino acid sequences; plasmid sequences; and primer sequences. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† A.C.M.: MedImmune, Gaithersburg, MD, 20878
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bibikova M.; Beumer K.; Trautman J. K.; Carroll D. Science 2003, 300, 764. [DOI] [PubMed] [Google Scholar]
- Urnov F. D.; Miller J. C.; Lee Y. L.; Beausejour C. M.; Rock J. M.; Augustus S.; Jamieson A. C.; Porteus M. H.; Gregory P. D.; Holmes M. C. Nature 2005, 435, 646. [DOI] [PubMed] [Google Scholar]
- Arnould S.; Chames P.; Perez C.; Lacroix E.; Duclert A.; Epinat J. C.; Stricher F.; Petit A. S.; Patin A.; Guillier S.; Rolland S.; Prieto J.; Blanco F. J.; Bravo J.; Montoya G.; Serrano L.; Duchateau P.; Paques F. J. Mol. Biol. 2006, 355, 443. [DOI] [PubMed] [Google Scholar]
- Ashworth J.; Havranek J. J.; Duarte C. M.; Sussman D.; Monnat R. J. Jr.; Stoddard B. L.; Baker D. Nature 2006, 441, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. C.; Tan S.; Qiao G.; Barlow K. A.; Wang J.; Xia D. F.; Meng X.; Paschon D. E.; Leung E.; Hinkley S. J.; Dulay G. P.; Hua K. L.; Ankoudinova I.; Cost G. J.; Urnov F. D.; Zhang H. S.; Holmes M. C.; Zhang L.; Gregory P. D.; Rebar E. J. Nat. Biotechnol. 2011, 29, 143. [DOI] [PubMed] [Google Scholar]
- Reyon D.; Tsai S. Q.; Khayter C.; Foden J. A.; Sander J. D.; Joung J. K. Nat. Biotechnol. 2012, 30, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L.; Ran F. A.; Cox D.; Lin S.; Barretto R.; Habib N.; Hsu P. D.; Wu X.; Jiang W.; Marraffini L. A.; Zhang F. Science 2013, 339, 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P.; Yang L.; Esvelt K. M.; Aach J.; Guell M.; DiCarlo J. E.; Norville J. E.; Church G. M. Science 2013, 339, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T.; Gersbach C. A.; Barbas C. F. III Trends Biotechnol. 2013, 31, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluccio A.; Miselli F.; Lombardo A.; Marconi A.; Malagoli Tagliazucchi G.; Goncalves M. A.; Pincelli C.; Maruggi G.; Del Rio M.; Naldini L.; Larcher F.; Mavilio F.; Recchia A. Mol. Ther. 2013, 21, 1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Foden J. A.; Khayter C.; Maeder M. L.; Reyon D.; Joung J. K.; Sander J. D. Nat. Biotechnol. 2013, 31, 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick T. J.; Fine E. J.; Antico C. J.; Bao G. Nucleic Acids Res. 2013, 41, 9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R.; Lombardo A.; Arens A.; Miller J. C.; Genovese P.; Kaeppel C.; Nowrouzi A.; Bartholomae C. C.; Wang J.; Friedman G.; Holmes M. C.; Gregory P. D.; Glimm H.; Schmidt M.; Naldini L.; von Kalle C. Nat. Biotechnol. 2011, 29, 816. [DOI] [PubMed] [Google Scholar]
- Pattanayak V.; Lin S.; Guilinger J. P.; Ma E.; Doudna J. A.; Liu D. R. Nat. Biotechnol. 2013, 31, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindley N. D.; Whiteson K. L.; Rice P. A. Annu. Rev. Biochem. 2006, 75, 567. [DOI] [PubMed] [Google Scholar]
- Gaj T.; Sirk S. J.; Barbas C. F. III Biotechnol. Bioeng. 2014, 111, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar I.; Hauber I.; Hauber J.; Buchholz F. Science 2007, 316, 1912. [DOI] [PubMed] [Google Scholar]
- Sclimenti C. R.; Thyagarajan B.; Calos M. P. Nucleic Acids Res. 2001, 29, 5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voziyanov Y.; Stewart A. F.; Jayaram M. Nucleic Acids Res. 2002, 30, 1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz F.; Stewart A. F. Nat. Biotechnol. 2001, 19, 1047. [DOI] [PubMed] [Google Scholar]
- Bolusani S.; Ma C. H.; Paek A.; Konieczka J. H.; Jayaram M.; Voziyanov Y. Nucleic Acids Res. 2006, 34, 5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. C.; Thorpe H. M. Mol. Microbiol. 2002, 44, 299. [DOI] [PubMed] [Google Scholar]
- Akopian A.; He J.; Boocock M. R.; Stark W. M. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordley R. M.; Smith J. D.; Graslund T.; Barbas C. F. III J. Mol. Biol. 2007, 367, 802. [DOI] [PubMed] [Google Scholar]
- Mercer A. C.; Gaj T.; Fuller R. P.; Barbas C. F. III Nucleic Acids Res. 2012, 40, 11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordley R. M.; Gersbach C. A.; Barbas C. F. III Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersbach C. A.; Gaj T.; Gordley R. M.; Mercer A. C.; Barbas C. F. III Nucleic Acids Res. 2011, 39, 7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T.; Mercer A. C.; Gersbach C. A.; Gordley R. M.; Barbas C. F. III Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T.; Mercer A. C.; Sirk S. J.; Smith H. L.; Barbas C. F. III Nucleic Acids Res. 2013, 41, 3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot C.; McPherson A. L.; Kolb A. F.; Stark W. M. PLoS One 2011, 6, e19537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirk S. J.; Gaj T.; Jonsson A.; Mercer A. C.; Barbas C. F. III Nucleic Acids Res. 2014, 10.1093/nar/gkt1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal D. J.; Dreier B.; Beerli R. R.; Barbas C. F. III Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier B.; Beerli R. R.; Segal D. J.; Flippin J. D.; Barbas C. F. III J. Biol. Chem. 2001, 276, 29466. [DOI] [PubMed] [Google Scholar]
- Dreier B.; Fuller R. P.; Segal D. J.; Lund C. V.; Blancafort P.; Huber A.; Koksch B.; Barbas C. F. III J. Biol. Chem. 2005, 280, 35588. [DOI] [PubMed] [Google Scholar]
- Isalan M.; Klug A.; Choo Y. Nat. Biotechnol. 2001, 19, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander J. D.; Dahlborg E. J.; Goodwin M. J.; Cade L.; Zhang F.; Cifuentes D.; Curtin S. J.; Blackburn J. S.; Thibodeau-Beganny S.; Qi Y.; Pierick C. J.; Hoffman E.; Maeder M. L.; Khayter C.; Reyon D.; Dobbs D.; Langenau D. M.; Stupar R. M.; Giraldez A. J.; Voytas D. F.; Peterson R. T.; Yeh J. R.; Joung J. K. Nat. Methods 2011, 8, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwell S.; Ridgway J. B.; Wells J. A.; Carter P. J. Mol. Biol. 1997, 270, 26. [DOI] [PubMed] [Google Scholar]
- Nohaile M. J.; Hendsch Z. S.; Tidor B.; Sauer R. T. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo-Sanchez E.; Stricher F.; Paques F.; Isalan M.; Serrano L. Nucleic Acids Res. 2008, 36, 2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. C.; Holmes M. C.; Wang J.; Guschin D. Y.; Lee Y. L.; Rupniewski I.; Beausejour C. M.; Waite A. J.; Wang N. S.; Kim K. A.; Gregory P. D.; Pabo C. O.; Rebar E. J. Nat. Biotechnol. 2007, 25, 778. [DOI] [PubMed] [Google Scholar]
- Szczepek M.; Brondani V.; Buchel J.; Serrano L.; Segal D. J.; Cathomen T. Nat. Biotechnol. 2007, 25, 786. [DOI] [PubMed] [Google Scholar]
- Guo J.; Gaj T.; Barbas C. F. III J. Mol. Biol. 2010, 400, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y.; Vo T. D.; Mendel M. C.; Greenberg S. G.; Wang J.; Xia D. F.; Miller J. C.; Urnov F. D.; Gregory P. D.; Holmes M. C. Nat. Methods 2011, 8, 74. [DOI] [PubMed] [Google Scholar]
- Eroshenko N.; Church G. M. Nat. Commun. 2013, 4, 2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson M. R.; Freemont P. S.; Rice P. A.; Goldman A.; Hatfull G. F.; Grindley N. D.; Steitz T. A. Cell 1990, 63, 1323. [DOI] [PubMed] [Google Scholar]
- Yang W.; Steitz T. A. Cell 1995, 82, 193. [DOI] [PubMed] [Google Scholar]
- Ritacco C. J.; Kamtekar S.; Wang J.; Steitz T. A. Nucleic Acids Res. 2013, 41, 2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenholtz R. A.; Rowland S. J.; Boocock M. R.; Stark W. M.; Rice P. A. Structure 2011, 19, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H. M. J. Biol. Chem. 1994, 269, 31134. [PubMed] [Google Scholar]
- Haykinson M. J.; Johnson L. M.; Soong J.; Johnson R. C. Curr. Biol. 1996, 6, 163. [DOI] [PubMed] [Google Scholar]
- Gersbach C. A.; Gaj T.; Gordley R. M.; Barbas C. F. III Nucleic Acids Res. 2010, 38, 4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz F.; Angrand P. O.; Stewart A. F. Nat. Biotechnol. 1998, 16, 657. [DOI] [PubMed] [Google Scholar]
- Thyagarajan B.; Olivares E. C.; Hollis R. P.; Ginsburg D. S.; Calos M. P. Mol. Cell. Biol. 2001, 21, 3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keravala A.; Lee S.; Thyagarajan B.; Olivares E. C.; Gabrovsky V. E.; Woodard L. E.; Calos M. P. Mol. Ther. 2009, 17, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesner R.; Zhang W.; Noske N.; Ehrhardt A. Human gene therapy 2010, 21, 1104. [DOI] [PubMed] [Google Scholar]
- Cornu T. I.; Cathomen T. Methods Mol. Biol. 2010, 649, 237. [DOI] [PubMed] [Google Scholar]
- Olivares E. C.; Hollis R. P.; Chalberg T. W.; Meuse L.; Kay M. A.; Calos M. P. Nat. Biotechnol. 2002, 20, 1124. [DOI] [PubMed] [Google Scholar]
- Moehle E. A.; Rock J. M.; Lee Y. L.; Jouvenot Y.; DeKelver R. C.; Gregory P. D.; Urnov F. D.; Holmes M. C. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn M. J.; Starker C. G.; McElroy A. N.; Webber B. R.; Riddle M. J.; Xia L.; DeFeo A. P.; Gabriel R.; Schmidt M.; von Kalle C.; Carlson D. F.; Maeder M. L.; Joung J. K.; Wagner J. E.; Voytas D. F.; Blazar B. R.; Tolar J. Mol. Ther. 2013, 21, 1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A.; Genovese P.; Beausejour C. M.; Colleoni S.; Lee Y. L.; Kim K. A.; Ando D.; Urnov F. D.; Galli C.; Gregory P. D.; Holmes M. C.; Naldini L. Nat. Biotechnol. 2007, 25, 1298. [DOI] [PubMed] [Google Scholar]
- Ellis B. L.; Hirsch M. L.; Porter S. N.; Samulski R. J.; Porteus M. H. Gene Ther. 2013, 20, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Burnight E. R.; Cooney A. L.; Malani N.; Brady T.; Sander J. D.; Staber J.; Wheelan S. J.; Joung J. K.; McCray P. B. Jr.; Bushman F. D.; Sinn P. L.; Craig N. L. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, E2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M.; Chylinski K.; Fonfara I.; Hauer M.; Doudna J. A.; Charpentier E. Science 2012, 337, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A. A.; Lu T. K. Annu. Rev. Biomed. Eng. 2012, 14, 155. [DOI] [PubMed] [Google Scholar]
- Friedland A. E.; Lu T. K.; Wang X.; Shi D.; Church G.; Collins J. J. Science 2009, 324, 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuti P.; Yazbek J.; Lu T. K. Nat. Biotechnol. 2013, 31, 448. [DOI] [PubMed] [Google Scholar]
- Bonnet J.; Yin P.; Ortiz M. E.; Subsoontorn P.; Endy D. Science 2013, 340, 599. [DOI] [PubMed] [Google Scholar]
- Bai H.; Sun M.; Ghosh P.; Hatfull G. F.; Grindley N. D.; Marko J. F. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez B.; Schwimmer L. J.; Fuller R. P.; Ye Y.; Asawapornmongkol L.; Barbas C. F. III Nat. Protoc. 2010, 5, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal D. J.; Crotty J. W.; Bhakta M. S.; Barbas C. F. III; Horton N. C. J. Mol. Biol. 2006, 363, 405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.