

Abstract
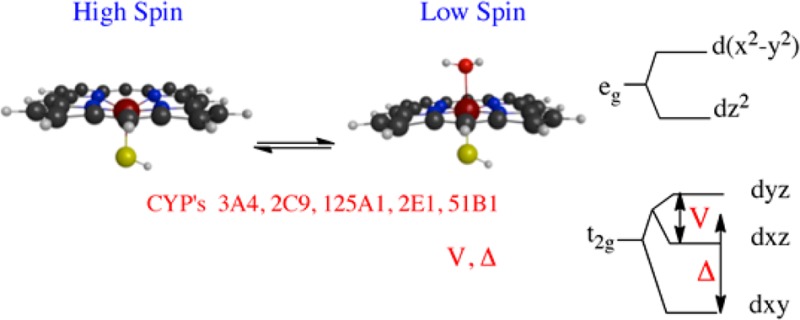
The heme-containing cytochrome P450s exhibit isoform-dependent ferric spin equilibria in the resting state and differential substrate-dependent spin equilibria. The basis for these differences is not well understood. Here, magnetic circular dichroism (MCD) reveals significant differences in the resting low spin ligand field of CYPs 3A4, 2E1, 2C9, 125A1, and 51B1, which indicates differences in the strength of axial water ligation to the heme. The near-infrared bands that specifically correspond to charge-transfer porphyrin-to-metal transitions span a range of energies of nearly 2 kcal/mol. In addition, the experimentally determined MCD bands are not entirely in agreement with the expected MCD energies calculated from electron paramagnetic resonance parameters, thus emphasizing the need for the experimental data. MCD marker bands of the high spin heme between 500 and 680 nm were also measured and suggest only a narrow range of energies for this ensemble of high spin Cys(S–) → Fe3+ transitions among these isoforms. The differences in axial ligand energies between CYP isoforms of the low spin states likely contribute to the energetics of substrate-dependent spin state perturbation. However, these ligand field energies do not correlate with the fraction of high spin vs low spin in the resting state enzyme, suggestive of differences in water access to the heme or isoform-dependent differences in the substrate-free high spin states as well.
The cytochrome P450s (CYPs) are thiolate-ligated heme-containing monooxygenases that oxidize many different functional groups or hydrocarbons, and they play critical roles in biosynthetic pathways, or detoxication, in virtually every organism.1,2 CYPs are both drug targets and major components of drug metabolism. In the ferric, resting enzyme, the heme cofactor of different CYPs equilibrates between the five-coordinate high spin state and the six-coordinate low spin state, defined by the constant Kspin = [high spin]/[low spin].3−5 A water molecule, or molecules, in the active site provide(s) the sixth ligand to the low spin heme iron.6,7 The Kspin in the resting state is CYP isoform dependent, although the molecular basis for the differences is unknown. In many cases, binding of substrates results in displacement of the water and conversion to higher fraction of five-coordinate high spin heme with a higher redox potential, and this allows for one electron reduction by NADPH-dependent protein redox partners to begin the catalytic cycle, as elegantly described originally by Sligar et al.3,8 Whereas biosynthetic CYPs typically exhibit tight coupling of substrate binding and spin state conversion, hepatic isoforms that dominate drug metabolism exhibit poor coupling; different drugs or substrates yield different fractions of high spin heme with variable spin state equilibria.8,9 Although the substrate-dependent spin state regulation for some CYPs is well documented, the molecular basis for the variability in the substrate-free ferric spin equilibrium has not been determined. For example, substrate-free CYP3A4 is only ∼19% high spin but exhibits an unexpectedly high rate of heme reduction in the absence of any bound substrate.10 In contrast, CYP2E1 and CYP125A1 are significantly high spin even in the absence of substrates.11,12 These examples amplify our incomplete understanding of the role of spin state in CYP catalysis, and they suggest that the local ligand field of the substrate-free low spin state is a source of variability in CYP isoform-dependent spin state control.
The ligand field of the substrate-free states is also important in drug design aimed to inhibit CYPs. Differences in axial ligand field of the substrate-free CYPs necessarily contribute to differential binding affinities of drugs that target the heme, such as antifungal azoles or anticancer drugs such as anastrozole or abiraterone.13,14 To the extent that axial water “competes” for heme ligation with these drugs, the energy of the water-iron bond contributes to the apparent binding affinity of the drug. This aspect of CYP inhibitor design has not been acknowledged. Here we demonstrate, for substrate-free states, significant differences in the energies of porphyrin-to-metal near-infrared (nIR) charge transfer (CT) bands among CYP51B1 and CYP125A1 from Mycobacterium tuberculosis, and partially solubilized forms of human hepatic CYP3A4, CYP2E1, and CYP2C9.
Materials and Methods
Protein Sample Preparation
Buffer salts were purchased from Sigma (St. Louis, MO, USA). Deuterium oxide (99% D2O) and glycerol (98%, -d8) were purchased from Cambridge Isotopes (Andover, MA). CYP3A4 was expressed and purified as previously described.15 CYP2C9 (hepta mutant) was constructed as previously described16 with the exception of a hexa His rather than a tetra His C-terminal sequence to facilitate purification. The enzyme was expressed in DL39 Escherichia coli and purified as described for CYP3A4. CYP2E1 was received as a generous gift from the lab of Dr. Emily Scott (University of Kansas). CYP51B1 from M. tuberculosis (Mtb) was purified as previously described.17 CYP125A1, also from Mtb, was prepared as previously described.18
For measurement of conventional absorbance spectra, protein samples were diluted directly from their respective storage buffer into 100 mM potassium phosphate (pH = 7.4) containing 10–20% glycerol. The final protein concentration ranged from 1 to 6 μM based on CYP450 content, as determined using the methods of Omura and Sato19 (note: to normalize the CYP51B1 spectrum, the published molar extinction coefficient at 419 nm of 134 mM–1 cm–1 was used17). For EPR spectral acquisitions, the proteins were concentrated to >100 μM CYP450 in the same buffer. For MCD samples, the protein sample buffer was sufficiently exchanged for deuterated phosphate to avoid contamination of unwanted C–H, N–H, and O–H vibrational overtones in the near-infrared spectral region. Deuterated buffers were prepared using 1 M potassium phosphate (pD = 7.4–7.5) stock solution that was dissolved and lyophilized three times in D2O. The proteins were concentrated (>250 μM) several times in deuterated buffer containing 20% glycerol-d8, using Centriprep (Millipore) 30 MWCO centrifugal filters before adding an additional 35% glycerol-d8 as a glassing agent to yield a final buffer concentration ranging from 100 and 200 mM phosphate depending on the solubility of the isoform, + 55% glycerol-d8. CYP450 content was measured for each sample solution immediately prior to acquisition of MCD spectra. The final protein concentrations utilized for MCD were 400 μM CYP3A4, 262 μM CYP2E1, 475 μM CYP2C9, 700 μM CYP125A1, or 770 μM CYP51B1.
Magnetic Circular Dichroism
MCD spectra were collected using an Aviv 40DS spectropolarimeter and a high-field superconducting magneto-optical cryostat (Cryo-Industries SMC-1659 OVT) equipped with a variable-temperature sample compartment. MCD intensities are measured as differential absorbance of left (σ–) and right (σ+) circularly polarized light (ΔA = AL – AR), following the sign convention of Piepho and Schatz.20 MCD spectra are measured in units of θ (mDeg) with the relationship, θ (mDeg) = 32982*ΔA. All spectra were baseline corrected via measurement and subtraction of a spectrum collected at zero field. For cryogenic temperature measurements (4.2–4.7 K), depolarization of the sample was verified to be less than 10% prior to sample data collection via CD measurement of a solution standard of nickel(II) tartrate. Near infrared absorption was detected with a nitrogen-cooled 1 × 1 mm InGaAs detector (Sciencetech Inc., USA). Visible spectra were collected from 500 to 850 nm in 2 nm steps using an integration time of 2 s and bandwidth of 4 nm; nIR spectra were collected from 900 to 1500 nm in 5 nm steps, 2 s integration time, and a bandwidth of 10 nm. Measurements at room temperature are expressed as Δε (M–1 cm–1 T–1) to account for the field dependence, while measurements at cryogenic temperature are expressed as Δε (M–1 cm–1). Preparation of a sample for MCD was performed by depositing 90 μL of protein solution upon a 1.5 cm diameter circular quartz plate placed on an in-house fabricated aluminum sample mount fitted with a 0.1 cm thick Teflon spacer. The solution was sealed between a second quartz plate and sandwiched between an aluminum housing with hand-tightened screws. The sample mount was secured to a rod containing a temperature sensor fit for the appropriate optical cryostat before being introduced in between the poles of the superconducting magnet.
EPR Spectroscopy and Calculation of Axial (Δ/λ) and Rhombic (V/λ) Crystal Field Terms
Conventional continuous-wave (CW) EPR spectra were measured on a Bruker ELEXSYS E540 X-band spectrometer with ER 4102 ST resonator and either a liquid nitrogen quartz insertion dewar or a Bruker ER 4112 HV helium flow cryostat. The samples were placed in 3 mm outer diameter (OD) EPR tubes and frozen in liquid nitrogen prior to acquisition at temperatures between 15 and 77 K. Accurate spectral g-values were obtained by fitting the CW-EPR spectra using EasySpin software, a toolbox contained within the MATLAB environment.21
Calculation of the axial (Δ/λ) and rhombic (V/λ) crystal field terms expressed in units of the spin–orbit coupling constant (λ) for ferric ion was performed via the methods of Taylor.22 Briefly, the fitted g-values obtained via least-squares regression analysis in EasySpin were applied to the following formalism:
UV/vis Absorbance Analysis and Determination of Kspin
Absorbance measurements were conducted at 25 °C on an Olis Modernized Aminco DW-2 (Olis, Inc., Bogart, GA) dual beam spectrophotometer equipped with a Julabo F30-C compact refrigerated circulator (Julabo USA, inc., Allentown, PA). Each binding experiment required 500 μL initial sample volume using a 0.1 × 1 cm path length quartz cuvette. The spectra were subjected to a least-squares fitting algorithm written in the Python programming language and adapted for use in IGOR Pro 6.1 (Wavemetrics, Lake Oswego, OR).
Results
UV/vis Spectra of Substrate-Free CYPs
Standard UV/vis spectra of several CYP isoforms, in the wavelength range typically used to monitor ligand binding, are shown in Figure 1. To calculate the fraction of enzyme in the high spin state and the value of Kspin for each ligand-free CYP, the absorbance spectra were fit to experimentally derived basis spectra for the pure high spin, low spin, and P420 species as determined by principle component analysis and described previously.23,24Kspin was then calculated by the relation: Kspin = [% high spin]/[% low spin]. The Kspin values are summarized in Table 1, along with MCD parameters determined in experiments described below. The UV/vis spectrum of each of these CYPs is essentially identical to previous results in the literature. The unusually high fraction of high spin heme with ligand-free CYP125A1 and CYP2E1 has been well established,11,18 as is the preponderance of low spin heme for the other CYPs. We report the results here for all of the proteins as the basis for direct comparison within a single data set.
Figure 1.
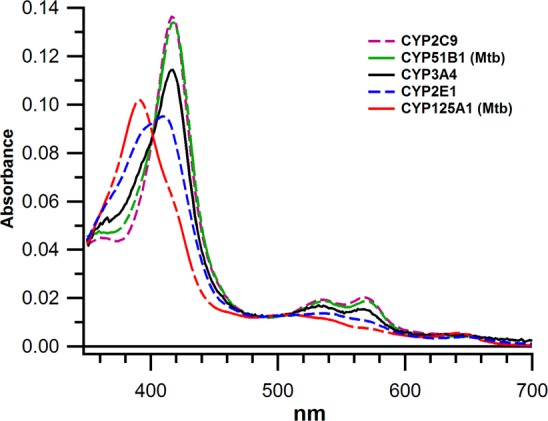
UV/vis absorbance of ligand-free CYPs at 298 K. The Soret and α,β bands at 520–680 nm are shown for CYP3A4, CYP2C9, CYP2E1, CYP51B1, and CYP125A1. The Soret of pure high spin heme is located at ∼390 nm and ∼417 for pure low spin. All spectra have been normalized to 1 μM CYP450.
Table 1. Spin State, Absorbance, and MCD Parameters.
| isoform | % HSa 298 K | Kspin 298 K | λmax nIR CT, 298 K (nm)b | nIR CT kcal/mol 298 K | λmax nIR CT, 4.2–4.7 K (nm)d | nIR energy kcal/mol | CT αβ, 298 K (nm) | Vis energy kcal/mol |
|---|---|---|---|---|---|---|---|---|
| CYP3A4 | 19 | 0.23 | 1065 | 26.9 | 1050 | 27.2 | 650 | 44.0 |
| CYP2C9c | 6 | 0.063 | 1085 | 26.4 | ||||
| CYP125A1 | 89 | 11 | n/a | n/a | 1050 | 27.2 | 650 | 44.0 |
| CYP51B1 | 9 | 0.10 | 1095 | 26.1 | 1095 | 26.1 | 652 | 43.9 |
| CYP2E1 | 58 | 1.6 | 1115 | 25.6 | 1130 | 25.3 | 658 | 43.4 |
R2-correlation coefficient for spectral fits ≥ 0.998.
CT = charge transfer, nIR = near-infrared.
No 298 K data available for CYP2C9.
CYP3A4, CYP2C9, CYP51 at 4.2 K; CYP2E1 and CYP125A1 at 4.7 K.
Magnetic Circular Dichroism (MCD)
MCD was used to compare the low spin states of these CYPs in greater detail. Although MCD has been used to characterize some soluble CYPs, microsomal CYPs have not been characterized, presumably due to difficulty in obtaining purified protein at the high concentrations required for investigation of the near-infrared (nIR) charge transfer (CT) transitions (∼400 μM). The temperature-dependent (C-term) nIR CT bands observed by MCD uniquely report on the axial and rhombic distortion of the heme iron ligand field in the low spin state, without overlapping bands from high spin electronic transitions.25−28 Unlike visible absorption bands, this spectroscopic feature reports exclusively on the low spin structure, and it is much more sensitive to rhombic strain.29 This MCD marker is distinct from, although similar to, the EPR gz values and derived axial (Δ/λ) and rhombic (V/λ) crystal field terms that describe the ferric low spin heme ligand field.22,30,31 The MCD spectra of substrate-free, partially solubilized, engineered variants of human CYPs and bacterial isoforms acquired between 4.2 and 4.7 K at 6 T field are shown in Figure 2. A striking difference in the wavelength maxima and overall spectral shape is apparent. The corresponding spectra at 298 K are also shown in Figure 2, and the data are summarized along with the absorbance data in Table 1. There is a remarkable absence of low spin signal for CYP125A1 at the higher temperature consistent with its large fraction of high spin heme, even with no ligand present. At the lower temperature, which is known to favor the low spin heme,32 we were able to detect the low spin nIR transition of CYP125A1. We were unable to obtain spectra at the high temperature for CYP2C9 for unknown reasons. Regardless, the increase in low spin heme observed at low temperatures by other methods is also observed in these MDC spectra, for several isoforms. Thus, at room temperature the differences in ligand field are less pronounced than at low temperatures.
Figure 2.
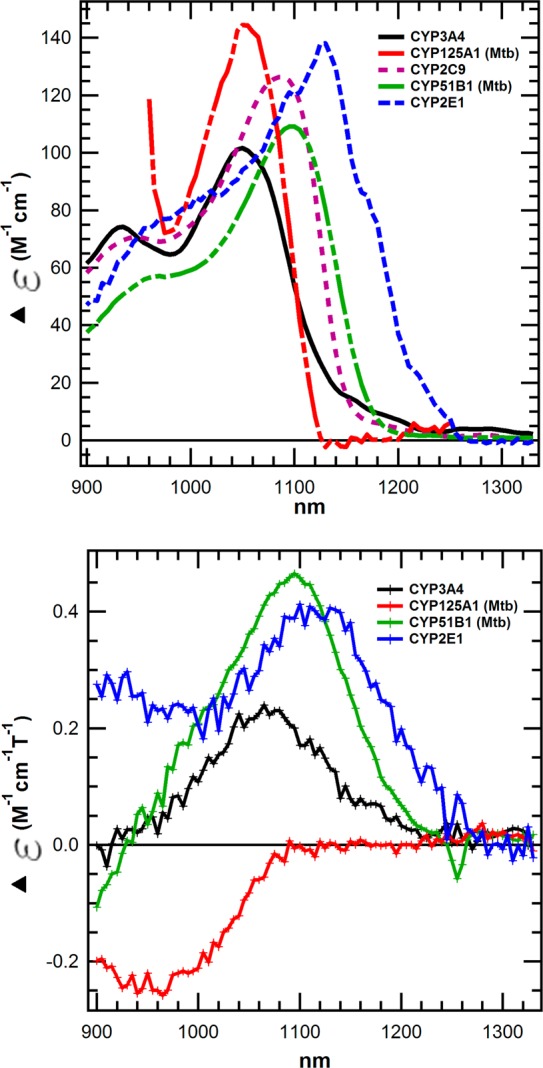
Top: Near IR MCD spectra of CYP3A4, CYP125A1, CYP2C9, CYP51B1, and CYP2E1 at 4.2–4.7 K and 6 T. Bottom: 6 T near nIR MCD spectra of CYP3A4, CYP125A1, CYP51B1, and CYP2E1 at 298 K. Note that the predominantly high spin nature of CYP125A1 at 298 K (red) precludes measurement of the low spin transition. All samples were prepared in 100–200 mM KPi (pH = 7.4) + 55% glycerol-d8.
The difference in energy of the CT transitions is substantial (range ΔΔG = 1.9 kcal/mol at ∼4.2 K; ΔΔG = 1.2 kcal/mol at 298 K) and is likely due to differences in energy of the low lying iron t2g (dyz) orbitals into which the pyrrole nitrogen π electrons are promoted from both a1u and a2u HOMOs25 (Scheme 1). It is unlikely that the differences result from changes in porphyrin molecular orbitals. The different energies of the MCD transition indicate that the sixth axial water ligand provides increasing ligand field strength33 in the order CYP2E1 > CYP51 > CYP2C9 > CYP3A4 ≈ CYP125A1 (Table 1). This is the first documentation of nIR MCD for any human CYPs and the rather remarkable differences in ligand field among substrate-free CYPs despite the common axial ligand, H2O. It is widely presumed that the thiolate axial ligand is sufficiently “dominant” to render the axial water ineffective in controlling the axial field strength. Mutations can modestly shift the nIR transitions of ligand-free CYPs, but shifts of 50–80 nm typically require ligand substitution.34,35
Scheme 1. Schematic Free Energy Profile of the Macroscopic Kspin and the Differences in the Low Spin States Revealed by MCD.

The right side includes the porphyrin HOMOs and the iron d-orbitals with ligand field parameters V and Δ indicated. The nIR charge transfer band energies for different CYPs correspond to different axial ligand field strengths in the relative order shown, with CYP2E1 and CYP125A1 having the strongest and weakest axial ligands, respectively. The lack of LFER between Kspin and these ligand field energies suggests a contribution of the high spin states to isoform-dependent differences in Kspin.
In addition to the nIR MCD data, we collected spectra in the region of the α,β bands near 650 nm at 298 K. In this region, a negative intensity MCD band at ∼650 nm is assigned as a LMCT (S(π) → Fe3+) transition (36) that is uniquely diagnostic for the presence of high spin heme. In addition to this MCD feature, the spectra shown in Figure 3 include several additional Cys thiolate (S–) → Fe3+ charge transfer bands originating from the high spin species that distinguish the two spin states in the CYP MCD spectrum.37 Clearly, the CYP2E1 and CYP125A1 yield the largest high spin intensity, as expected. Interestingly, the spectral minima for these isoforms are significantly different with CYP125A1 and CYP2E1 having minima at 652 and 658 nm, respectively. The weak signals from CYP3A4 and CYP51B1 yield minima at ∼650 and 652 nm, respectively. Thus, the apparent strength of the thiolate ligand in the high spin species increases in the order CYP2E1 > CYP51B1 > CYP3A4 ≈ CYP125A1. Because of the weak high spin signals for some of these isoforms, we note that this ranking is approximate. However, it is clear that the ligand field of these isoforms is also modestly different in the high spin state. The range of energies for these isoforms at 298 K is ΔΔG = 0.5 kcal/mol, which is much less than the range observed for the low spin axial field strength.
Figure 3.

The 6 Tesla MCD spectra of the α,β region acquired at 298 K for CYP3A4, CYP2E1, CYP51B1, and CYP125A1 reveals large differences in high spin content. The negative MCD feature centered ∼650 (inset) is only present in high spin CYPs and represents thiolate → Fe(III) LMCT for the high spin enzyme.
EPR Spectra and Comparison to MCD
The CW EPR spectra of the substrate free CYPs 3A4, 51B1, 125A1, and 2C9 were measured. The normalized spectra are shown in Figure 4. We observed results directly analogous to those previously reported for many CYPs, and the data are summarized in Table 2. The EPR spectra clearly indicate low spin, H2O-ligated heme in each case. The crystal field parameters for rhombic (Δ/λ) and axial (V/λ) distortion are included.
Figure 4.
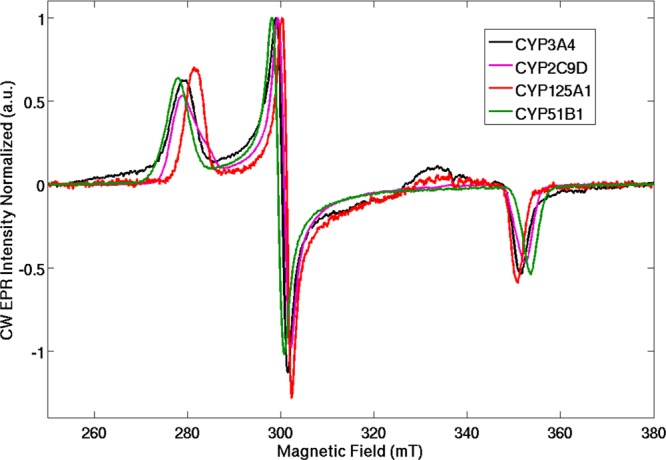
CW EPR spectra of ligand-free CYPs 3A4, 2C9, 125A1, and 51B1. The EPR spectral g-values were fit using EasySpin as described in the text to extract accurate values that were used in the calculation of crystal field terms.
Table 2. EPR Parametersa.
| CYP isoform | gz | gy | gx | Δ/λ | V/λ | V/Δ | Eyz/λ | ECT (calc) cm–1 | 4.2 K predicted nIR CT kcal/mol | 4.2 K predicted nIR CT nm |
|---|---|---|---|---|---|---|---|---|---|---|
| 3A4 | 2.421 | 2.249 | 1.921 | 5.338 | 4.909 | 0.920 | 4.234 | 9538 | 27.3 | 1048 |
| 2C9 | 2.429 | 2.252 | 1.917 | 5.256 | 4.808 | 0.915 | 4.156 | 9432 | 27.0 | 1060 |
| 125A1 | 2.400 | 2.243 | 1.926 | 5.412 | 5.147 | 0.951 | 4.378 | 9735 | 27.8 | 1027 |
| 51B1 | 2.436 | 2.260 | 1.913 | 5.063 | 4.729 | 0.934 | 4.052 | 9289 | 26.6 | 1077 |
EPR data for CYP2E1 not available.
In addition, the EPR parameters were used to calculate the expected energies for the nIR MCD transitions, according to the method of Gadsby and Thomson.33 The predicted and experimentally measured energies are compared in Figure 5. The results emphasize the discrepancy between the predicted MCD transitions based on EPR parameters and these experimental results, for these thiolate-heme proteins. The experimental EPR parameters poorly predict the MCD transitions for all the isoforms except CYP3A4.
Figure 5.
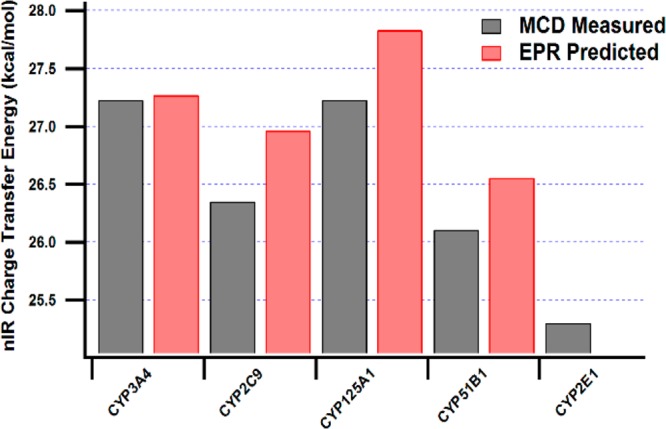
Measured MCD nIR CT energies for the CYPs studied here versus those predicted based on the EPR-derived axial (Δ/λ) and rhombic (V/λ) crystal field terms as described by Gadsby and Thomson.33 Note: no EPR data are available for CYP2E1. The results emphasize the inaccuracy of low spin ligand field energy calculations based on EPR correlations for these CYPs and the necessity for direct measurement of the nIR CT transition by MCD.
Discussion
This work extends a vast array of previous spectroscopic work by many laboratories to characterize the ferric spin states of CYPs. The major new information here includes a direct comparison by MCD and EPR of several CYP isoforms including human CYPs that historically have been inaccessible to study by MCD spectroscopy. Although low spin complexes of CYPs with heteroatomic ligands such as azoles, thiols, or alcohols in the sixth axial position have been characterized by MCD and EPR, the H2O-bound structures have not received detailed scrutiny. In fact, these MCD spectra are the first of any human CYP and provide the first clear evidence that their ligand fields in the resting states differ from each other and from some bacterial CYPs, corresponding to differences in the strength of the axial Fe3+–H2O bond. The temperature dependence of the CT transition further amplifies the differences in resting states of these isoforms. Whereas CYP3A4 and CYP51B1 exhibit either a spectral blue shift or no shift upon increasing the temperature from 4.2 to 298 K, CYP2E1 yields an unusual red shift. Further studies are needed to explain the differential temperature dependence, but the temperature effects highlight the fundamental difference of water-ligated states.
These differences have functional implications for the energetics of substrate-dependent or inhibitor-dependent shifts in the ferric spin equilibrium. A common strategy for the design of CYP inhibitors is to incorporate azole or pyridine fragments aimed to displace the axial H2O and form iron–nitrogen coordinate bonds. The net gain in free energy of inhibitor binding depends not only on the strength of the resulting iron–nitrogen bond but also on the energy that is forfeited in the Fe3+–H2O that must be displaced. The results here indicate that identical fragments could have modestly different intrinsic binding affinities for different CYP isoforms simply because the resting states of these enzymes vary in the strength of the starting Fe3+–H2O bond. It will be important to understand further the mechanism for these significant differences in ligand field and how the protein architecture and solvation contribute.
Differences in the ligand field in the high spin state of the substrate-free enzymes are also evident, albeit with a much narrower range of energies in the proximal Cys (S–) → Fe(III) bond. This is evident in Figure 3. The range in energies for these high spin transitions is only ∼0.4 kcal/mol, and presumably reflects subtle differences in second sphere hydrogen bonding to the proximal cys thiolate as well as other subtle environmental factors. Because of the low intensity of the MCD bands in the α,β region, and the contribution of multiple electronic transitions to these spectral features, we are cautious to not assign relative energies of the axial thiolate ligand on their basis. However, the very modest differences in ligand field we observe for these high spin transitions are unlikely to drive the larger differences in ferric spin equilibrium between these CYP isoforms.
It is, however, striking that the overall Kspin values do not correlate with the strength of the axial Fe3+–H2O bonds of the low spin states (Table 1); there is no linear free energy relationship (LFER) between Kspin and the low spin axial ligand field energy. For example, CYP2E1 has an unusually high fraction of high spin ferric heme in the absence of substrate, and the lowest energy CT transition, but CYP125A1 is also largely high spin with the highest energy CT band. Together, these observations emphasize that the ferric spin equilibrium is not controlled solely by the strength of the axial water interaction in the low spin state. Apparently, modest compensatory differences in other features of the low spin or high spin states contribute to the macroscopic Kspin. Possibly, differences in Kspin reflect differences in solvent accessibility to the heme. For example, facile water access to the heme in CYPs with low Kspin values may push the ferric spin equilibrium to the low spin state even though the resulting Fe3+–H2O bond may be less stable. Alternatively, isoforms with restricted access to very hydrophobic active sites may favor a high Kspin value even though the resulting Fe3+–H2O bond is very strong. Speculatively, although, the microenvironment differentially tunes the water-dependent ligand field when water is present, differences in protein architecture and solvation control access of water to the heme and hence the ferric spin equilibrium.
It is important to mention a potential caveat of this work. Whereas the CYP125A1 was purified by methods previously shown to yield ligand free enzyme by crystallography, the CYP2E1 preparation conceivably includes a ligand carried through the purification procedure, since we do not have a crystal structure from that preparation. However, because several other laboratories have documented a large fraction of high spin enzyme from CYP2E1 purified by different methods, we suggest that our preparation is ligand free and that it has an inherently high fraction of high spin heme, as observed by others. We acknowledge the possibility that our preparation includes some ligand that induces a high spin component, although we observe no other indication of an active site bound ligand. Regardless, even if some ligand was bound to CYP2E1, it would not alter the conclusion that ferric spin equilibrium is not correlated with strength of water ligation. Similarly, regarding the results with the CYP2C9D mutant, we emphasize that this mutation is modestly structurally different than the wild type CYP2C9 in crystal structures, although the mutant binds and metabolizes ligands with similar catalytic properties. Thus, the details of the ligand field of the mutant may not be identical to the wild type. However, the mutant provides a valuable additional variant with a ferric spin equilibrium distinct form other isoforms used here, and it is expressed and soluble at sufficient levels to obtain the high concentrations of protein required for MCD.
Acknowledgments
The authors acknowledge Professor Emily Scott, University of Kansas, for supplying CYP2E1 (NIHR01GM076343), and Professor Daniel R. Gamelin, University of Washington, for access to the MCD spectrometer. Additionally, the authors would like to thank Dr. Arthur Roberts (University of Georgia) for his assistance with determination of CYP spin equilibria.
Glossary
Abbreviations
- CYP
cytochrome P450
- MCD
magnetic circular dichroism
- EPR
electron paramagnetic resonance
- nIR
near-infrared
- UV
ultraviolet
- LMCT
ligand-to-metal transfer
The authors declare no competing financial interest.
Funding sources: NIH 1R01 GM110790 (WMA), NIH R01GM25515 (POM), and NSF DGE1256082 (AMS).
Funding Statement
National Institutes of Health, United States
References
- Sono M.; Roach M. P.; Coulter E. D.; Dawson J. H. (1996) Heme-containing oxygenases. Chem. Rev. 96, 2841–2888. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. (2001) Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 14, 611–650. [DOI] [PubMed] [Google Scholar]
- Sligar S. G. (1976) Coupling of spin, substrate, and redox equilibriums in cytochrome P450. Biochemistry 15, 5399–5406. [DOI] [PubMed] [Google Scholar]
- Fisher M. T.; Sligar S. G. (1985) Control of heme protein redox potential and reduction rate: linear free energy relation between potential and ferric spin state equilibrium. J. Am. Chem. Soc. 107, 5018–5019. [Google Scholar]
- Roberts A. G.; Campbell A. P.; Atkins W. M. (2005) The thermodynamic landscape of testosterone binding to cytochrome P450 3A4: ligand binding and spin state equilibria. Biochemistry 44, 1353–1366. [DOI] [PubMed] [Google Scholar]
- Thomann H.; Bernardo M.; Goldfarb D.; Kroneck P. M. H.; Ullrich V. (1995) Evidence for water binding to the Fe center in cytochrome P450cam obtained by 17O electron spin-echo envelope modulation spectroscopy. J. Am. Chem. Soc. 117, 8243–8251. [Google Scholar]
- Goldfarb D.; Bernardo M.; Thomann H.; Kroneck P. M. H.; Ullrich V. (1996) Study of water binding to low spin Fe(III) in cytochrome P450 by pulsed ENDOR and four-pulse ESEEM spectroscopies. J. Am. Chem. Soc. 118, 2686–2693. [Google Scholar]
- Sligar S. G.; Cinti D. L.; Gibson G. G.; Schenkman J. B. (1979) Spin state control of the hepatic cytochrome P450 redox potential. Biochem. Biophys. Res. Commun. 90, 925–932. [DOI] [PubMed] [Google Scholar]
- Hlavica P. (2007) Control by substrate of the cytochrome P450-dependent redox machinery: mechanistic insights. Curr. Drug Metab. 8, 594–611. [DOI] [PubMed] [Google Scholar]
- Grinkova Y. V.; Denisov I. G.; McLean M. A.; Sligar S. G. (2013) Oxidase uncoupling in heme monooxygenases: Human cytochrome P450 CYP3A4 in Nanodiscs. Biochem. Biophys. Res. Commun. 430, 1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillam E. M. J.; Guo Z. Y.; Guengerich F. P. (1994) Expression of modified human cytochrome P450 2E1 in Escherichia coli, purification, and spectral and catalytic properties. Arch. Biochem. Biophys. 312, 59–66. [DOI] [PubMed] [Google Scholar]
- Ouellet H.; Guan S.; Johnston J. B.; Chow E. D.; Kells P. M.; Burlingame A. L.; Cox J. S.; Podust L. M.; De Montellano P. R. O. (2010) Mycobacterium tuberculosis CYP125A1, a steroid C27 monooxygenase that detoxifies intracellularly generated cholest-4-en-3-one. Mol. Microbiol. 77, 730–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVore N. M.; Scott E. E. (2012) Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 482, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurelli S.; Chiesa M.; Giamello E.; Di Nardo G.; V. Ferrero V. E.; Gilardi G.; Van Doorslaer S. (2011) Direct spectroscopic evidence for binding of anastrozole to the iron heme of human aromatase. Peering into the mechanism of aromatase inhibition. Chem. Commun. 47, 10737–10739. [DOI] [PubMed] [Google Scholar]
- Woods C. M.; Fernandez C.; Kunze K. L.; Atkins W. M. (2012) Allosteric activation of cytochrome P450 3A4 by alpha-naphthoflavone: branch point regulation revealed by isotope dilution analysis. Biochemistry 50, 10041–10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P. A.; Cosme J.; Ward A.; Angove H. C.; Matak Vinkovic D.; Jhoti H. (2003) Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 424, 464–468. [DOI] [PubMed] [Google Scholar]
- McLean K. J.; Warman A. J.; Seward H. E.; Marshall K. R.; Girvan H. M.; Cheesman M. R.; Waterman M. R.; Munro A. W. (2006) Biophysical characterization of the sterol demethylase P450 from Mycobacterium tuberculosis, its cognate ferredoxin, and their interactions. Biochemistry 45, 8427–8443. [DOI] [PubMed] [Google Scholar]
- Ouellet H.; Lang J. r. m.; Couture M.; Ortiz de Montellano P. R. (2009) Reaction of Mycobacterium tuberculosis cytochrome P450 enzymes with nitric oxide. Biochemistry 48, 863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T.; Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes: I. Evidence for its hemoprotein nature. J. Biol. Chem. 239, 2370–2378. [PubMed] [Google Scholar]
- Piepho S. B., and Schatz P. N. (1983) Group Theory in Spectroscopy with Applications to Magnetic Circular Dichroism, Wiley, New York. [Google Scholar]
- Stoll S.; Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55. [DOI] [PubMed] [Google Scholar]
- Taylor C. P. S. (1977) The EPR of low spin heme complexes relation of the t2g hole model to the directional properties of the g tensor, and a new method for calculating the ligand field parameters. Biochim. Biophys. Acta - Protein Struct. 491, 137–148. [DOI] [PubMed] [Google Scholar]
- Renaud J. P. (1996) Thermodynamic studies of substrate binding and spin transitions in human cytochrome P-450 3A4 expressed in yeast microsomes. Biochem. J. 319, 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A. G.; Yang J.; Halpert J. R.; Nelson S. D.; Thummel K. T.; Atkins W. M. (2011) The structural basis for homotropic and heterotropic cooperativity of midazolam metabolism by human cytochrome P450 3A4. Biochemistry 50, 10804–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheesman M. R., Greenwood C., Thomson A. J., and Sykes A. G. (1991) Magnetic Circular Dichroism of Hemoproteins, In Adv. Inorg. Chem., pp 201–255, Academic Press. [Google Scholar]
- Cheng J. C.; Osborne G. A.; Stephens P. J.; Eaton W. A. (1973) Infrared magnetic circular dichroism in the study of metalloproteins. Nature 241, 193–194. [Google Scholar]
- Walker F. A. (1999) Magnetic spectroscopic (EPR, ESEEM, Mössbauer, MCD and NMR) studies of low spin ferriheme centers and their corresponding heme proteins. Coord. Chem. Rev. 185–186, 471–534. [Google Scholar]
- McKnight J.; Cheesman M. R.; Thomson A. J.; Miles J. S.; Munro A. W. (1993) Identification of charge-transfer transitions in the optical spectrum of low spin ferric cytochrome P-450 Bacillus megaterium. Eur. J. Biochem. 213, 683–687. [DOI] [PubMed] [Google Scholar]
- Thomson A. J.; Gadsby P. M. A. (1990) A theoretical model of the intensity of the near-infrared porphyrin-to-iron charge-transfer transitions in low spin iron(III) haemoproteins. A correlation between the intensity of the magnetic circular dichroism bands and the rhombic distortion parameter of iron. J. Chem. Soc., Dalton Trans. 0, 1921–1928. [Google Scholar]
- Griffith J. S. (1957) Binding in haemolglobin azide as determined by electron resonance: theory of electron resonance in ferrihaemoglobin azide. Nature 180, 30–31. [DOI] [PubMed] [Google Scholar]
- Peisach J.; Stern J. O.; Blumberg W. E. (1973) Optical and magnetic probes of the structure of cytochrome P-450s. Drug Metab. Dispos. 1, 45–61. [PubMed] [Google Scholar]
- McLean K. J.; Lafite P.; Levy C.; Cheesman M. R.; Mast N.; Pikuleva I. A.; Leys D.; Munro A. W. (2009) The structure of Mycobacterium tuberculosis CYP125: molecular basis for cholesterol binding in a P450 needed for host infection. J. Biol. Chem. 284, 35524–35533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby P. M. A.; Thomson A. J. (1990) Assignment of the axial ligands of ferric ion in low spin hemoproteins by near-infrared magnetic circular dichroism and electron paramagnetic resonance spectroscopy. J. Am. Chem. Soc. 112, 5003–5011. [Google Scholar]
- Girvan H. M.; Seward H. E.; Toogood H. S.; Cheesman M. R.; Leys D.; Munro A. W. (2007) Structural and spectroscopic characterization of P450 BM3 mutants with unprecedented P450 heme iron ligand sets: new heme ligation states influence conformational equilibria in P450 BM3. J. Biol. Chem. 282, 564–572. [DOI] [PubMed] [Google Scholar]
- Girvan H. M.; Toogood H. S.; Littleford R. E.; Seward H. E.; Smith W. E.; Ekanem I. S.; Leys D.; Cheesman M. R.; Munro A. W. (2009) Novel haem co-ordination variants of flavocytochrome P450 BM3. Biochem. J. 417, 65–76. [DOI] [PubMed] [Google Scholar]
- Clay M. D.; Jenney F. E.; Hagedoorn P. L.; George G. N.; Adams M. W. W.; Johnson M. K. (2001) Spectroscopic studies of Pyrococcus furiosus superoxide reductase: implications for active-site structures and the catalytic mechanism. J. Am. Chem. Soc. 124, 788–805. [DOI] [PubMed] [Google Scholar]
- Lehnert N. (2012) Elucidating second coordination sphere effects in heme proteins using low-temperature magnetic circular dichroism spectroscopy. J. Inorg. Biochem. 110, 83–93. [DOI] [PubMed] [Google Scholar]