

Abstract
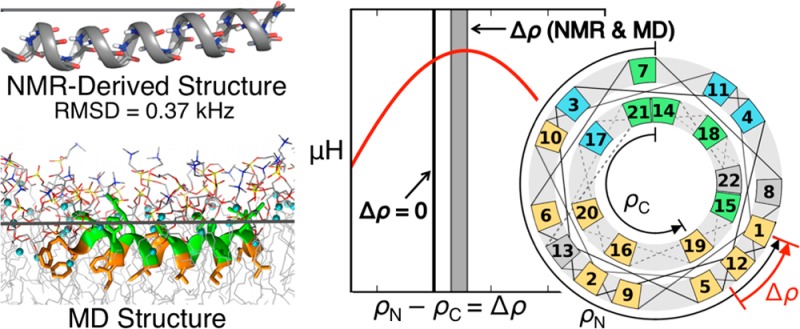
While antimicrobial peptides (AMPs) have been widely investigated as potential therapeutics, high-resolution structures obtained under biologically relevant conditions are lacking. Here, the high-resolution structures of the homologous 22-residue long AMPs piscidin 1 (p1) and piscidin 3 (p3) are determined in fluid-phase 3:1 phosphatidylcholine/phosphatidylglycerol (PC/PG) and 1:1 phosphatidylethanolamine/phosphatidylglycerol (PE/PG) bilayers to identify molecular features important for membrane destabilization in bacterial cell membrane mimics. Structural refinement of 1H–15N dipolar couplings and 15N chemical shifts measured by oriented sample solid-state NMR and all-atom molecular dynamics (MD) simulations provide structural and orientational information of high precision and accuracy about these interfacially bound α-helical peptides. The tilt of the helical axis, τ, is between 83° and 93° with respect to the bilayer normal for all systems and analysis methods. The average azimuthal rotation, ρ, is 235°, which results in burial of hydrophobic residues in the bilayer. The refined NMR and MD structures reveal a slight kink at G13 that delineates two helical segments characterized by a small difference in their τ angles (<10°) and significant difference in their ρ angles (∼25°). Remarkably, the kink, at the end of a G(X)4G motif highly conserved among members of the piscidin family, allows p1 and p3 to adopt ρ angles that maximize their hydrophobic moments. Two structural features differentiate the more potent p1 from p3: p1 has a larger ρ angle and less N-terminal fraying. The peptides have comparable depths of insertion in PC/PG, but p3 is 1.2 Å more deeply inserted than p1 in PE/PG. In contrast to the ideal α-helical structures typically assumed in mechanistic models of AMPs, p1 and p3 adopt disrupted α-helical backbones that correct for differences in the amphipathicity of their N- and C-ends, and their centers of mass lie ∼1.2–3.6 Å below the plane defined by the C2 atoms of the lipid acyl chains.
Introduction
Cationic α-helical antimicrobial peptides (AMPs) are essential components of the innate immune system and have garnered much interest in the search for novel antimicrobial agents due to their direct antimicrobial effects, broad-spectrum activity, rapid eradication of their targets, potency against drug-resistant bacteria, and low incidence of induced bacterial resistance.1−8 Their therapeutic potential is multifaceted since their functions include bactericidal, fungicidal, virucidal, and tumorocidal activities as well as immunomodulatory effects that allow them to coordinate responses from both the innate and adaptive immune systems.2,3,6 Their antimicrobial activity, which is generally due to membrane disruption and permeabilization, correlates more with their amphipathicity and ability to induce nonbilayer phases than a specific motif in their amino acid sequence.9,10 The amphipathic α-helix adopted by many AMPs is an advantageous membrane-binding motif since it matches the interfacial nature of phospholipid bilayers and enables both the hydrophilic and hydrophobic sides of the helix to be involved in membrane binding. Furthermore, secondary-structure formation upon membrane binding is thermodynamically favorable.11
The direct relationships between the amino acid composition of AMPs, their mechanism of action, and the bilayer composition of their targets are not well understood. On one hand, amphipathicity has become a major consideration in engineering therapeutically useful AMPs since it relates to manipulable properties of the α-helix, such as the amino acid composition, hydrophobic moment, charge distribution, orientation of large residues, and relative sizes of the hydrophobic and hydrophilic sectors.1,3,12−16 On the other hand, mounting evidence suggests that the more potent AMPs do not have perfectly amphipathic secondary structures and the balance between hydrophobic and electrostatic interactions involving the peptide, lipids, and water determines the AMP’s membrane-destabilizing activity. To discern molecular determinants (e.g., deviations from idealized amphipathic structures) that generate intermolecular forces conducive to membrane destabilization and to design analogs with predictable therapeutic effects, rigorous structural and topological analyses of AMPs with sequence homology but distinct potencies must be performed under biologically relevant conditions.
The Antimicrobial Peptide Database (APD),17 which contains more than 2200 AMPs, indicates that 44% have known secondary structures (41% have α-helical content; 18% contain β-sheets) and only 14% have known 3D structures, 90% of which were solved by NMR. Whereas solid-state NMR (ssNMR) is particularly well suited to investigate the structures of AMPs under native-like conditions and the structure of an α-helical AMP, mastoparan-X, was solved by ssNMR in gel-state bilayers,18 α-helical AMP high-resolution structures in liquid-crystalline (“fluid”) phospholipid bilayers are lacking. In this research, four high-resolution structures of two 22-residue long AMPs from the piscidin family, piscidin 1 (p1) and piscidin 3 (p3), were determined in fluid 3:1 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC)/1,2-dimyristoyl-sn-glycero-3-phosphatidylglycerol (DMPG) and 1:1 1-palmitoyl-2-oleoyl-sn-glycero-phosphatidylethanolamine (POPE)/1-palmitoyl-2-oleoyl-sn-glycero-phosphoglycerol (POPG) lipid bilayers to model the lipid composition of bacteria such as Staphylococcus aureus (Gram-positive) and Escherichia coli (Gram-negative). The peptides, which are kinked at a central glycine, are immersed in the lipids resulting in local bilayer distortion and global thinning. The substantial structural divergences between the structures of p1 in bilayers and those previously determined in SDS (sodium dodecyl sulfate) and DPC (dodecylphosphocholine) micelles19,20 highlight the impact of the environment on AMP structures and the need for structural studies of AMPs with native-like lipids as presented here.
Piscidins, found widely in teleost fish and first discovered in mast cells,21−23 have broad-spectrum activity against a large number of Gram-positive and -negative bacteria, including methicillin-resistant S. aureus (MRSA),24 viruses such as HIV-1,25 fungi,26 yeasts, and cancer cells.27 The piscidin family contains homologues that are ideal for probing the relationships between AMP structure and function. In particular, p1 and p3 have highly conserved N-terminal segments and varied C-terminal segments. The respective minimum inhibitory concentrations (MICs) for p1 and p3 are 2–10 and 10–20 μM against E. coli; they are ≤2 and 2–10 μM against Bacillus cereus.(21,22) Both peptides have an MIC ≤ 2 μM against S. aureus. P1, which has known membrane permeabilizing capability,19,20 is not only more antimicrobial but also more hemolytic than p3 since the concentration of peptide required to lyse 50% of red blood cells (EC50) is much lower for p1 (∼50 μg/mL) than p3 (≫100 μg/mL).28 Significantly, a recent screening of the APD by Wang6 indicated that p1, which has anticancer properties,27 is one of the top six potent AMPs with anti-HIV-1 activity. With ascaphin-8, p1 is the only AMP that is active against both a broad spectrum of bacteria (including MRSA) and HIV-1.6 These compelling properties translate into a strong motivation to use piscidin as a template to develop new therapeutics. Members of the piscidin family contain up to 10 times as many histidines as do other AMPs in the APD (20% in p1 versus 2% in a group of over 2200 AMPs).17 These residues may act as pH-sensitive switches that modulate the amphipathic character and antimicrobial activity of piscidin.29 Both p1 and p3, which contain a helix-disrupting G(X)4G motif between G8 and G13, are intrinsically disordered molecules that are unstructured in water but form highly α-helical structures in the presence of lipid bilayers.30−32 Since the adsorption of p1 and p3 onto lipid bilayers is a precursor to dramatic lipid bilayer perturbation and permeabilization under nonequilibrium conditions and therefore so central to their mechanism of action, the equilibrium structures and orientations of these piscidins bound to bilayers could reveal characteristics that correlate with their distinct biological activities.
When p1 is bound to anionic SDS micelles, the full length of the peptide backbone is a straight α-helix. This structure and that of mutants led to a proposal that a hydrophobic patch at the N-end facilitates insertion in bilayers.19 In zwitterionic DPC micelles, p1 adopts an α-helical conformation from residues 8 through 17, one β-turn at the N-end and two β-turns at the C-end.19 Oriented sample solid-state NMR (OS ssNMR) studies of partially labeled p1 and p3 in phospholipid bilayers that were either mechanically or magnetically aligned indicated that the peptides adopt an in-plane orientation.30,31,33 In the ssNMR studies, 3:1 DMPC/DMPG bilayers (25% anionic) were used to mimic negatively charged bacterial membranes such as Gram-positive S. aureus that have low intrinsic curvature and are highly susceptible to p1 and p3. Accompanying dynamic studies in 3:1 PC/PG identified two possible types of backbone motions, including a fast diffusion in the plane of the bilayer and a rocking motion about the helical axis.33 None of the above-referenced studies provided the high-resolution structures of p1 and p3. Previously, molecular dynamics (MD) simulations were used to characterize the dynamics of p1 in implicit bilayers of different geometries34 and atomistic lipid bilayers.35,36 Notably, p1 was found to insert more deeply into zwitterionic PC than anionic PG bilayers. These MD studies are an important first step in characterizing the interactions of p1 with lipid bilayers. However, MD simulations on the protonated forms of p1 were not performed, and p3 was not considered. Furthermore, the scope of these MD simulations was limited because no experimentally measured orientations of the peptide existed to inform the simulations.
OS ssNMR is a powerful technique for the structural studies of AMPs bound to membranes; however, rigorous structural characterization relies on incorporating the effect of the peptide dynamics on the NMR observables.37−40 There are two primary routes to obtain the three-dimensional (3D) structures and orientations of amphipathic peptides from OS ssNMR data. The first is based on the sinusoidal patterns of 1H–15N dipolar coupling (DC) or 15N chemical shift anisotropy (CSA) restraints as a function of residue number, commonly denoted as “dipolar waves” or “chemical shift (CS) waves”.41,42 For ideal α-helices, the period and amplitude of the wave are simply related to the tilt, τ (orientation of the helical axis with respect to the bilayer normal), and azimuthal rotation angle, ρ (rotation of the helix around its helical axis) (Figure 1A). Alternatively, structure determination programs (e.g., Xplor-NIH)43 can use ssNMR DC and CSA as restraints in conjunction with a molecular mechanics force field to generate 3D structures by simulated annealing.44−46 The balance of CSA, DC, and ϕ/ψ restraints is important and can greatly affect the refined structure.45 These programs typically yield an ensemble of structures, which are presumed to be representative of the natural structure and are a starting point of structural analysis. The structures in the ensemble usually differ from each other because of numerous local minima on the energy surface. This is usually not a serious drawback, but when the experimentally obtained data are an average of substantially different conformations and orientations, a single structure may be an inaccurate representation. For example, the significant differences in the average tilt of a designed transmembrane helix (WALP23) determined by NMR and MD simulations were attributed to bias averaging in the former.47 MD simulations can resolve questions on bias averaging since they provide a time series of the individual orientations that compose the average. Moreover, if satisfactory agreement between MD and OS ssNMR is achieved, the atomistic description obtained from an MD simulation can be relied on to accurately determine details such as the peptide’s depth of insertion not easily measured experimentally with OS ssNMR in a membrane environment.
Figure 1.
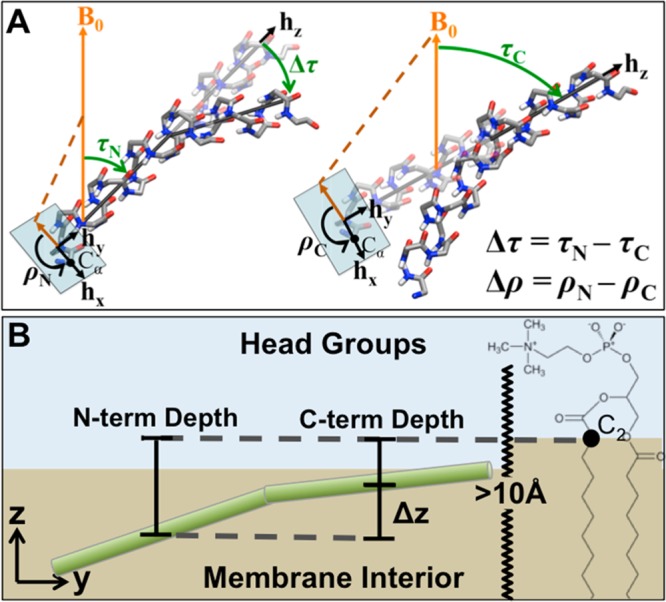
Orientation and bilayer position of an interfacially aligned peptide. (A) Orientation of a peptide that is kinked in the plane perpendicular to the bilayer surface. The angles (τN,ρN) and (τC,ρC) are used to characterize the orientations of the helical segments on the amino (left) and carboxyl (right) sides of the kink. (B) Bilayer location of the kinked peptide. The depth of insertion of the peptide is defined as the distance between the center of mass (CM) of the peptide backbone and the hydrophobic interface defined by the C2 of the bulk lipids (>10 Å from the nearest peptide). Since the peptide is kinked, a CM is defined for each helical segment before and after the kink (see Materials and Methods for details).
As subsequently discussed here, the combination of CD and OS ssNMR data was used to determine the general conformation and orientation of p1 and p3 in 3:1 DMPC/DMPG and 1:1 POPE/POPG. Peptide structures for the four systems corresponding to the two peptides in the two lipid mixtures were independently generated by simulated annealing refinement of the NMR data and by MD simulations. Excellent agreement between structures obtained by both methods strengthened confidence in the depth of insertion and side-chain orientations determined solely from MD. The rigorous comparisons of experimental and simulated structures and orientations of both p1 and p3 in liquid-crystalline lipid bilayers were analyzed to provide the details necessary to confidently identify molecular features of the peptides that may affect the lipid specificity, bilayer location, membrane-destabilizing capability, and antimicrobial potency of the peptides.
Materials and Methods
Materials
The synthesis and purification of carboxyamidated p1 (MW 2571) and p3 (MW 2492) have been previously reported.31
Circular Dichroism
CD experiments for p1 and p3 added to suspensions of unilamellar phospholipid vesicles at a peptide-to-lipid ratio of 1:60 were performed following a standard protocol described previously28 and briefly outlined in the SI. Percent helical content given in Table S1 were calculated as explained in the SI.
Preparation of Samples for Solid-State NMR
Oriented samples were prepared following a procedure previously reported.28
15N Solid-State NMR
Two-dimensional heterocorrelation (2D HETCOR)32 NMR experiments were performed at the National High Magnetic Field Laboratory on an ultrawide bore superconducting 21.1T magnet with a Bruker Avance 900 MHz NMR console (Larmor frequencies of 897.11 and 90.92 MHz for 1H and 15N, respectively) and on a 14.1 T Bruker Avance WB600 NMR spectrometer (Larmor frequencies of 600.13 and 60.82 MHz for 1H and 15N, respectively) and at the Rennselaer Polytechnic Institute on a Bruker Avance WB600 NMR spectrometer (Larmor frequencies of 600.36 and 60.84 MHz for 1H and 15N, respectively). Spectra were obtained using low electrical field PISEMA probes48 and previously reported parameters32 that are summarized in the SI. The respective CS of 1H and 15N were referenced to the proton water peak at 4.7 ppm and the 15N signal of an 15N-labeled ammonium sulfate aqueous solution (5%, pH 3.1) at 0 ppm.
Ss-NMR samples featured multilamellar vesicles and a peptide-to-lipid ratio of 1:20, which reflects the conditions under which AMPs similar to piscidin are active49 and allows for optimal sensitivity of the NMR experiments. 2D HETCOR experiments were applied to samples of p1 and p3 oriented with the bilayer normal parallel to the static magnetic field, B0. The experiments consisted of both HETCOR and dipolar-encoded HETCOR (de-HETCOR) experiments32 since de-HETCOR OS ssNMR spectra provide both the 1H and 15N CSA, and their associated 1H–15N DC (i.e., half the dipolar splitting, DNH). Because DC orientational restraints are very sensitive to the orientations of peptide planes with respect to B0 and the bilayer normal, they can be used along with the 15N CSA restraints to determine the peptide structure and topology. Molecular motions of piscidin include the diffusion of the peptide about the bilayer normal and the rocking of the peptide about the helical axis.30,31,33 In the oriented preparations used here, the bilayer normal is parallel to B0, and therefore motional averaging is mostly due to rotational diffusion about the bilayer normal. Aside from the helical “fraying” characterized at the terminal region of the peptides, which may be attributed to additional local dynamics at the terminal residues, there is no strong evidence of other possible dynamics, which, if present, have only a negligible impact on the NMR observables. Indeed, as revealed by the MD simulations presented in this work, these NMR observables are reliable restraints to obtain the structure of piscidin with high precision and accuracy.
To facilitate the assignments of NMR signals from the 21 peptide planes of piscidin, a collection of 15N-amide singly and multiply labeled samples were used. To ensure that correct assignments were made when multiply labeled samples were analyzed, assignments of p1 in 3:1 PC/PG were done with a large number of singly labeled peptides (H3, H4, F6, R7, V10, V12, K14, H17, R18, V20). Complete resonance assignments for the two peptides in the two lipid mixtures are explained in the SI and summarized in Table S2. Assignments of signals from each peptide were done using an iterative process where the experimental data were fitted with dipolar waves as described under Dipolar and Chemical Shift Wave Analysis. Various permutations of assignments in multiply labeled samples were tested. Based on the chemical shifts range of 37–73 ppm and polarity index slant angle (PISA) wheel simulations previously done on a 10-site labeled p1 sample in 3:1 PC/PG,32 the tilt angle was determined to be close to 90°. Therefore, waves were generated with this starting value and changed until a combination of τ and ρ yielded a good fit between experimental data and simulated waves. Due to ambiguities that arise from the symmetry of nuclear spin interactions, two peptide orientations were consistent with the wave analysis: 180° – τ/ρ and τ/180° + ρ, with 0° < τ < 180° and 180° < ρ < 360°. These orientations are magnetically equivalent but chemically nonequivalent when a peptide is considered in a given leaflet of the bilayer. Considering the upper leaflet where by convention the bilayer normal is pointing up along the z-axis and B0, the τ/180° + ρ combination corresponds to a peptide that directs its hydrophobic side chains toward the hydrophobic bilayer, while the other combination of 180° – τ/ρ points the hydrophilic side chains toward the core of the bilayer, which is energetically unfavorable. As a result, it is possible to identify a single orientation for each piscidin. Corresponding helical wheel diagrams are shown in Figure 2.
Figure 2.

Helical wheel diagrams rotated to match the ρN and ρC of the refined NMR structures (NMRr; left set) or the MD structures (right set). In each set, p1 (left) and p3 (right) are shown in 3:1 PC/PG (top) and 1:1 PE/PG (bottom). Residues in green indicate more polar groups, while those in light orange are hydrophobic; histidines and glycine are in blue and gray, respectively. The orientation of the hydrophobic moment (μH) is identified with an orange arrow. The coordinate system that defines ρ is given in the Orientation from NMRr and MD Structures section.
Structure Determination
Refined NMR (NMRr) structures were obtained using a simulated annealing protocol within Xplor-NIH43 with torsion angle MD in the presence of the experimental restraints.45 Simulated annealing was performed by reducing the temperature from the initial value of 2000 to 50 K in steps of 12.5 K. Ideal ϕ/ψ angle restraints (−65°/–40°) with ±20° variation were used for all residues with kta ramped from 300 to 100 kcal·mol–1·rad–2. krdc was gradually increased from 2 to 5 kcal·mol–1·s2, and kcsa was set constant at 0.1 kcal·mol–1·s2 in order to be consistent with the experimental error. These force constants, which correspond to a CSAscale/DCscale < 0.1, were chosen to obtain the optimal balance between the effects of the DC and CSA restraints in the structure calculations.45,46 The NMR restraints were taken from bilayer samples. To match the experimental conditions, the orientation tensor axial component Da was set to an initial value of 10.4 kHz and refined to ∼9.8 kHz. Rhombicity (Rh) was fixed at zero for all calculations. The calculation also included the Xplor-NIH potential for knowledge-based torsion angles with ramped force constants of 0.002 to 1 kcal·mol–1·rad–2. kvdW and svdW were geometrically increased from 0.004 to 4 kcal·mol–1·Å–4 and 0.9 to 0.8, respectively. Routine terms ANGL, BOND, and IMPR were also added to the calculation. A total of 100 structures were generated, and the 10 lowest-energy structures were accepted for analysis and representation. The same setup was employed for each structure calculation. The atomic coordinates for the 10 lowest-energy structures of the four systems have been deposited in the Protein Data Bank under the file names 2MCU, 2MCV, 2MCW, and 2MCX.
Charge State of the Histidine Side Chains
Investigating the charged state of the histidine side chains in membrane-bound piscidin was done by performing OS ssNMR on aligned samples of p1 in 3:1 PC/PG at the higher pH of 8.8 to be above the pKa of the histidine side chains and provide conditions to study the peptide’s structure when the histidine side chains are neutral.
MD Simulations
Table 1 lists the MD simulations of p1 and p3. Simulations were performed using CHARMM 36b250 with CHARMM 22 protein parameters51 and CHARMM 36 lipid parameters52 with modified Lennard-Jones pairwise distances for sodium ions interacting with select lipid oxygens.53 Each system was composed of 80 lipids, two peptides (1 per leaflet), a water layer ∼16 Å thick above and below the bilayer, 16 sodium ions, and chloride ions to neutralize the system. The 1:40 peptide:lipid ratio (half that of the NMR experiments) was chosen to study the peptides at low concentration, before they interact with each other along the bilayer plane.
Table 1. MD Simulations Presented in This Studya.
| peptide | lipid | charged histidines |
|---|---|---|
| p1 | 3:1 DMPC/DMPG | none |
| p3 | 3:1 DMPC/DMPG | none |
| p1 | 1:1 POPE/POPG | none |
| p3 | 1:1 POPE/POPG | none |
| p1 | 3:1 DMPC/DMPG | 3, 4, 11 |
| p1 | 3:1 DMPC/DMPG | 3, 4, 11, 17 |
| p3 | 3:1 DMPC/DMPG | 3, 4, 11 |
All simulations were run with a protein/lipid ratio of 2:80.
Initial structures of p1 and p3 were generated with ϕ/ψ angles of −61° and −45°, respectively, and extended side chains. One peptide was aligned with the center of mass (CM) of its heavy backbone atoms 14 Å above the center of the bilayer, while the second peptide was 14 Å below the bilayer. This initial position yields a bilayer location of the peptide that is no deeper than the C2 atoms of the lipids. To minimize interaction across the bilayer, the peptides were rotated to be perpendicular to each other with respect to the bilayer normal. Each peptide was rotated around its helical axis to ensure that hydrophobic residues were facing the bilayer core. Systems were assembled using the CHARMM-GUI interface.54 Separate simulations of p1 and p3 were performed with different charge states of the histidine side chains as shown in Table 1. Peptides were simulated with neutral histidine side chains unless otherwise indicated in the text.
In all simulations, specific Lennard-Jones cutoffs and particle-mesh Ewald55 electrostatics were required for the preceding force fields.52 The Lennard-Jones potential terminated at 12 Å, with a smoothing function operating between 8 and 12 Å. The Nosé-Hoover algorithm with thermal piston value of 10 000 kcal/mol·ps2 was used to control temperature.56,57 A Langevin piston with a piston mass of 1000 amu maintained a constant normal pressure of 1 atm and zero surface tension.58 The temperature was held constant at 313 K. Trajectories were generated with a leapfrog Verlet algorithm with a time step of 1 fs. All bonds to hydrogen atoms were constrained using the SHAKE algorithm.59 The initial 10 ns of each trajectory were removed to allow for equilibration, and the final 90 ns were used for analysis. Peptide orientations and depths of insertion were calculated as averages over 10 ns blocks of simulation time.
Dipolar and Chemical Shift Wave Analysis
The 15N–1H DC and 15N CSA restraints were fitted to waves as described by Perrin et al.60 and summarized briefly in the SI.
Orientation from NMRr and MD Structures
In the oriented samples analyzed here, the z-axis is parallel to the bilayer normal and B0, which by convention points up along the z-axis. As shown in Figure 1A, the molecular frame is defined using hx, hy, and hz; hx is the radial vector going through the center of the helix and the Cα of the first residue in the peptide sequence; hz is the helical axis; and hy is orthogonal to hx and hz. The orientation of each peptide is characterized by τ and ρ (Figure 1A). These orientations correspond to the Euler angles (α, β, γ), where τ(β) is the tilt of the peptide and ρ(γ) is the rotation of the Cα from residue 1 around the helical axis. The tilt, which is equal to zero when hz is parallel to the z-axis (Figure 1A), is measured as a clockwise rotation of the helix away from B0. Following the convention of Cross and co-workers,61 ρ, which is zero when hx is aligned along the positive direction of the z-axis, is measured as a counterclockwise rotation about hz (Figure 1A). The orientation for the simulated piscidin at each time step was calculated relative to a piscidin structure with known orientation (ϕ/ψ angles of −61°/-45° and all Euler angles set to zero). The depth of insertion of a peptide was calculated as the distance between the CM for heavy backbone atoms and the C2 atom of chain 2 of each bulk lipid molecule in the same leaflet. Bulk lipids were defined as any lipid at least 10 Å away from the nearest peptide. Separate CMs for residues 3–10 and 14–20 were used to avoid artifacts due to fraying of terminal residues and kinking at G13 and obtain reliable depths of insertion for the N and C segments (Figure 1B).
Statistical Analysis
A subsection that describes the statistical analysis used for the refined NMR structures and MD simulations is provided in the SI.
Results and Analysis
Circular Dichroism
The CD spectra of p1 and p3 collected in the presence of 3:1 PC/PG and 1:1 PE/PG in phosphate buffer at pH 7.4 (Figure S1) and 6.0 display the two minima of 222 and 208 nm that are characteristic of α-helical structures, therefore both peptides become highly α-helical in the presence of these phospholipids (Table S1).
High-Resolution Solid-State NMR of p1 and p3 in Lipid Bilayers
Both p1 and p3 contain 21 peptide planes. To investigate the structure and orientation of each peptide in a given oriented lipid mixture, the 21 peptide planes were 15N-labeled and subjected to OS ssNMR spectroscopy. In the de-HETCOR spectra (Figure 3), 17–21 DC and 17–21 15N CS values were detected and summarized in Table S2. The sharp signals indicate structural homogeneity and stability of the corresponding labeled amide sites. Importantly, these spectra show no indication of signal from isotropically mobile residues or underlying powder pattern signal that would indicate a subset of static or unoriented peptide molecules. The 15N resonances are clustered between 37 and 73 ppm, as expected for α-helical amphipathic structures with an in-plane bilayer orientation.37−39 The single DC restraint associated with each labeled amide site reflects the orientation of the peptide with respect to B0 and therefore is crucial to structural determination. DC and/or CSA orientation restraints that are missing or close to isotropic values are consistent with structural heterogeneity and helical fraying on the NMR time scale (μs for the 15N CSA) and a more dynamic nature of terminal residues where some of the main chain carbonyl and amide groups are not hydrogen-bonded. Accordingly, no DC could be obtained for position 1 of p1 and p3 in all lipid systems, and orientational restraints for p3 were detected at neither position 2 in both lipid systems nor position 3 in PE/PG. These results indicate that p3 has more structural heterogeneity than p1 at the amino end, and 1:1 PE/PG induces more structural fluctuations in p3 than 3:1 PC/PG does. Further scrutiny reveals that the NMR data, which are consistent with the highly yet partial helical conformation determined by CD spectroscopy, help assign the small deviation in helical content to the terminal residues where dynamic fraying is detected on the NMR time scale.
Figure 3.

2D de-HETCOR NMR spectra of p1 (left) and p3 (right) in oriented, hydrated 3:1 DMPC/DMPG (top) and 1:1 POPE/POPG (bottom). Spectral superimpositions of singly to triply labeled samples are shown. The peptide-to-lipid ratio was 1:20 (molar). The spectra were collected at 305 (1:1 POPE/POPG) and 313 K (3:1 DMPC/DMPG), above the phase transition temperature of the lipids. Each pair of 15N/1H splittings (DNH) is shown using a single color. In addition to 15N backbone amide labels, the arginine side chains also contained 15N sites, some of which were detected in the de-HETCOR spectra. Only the backbone signals are annotated. The de-HETCOR data for G22 p1, which are outside the range of the spectra shown here, and the PISEMA data for F6I9A12 p3 in DMPC/DMPG are included in Figure S2.
Use of Dipolar Waves to Characterize the Local Backbone Structure of p1 and p3
To characterize the local backbone structure of p1 and p3, the experimental 15N–1H DC restraints obtained from the aligned samples were plotted as a function of residue number and shown in Figure 4. Three structural features are obtained from these plots.41,60 First, α-helical segments are revealed between residues 3–10 and 14–20 since the corresponding experimental data points can be fitted to a sinusoidal wave (gray line) with a periodicity of 3.6.41 Second, the fact that some experimental points at the N- and C-ends cannot be fitted with such a wave indicates that the corresponding residues are experiencing helical fraying. Third, the necessity to fit residues 3–10 and 14–20 separately demonstrates the presence of a kink at G13. These three findings are confirmed by the 15N CSA restraints plotted in Figure S3. While dipolar and chemical shift waves have been established as a useful tool for structural analysis,41,42 they have largely been superseded by structure determination programs like Xplor-NIH that combine both the DC and CSA restraints along with aspects of covalent bond geometry.44−46 The next section covers the 3D structural determination of p1 and p3 by Xplor-NIH.
Figure 4.
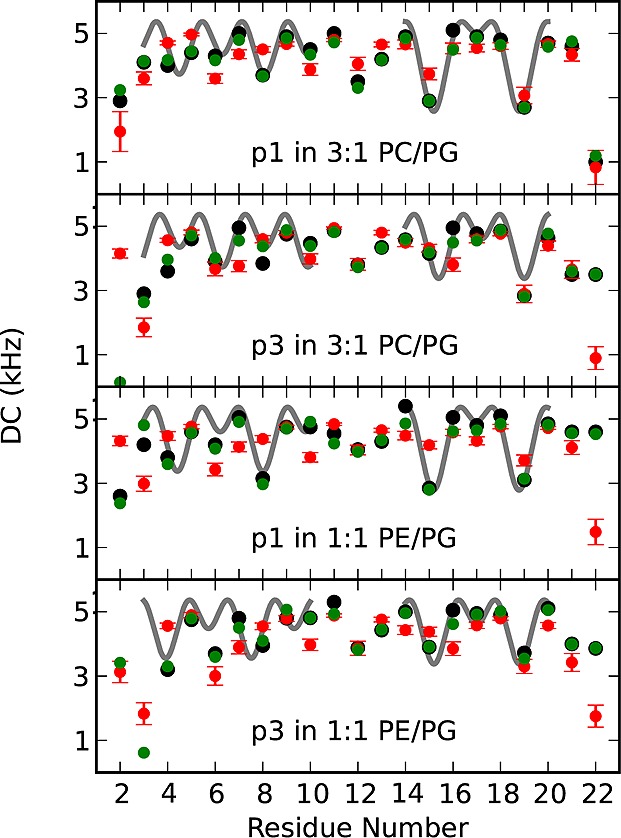
DC restraints experimentally observed (black), calculated from the refined NMR structures (green) and calculated from the MD simulations (red): p1 in 3:1 DMPC/DMPG (top) and 1:1 POPE/POPG (bottom-middle) and p3 in 3:1 DMPC/DMPG (top-middle) and 1:1 POPE/POPG (bottom). Absolute values of the dipolar couplings are plotted. Dynamics on the NMR time scale led to a lack of de-HETCOR signals for some of the terminal residues (Table S2). Dipolar waves fitted to the experimentally observed DC between residues 3–10 and 14–20 are shown in gray for an ideal α-helix with dihedral angles (ϕ = −61°, ψ = −45°). These α-helical regions are identified on the basis of having low fitting errors (average error per residue that is less than or similar to the experimental error of ±0.5 kHz).
3D Structures of p1 and p3 Bound to Lipid Bilayers
15N–1H DC and 15N CSA values obtained from the de-HETCOR spectra were used as restraints in Xplor-NIH to calculate the atomic-level structures of p1 and p3 in 3:1 PC/PG and 1:1 PE/PG. The excellent agreement between the experimental dipolar splittings and CSA values and those calculated from the four refined structures is demonstrated in Figure 5 by the low rmsd of 0.36 to 0.51 kHz and 3.0 to 3.9 ppm, respectively. For each system, Figure 6 shows an overlay of the top 10 structures (ensemble) and the lowest-energy structure that were obtained upon refinement of the NMR restraints. Table S5 summarizes the NMR structure statistics, and Figure S4 shows the surface of each peptide based on amino acid type. Considering the backbone heavy atoms of residues 3–20 that exhibit α-helicity (Figure 4), the rmsd between the top 10 peptide structures of each ensemble and their mean structure is in the range of 0.35–0.39 Å as shown in Table S5 (the table also gives rmsd values between the top 10 structures of each ensemble). This precision, which makes these structures high resolution,62 is better than values typically obtained for membrane protein structures solved by ssNMR63 and substantiates the results from wave analyses.
Figure 5.
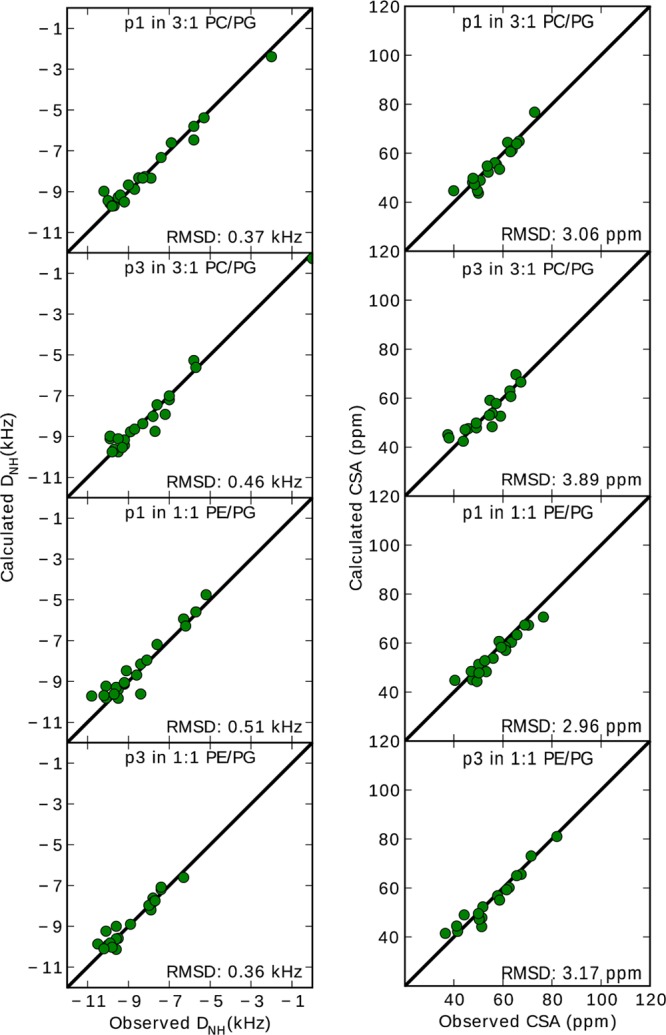
Correlation plots between calculated and experimental dipolar splittings, DNH (left) and CSA (right). Values calculated from the refined NMR structures are plotted as a function of the original NMR values. The rmsd for residues 3–20 are shown.
Figure 6.
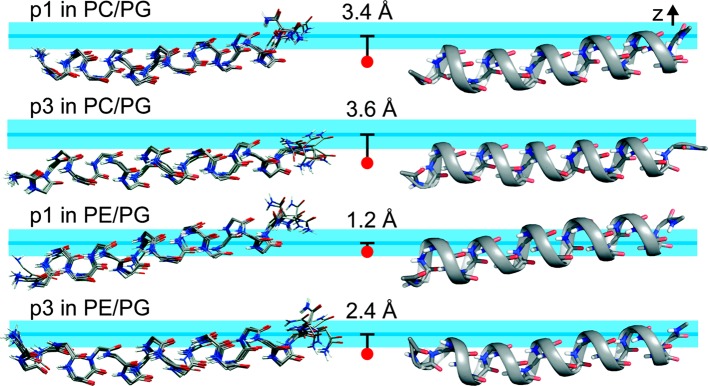
Ten lowest-energy backbone structures (left) and ribbon structures for the lowest-energy conformations (right) of p1 and p3 in 3:1 DMPC/DMPG and 1:1 POPE/POPG. These Xplor-NIH refined structures were calculated using ssNMR 15N chemical shifts and 15N/1H dipolar couplings. Peptides are aligned with the N-termini on the left. Considering residues 3–20 that show α-helicity, the rmsd values between the top 10 structures of each ensemble and their mean structures are 0.39 and 0.37 Å for p1 in PC/PG and PE/PG, respectively, and 0.39 and 0.35 Å for p3 in PC/PG and PE/PG, respectively (Table S5). Cross-correlation plots between experimental and calculated 1H–15N DNH and 15N CSA are shown in Figure 5. Three properties obtained from the MD simulations are included to show the average position of the C2 plane of the bulk lipids (horizontal blue line) relative to the fixed peptide: the z-position of the CM for backbone atoms of residues 3–10 and residues 14–20 (red dot); the average depth of insertion of the peptide (the distance between the blue line and red dot); and the fluctuations of the C2 plane with respect to the CM of the peptide (light blue horizontal band with thickness ±2rmsf = ±1.8 Å). Hence, the C2 plane fluctuates within the blue band with respect to the peptide.
MD Simulations
In all simulated systems, piscidin remained predominantly α-helical and interfacially oriented throughout the 90 ns trajectory. The MD structures are consistent with the experimental NMR restraints and structural features of the refined NMR structures. Indeed, the DC and CSA calculated from the MD structures (Figure 4) have an rmsd of 1.05–1.46 kHz and 7.31–10.94 ppm from the original NMR values for residues 3–20 (4–22 for p3 in PE/PG). This comparison cross-validates the NMR and MD data and demonstrates the accuracy of the structures. Furthermore, fraying of terminal residues is indicated by smaller DC values simulated for residues 2, 3, and 22 (Figure 4). On this basis, N-terminal fraying is greater in p3 than p1 in agreement with the NMR data. Additionally, kinking at G13 is manifested by a change in the pattern of the MD data points that coincides well with that observed by NMR (Figure 4).
Comparison of p1 and p3 in Terms of Orientations Determined from the Refined NMR and MD Structures
Characterizing accurate peptide orientations is critical to further interpretation of the results in terms of structure–function relationships. Here, the agreement demonstrated between the high-precision refined NMR and average MD structures and orientations is seen as accuracy in the descriptions of the piscidin peptides in terms of conformation and topology. The tilt angles τ and azimuthal rotation angles ρ were calculated separately for residues 3–10 and 14–20 due to the kink at G13, and their respective differences are Δτ (τN – τC) and Δρ (ρN – ρC), where the subscripts N and C denote the N- and C-ends, respectively. The orientations, summarized in Table 2, have average τ near 88° (parallel to the surface) and average ρ around 235°. With the exception of p1 in PE/PG, τ values from MD are within 2 standard errors (SE) of those from NMRr, indicating statistical equivalence. Figure 7 and Table 2 show that the root-mean-square fluctuations (rmsf) from simulations are 7 to 9° based on the MD data. This reflects the highly dynamic nature of the interfacially bound piscidin peptides. All of the τ from NMR are within ±1.3 rmsf of the MD values, further indicating that the deviations are of minor consequence. A more rigorous statistical comparison is difficult because the standard deviations in τ from the NMR structures (1–3°) are incomplete measures of uncertainty; i.e., SE obtained from a larger set of independent structure determinations are likely higher than the present standard deviations. The ρ from MD are mostly higher than those from NMR, and the differences are usually more than 2 SE. Nevertheless, the trends are comparable, and the values from NMRr are within 2.5 rmsf in all cases; i.e., the structures from MD are frequently sampling the values of ρ from the NMR structures. Overall, p1 has larger average ρ than p3 (average ρC derived from the NMR structures are similar for both peptides, but the corresponding MD values are more reliable since they are not susceptible to bias averaging). The finding that ρ is the orientational quantity most pertinent to differentiate p1 from p3 is illustrated in Figure 2, where helical wheels for each peptide are rotated to their corresponding values of ρ (Table 2). Rotations are performed using a convention where B0 and the bilayer normal are pointing up along the z-axis and the angle ρ, which is equal to zero when the first residue in the sequence is at the 12 o’clock position, is measured as a counterclockwise rotation of the helical wheel about the helical axis. From these diagrams, it is evident that ρ ∼ 235° evenly distributes the hydrophilic residues at the water-bilayer interface, while the hydrophobic residues face the hydrocarbon core of the bilayer. This hallmark orientation of piscidin is illustrated in Figure 8 that shows a MD snapshot of each system with orientation and depth of insertion characteristic of the average structure.
Table 2. Tilt Angles τ (°) and Azimuthal Rotation Angles ρ (°) Obtained from the Refined NMR Structures (NMRr) and MD Simulations and Depth of Insertion z (Å) from MD for the N- (residues 3–10) and C- (residues 14–20) Helical Segments of Each Peptidea.
| residues 3 to 10 |
residues 14 to 20 |
kink |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| NMRr | MD |
NMRr | MD |
NMRr | MD |
||||
| avg | avg | rmsf | avg | avg | rmsf | avg | avg | rmsf | |
| τN | τC | Δτ = (τN – τC) | |||||||
| p1 PC/PG | 90 (2) | 91 (1) | 7 | 86 (1) | 87 (2) | 9 | 4 (2) | 5 (1) | 8 |
| p3 PC/PG | 91 (1) | 93 (2) | 7 | 84 (1) | 85 (2) | 8 | 7 (2) | 7 (1) | 8 |
| p1 PE/PG | 83 (3) | 92 (2) | 8 | 86 (1) | 90 (2) | 9 | –3 (3) | 2 (1) | 7 |
| p3 PE/PG | 92 (1) | 93 (2) | 7 | 83 (2) | 86 (2) | 8 | 9 (3) | 7 (1) | 7 |
| ρN | ρC | Δρ = (ρN – ρC) | |||||||
| p1 PC/PG | 246 (3) | 265 (3) | 11 | 213 (1) | 245 (3) | 13 | 33 (4) | 21 (1) | 8 |
| p3 PC/PG | 236 (2) | 245 (3) | 11 | 215 (2) | 225 (3) | 11 | 20 (2) | 20 (1) | 8 |
| p1 PE/PG | 253 (2) | 256 (3) | 13 | 218 (2) | 236 (3) | 12 | 35 (4) | 20 (1) | 8 |
| p3 PE/PG | 250 (2) | 241 (3) | 11 | 220 (2) | 223 (2) | 10 | 30 (3) | 18 (1) | 8 |
| zN | zC | Δz = (zN – zC) | |||||||
| p1 PC/PG | – | 3.4 (0.1) | 0.8 | – | 3.3 (0.1) | 0.8 | 1.7 (0.1) | 0.1 (0.3) | 1.3 |
| p3 PC/PG | – | 3.7 (0.1) | 0.9 | – | 3.4 (0.1) | 0.9 | 2.2 (0.1) | 0.3 (0.3) | 1.4 |
| p1 PE/PG | – | 1.0 (0.1) | 0.8 | – | 1.4 (0.1) | 0.8 | 0.9 (0.1) | –0.4 (0.4) | 1.6 |
| p3 PE/PG | – | 2.4 (0.1) | 0.9 | – | 2.3 (0.1) | 0.9 | 1.6 (0.1) | 0.1 (0.4) | 1.5 |
To characterize the kink, differences in tilt (Δτ), azimuthal rotation (Δρ), and depth (Δz) between the N- and C-segments are also provided. For each system, the uncertainties indicated in parentheses by the NMRr and MD angles correspond to standard deviations among the top 10 NMR structures and SE of the MD data calculated from 10 ns blocks, respectively. The rmsf from MD are also included.
Figure 7.
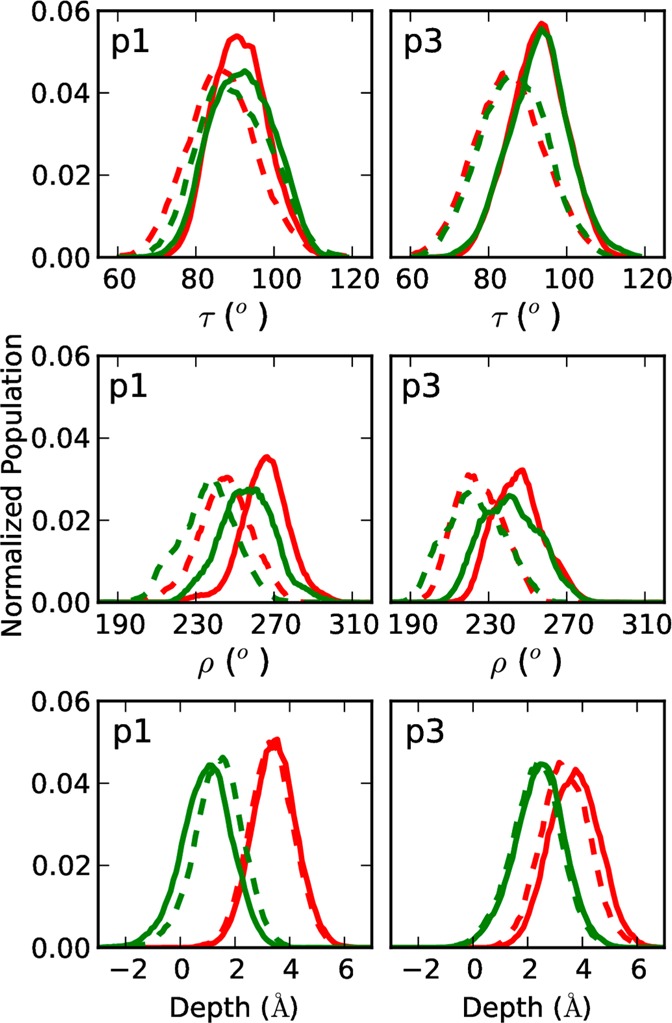
Distribution of tilt angles (top), azimuthal rotation angles (middle), and depths of insertion (bottom) sampled by p1 (left) or p3 (right) in PC/PG (red) or PE/PG (green) during the entire MD simulation (90 ns). Curves for N- and C-terminal residues are shown as solid and dashed lines, respectively.
Figure 8.
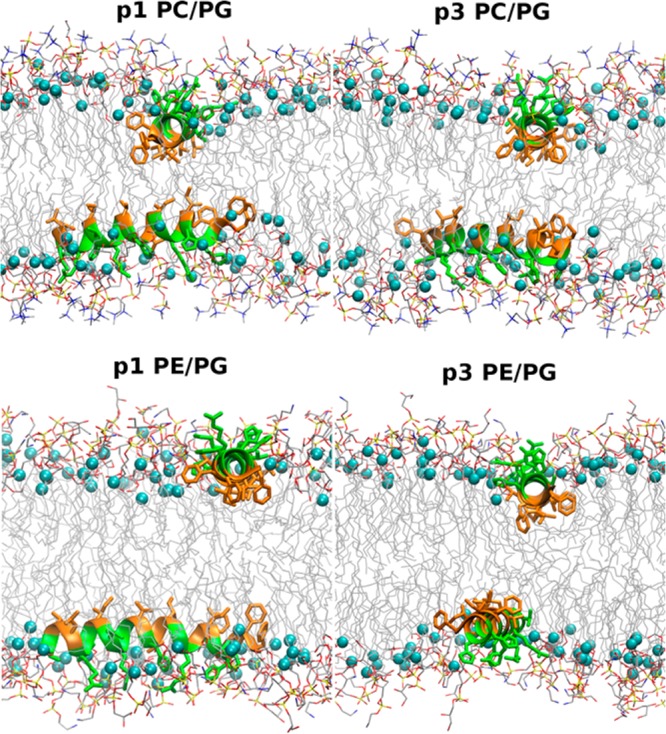
Snapshots of p1 and p3 in lipid bilayers from the MD simulations at a time step in which τN, τC, ρN, and ρC corresponded to the average orientations listed in Table 2. Hydrophobic residues are colored in orange, polar and charged residues in green, and the C2 atoms of the acyl chain in the lipids are indicated by cyan spheres. Two peptides were included in the simulations such that there was one peptide per leaflet, and the two peptides were oriented perpendicular to each other at the beginning of each simulation.
Characterization of the Kinked Structures and their Hydrophobic Moment
The availability of high-resolution and accurate structures of p1 and p3 enables detailed conformational comparisons. As illustrated in Table 2 and Figures 2 and 4, both the NMR and MD data show that p1 and p3 have a slight bend or kink characterized by a small change in τ (Δτ < 10°) and a larger change in ρ (Δρ ≈ 20° for MD and ≈30° for the refined NMR structures). Staggered helical wheels can be used to illustrate Δρ, as shown in Figure 2. Residues 1–13 and 14–22 are plotted as two different concentric circles, with residues 14–22 having a smaller radius. When the uncertainties in ρ of 2–4° for the NMR structures and rmsf in ρ of 10–13° for the MD structures are considered, the helical wheels derived from the refined NMR and MD structures and shown in Figure 2 have consistent helical rotations within the range of fluctuations obtained by MD. This corroborative structural determination increases confidence in the details of the geometric configurations.
To determine the driving force behind the different helical rotations on each side of G13, the peptide’s amphipathicity and more specifically the hydrophobic moment (μH) of each peptide as a function of Δρ were considered. This is illustrated in Figure 9 using the White and Wimley interfacial hydrophobicity scale.64 On that scale, p1 has a slightly larger hydrophobic moment than p3. Figure 9 shows that the helical segments on each side of the kink adopt distinct mean orientations that maximize the overall hydrophobic moment of the peptide and therefore its hydrophobic contacts with the bilayer hydrocarbon core. Overall, relative values of ρ are different for p1 and p3, but the peptides flex similarly as measured by Δρ to optimize their hydrophobic moment. Thus, the different amino acid compositions of p1 and p3 translate into different absolute helical rotations, but the amount of flexing at the conserved G13 is the same for both peptides.
Figure 9.
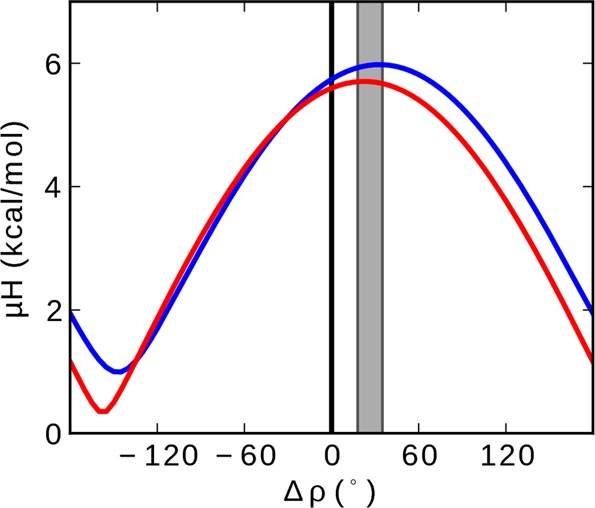
Hydrophobic moment (μH) versus Δρ for p1 (blue) and p3 (red) determined by rotating a helical wheel for residues 13–22 relative to a helical wheel for residues 1–12. Energies are from the White and Wimley interfacial hydrophobicity scale.64 Vertical gray band represent the range of Δρ calculated from the refined NMR and MD structures. The black line is positioned at Δρ = 0 (peptide with no kink).
Positioning of p1 and p3 in Lipid Bilayers
MD simulations were used to position the peptides in the bilayers and identify peptide–lipid interactions. These interactions are likely to play an important role in the peptide’s function since they provide a framework from which the peptide can remodel and disrupt the bilayer. In each simulation, the CM of the peptide lies within the hydrophobic section of the membrane and just below the C2 atom of the second acyl chains of the lipids (Tables 2 and S3). Based on these data, the CM of both p1 and p3 fluctuates between 2 and 5 Å below the C2 atoms in 3:1 PC/PG (Figure 7), and the depths of insertion are indistinguishable. The situation is different in PE/PG since the peptides are significantly less inserted with p1 and p3 being 2 and 1 Å closer to the hydrophilic medium, respectively. The change in the peptide’s depth of insertion between 3:1 PC/PG and 1:1 PE/PG correlates well with the greater propensity for interactions of the peptides with the headgroup region of 1:1 PE/PG probably because PE is smaller and more accommodating than PC and/or the ionic content is greater in 1:1 PE/PG than 3:1 PC/PG. For reference, the depths of insertion from MD are added to the diagrams of the refined NMR structures in Figure 6. Table 2 lists the difference in depth of insertion between the N- and C-helical segments of each peptide (Δz) for both the NMRr and MD structures. This quantity ranges from 0.9 to 2.2 Å for the NMR structures, indicating a slight burial of the N-terminal segment with respect to the C-terminus. In contrast, Δz is smaller for the MD structures (Δz ± SE brackets zero in all cases) indicating that on average the N- and C-terminal segments experience similar depths of insertion. However, the rmsf for Δz is ∼1.5 Å from MD (Table 2), so the instantaneous Δz values from the simulations sample those from the NMR structures, as do the τ and ρ values.
As illustrated in Figure 8, phenylanalanines at the N-end reach well below the C2 atoms. For instance, F1 and F2 of p1 are buried 5.3 and 6 Å below the C2 atoms in PC/PG, respectively. While histidine side chains form a ridge at the interface between the polar and nonpolar regions of the membrane, the tips of the arginine and lysine residues reside in the headgroup region of the lipids where they snorkel to hydrogen bond and form salt bridges with the glycerol and phosphate groups of the lipids. On the N-side of the kink at G13, multiple phenylalanines facilitate deep insertion into the membrane whereas on the C-side, the electrostatic interactions between the lipid headgroups and the arginine and lysine residues at positions 14 and 18 drive the peptide to point toward the bilayer surface.40,65,66
Charge State of the Histidine Side Chains in 3:1 PC/PG Bicelles
Peptide charge is an important consideration in mechanistic studies of AMPs since it plays a major role in defining AMP–lipid interactions that the peptide relies on for membrane activity. Membrane-active peptides rely on arginine and lysine side chains for cationicity since their very high pKa favors the protonated and positively charged state in a membrane environment.66 However, histidine side chains have a much lower pKa (with an average of ∼6), therefore their charge state is most reliably determined in situ. To investigate their charge state in piscidin bound to lipid bilayers, OS ssNMR experiments were performed on p1 in PC/PG and supplemented with MD simulations done with different charge states of the histidine side chains. The ssNMR experiments offer the following two advantages: (1) only the bound state is detected since the unbound form is too dynamic to cross polarize; and (2) if a bound peptide exists in different charged states, their structures, orientations, and membrane locations will most likely differ (see depth from MD in Table S3) and lead to broad or even split NMR signals. At pH 6.0, single and sharp signals are obtained for each 15N resonance throughout the helical backbone of piscidin in both PC/PG and PE/PG (Figure 3, Table S2), therefore a single charge state of the peptide with each histidine side chain being either neutral or protonated must exist. As seen in Table S4, NMR structural parameters collected at pH 6.0 and 8.8 within the helical core (between positions 5 and 20) of p1 in 3:1 PC/PG are pH stable. This indicates that the same protonation state of the peptide is detected at both pH values and the neutral form of the four histidine side chains present at the higher pH of 8.8 must also be the one observed at pH 6.0. At position 2, the NMR restraints indicate that the fraying detected at pH 6.0 remains at pH 8.8. However, these effects are small since no major change in helicity was detected by CD when the pH was dropped from pH 7.4 to 6.0 (Table S1).
These ssNMR results are corroborated by the MD simulations performed on p1 and p3 with neutral histidine side chains since the structures and orientations derived from NMR and MD are in good agreement, as presented above. Furthermore, the sharp NMR signals indicate that the peptides are strongly bound to the bilayer, which is consistent with the MD results that each peptide’s CM inserts below the C2 atoms (Table S3). In contrast, the MD simulations done in PC/PG for p1 and p3 with charged histidine side chains show significant decreases in the peptide burial compared to those with neutral histidines. The protonation of H17 in addition to H3, H4, and H11 in p1 has more dramatic effects in two ways: it causes a 2.9 Å decrease in burial of the C-terminal segment of peptide (Table S3) and a 5° decrease in both τN and τC. This altered equilibrium bilayer orientation and positioning of p1 with four charged histidines is not consistent with the orientations derived from OS ssNMR data (Table 2) and therefore supports the neutral state of the histidine side chains of p1.
Discussion
Detailed Structural and Topological Determination of Amphipathic Helices Bound to Lipid Bilayers
AMPs are powerful multifunctional molecules that have inspired the design of new antimicrobial agents and methods of treatments. To improve the rational design of AMPs and to refine the overly simplified structures used in mechanistic models of AMPs, atomic-level information has been collected on p1 and p3. As noted in the Introduction, p1 is fully helical in SDS micelles, while only 45% of the peptide is helical in DPC micelles.19,20 The use of native-like bilayers reveals an intermediate structural behavior since the peptides are highly helical and generally straight but are frayed at their extremities and have a kink described by a 25° change in ρ between their N- and C-terminal residues. Formation of secondary structure, in this case helical, was expected since it is a main energetic driving force for membrane binding of amphipathic peptides and corresponds to a nonclassical hydrophobic effect for which binding is accompanied by negative enthalpy and entropy changes.67 Since the flexing at G13 enhances the hydrophobic moment of the peptides, it also improves the amphipathicity of the α-helix and therefore provides an energetic advantage. The piscidin peptides are less helical and inserted in 1:1 POPE/POPG than 3:1 DMPC/DMPG (Tables S1 and S3). Similar effects have been observed for other AMPs studied in the presence of bilayers containing PE and/or higher anionic content.68−76
The process of cross-validating the NMR and MD results as performed here establishes a rigorous approach for the detailed determination of in-plane helical structures and allows for the examination of specific details such as helical kinks, rotations, and fluctuations, all of which may be important for function. Four primary points are learned from approaching structural work in this manner. First, CD and MD are necessary to reinforce the need for greater priority on the DC over the CSA in refining the piscidin structures. The lower reliability of the CSA for this structural work may come from using a common 15N chemical shift tensor for all nonglycine residues;45,77 this approximation does not accommodate the possibility of small variations in rhombicity of the 15N CS tensors, which could be an important consideration for peptides that take on orientations nearly perpendicular to B0. In contrast, transmembrane helices may have smaller site to site variations in the axial components of their chemical shift tensors.78 Second, in the case where the peptide fluctuates with τ around 90° ± 10° and ρ around 240° ± 20° as determined by MD, the orientation of the time-average structure determined by refinement of the NMR data is very close to the average orientation directly obtained by MD, i.e., without bias averaging. This is consistent with previous studies that have shown insignificant experimental bias in the average τ when fluctuations in τ are <20°.47 Third, the rmsf for the azimuthal angles ρN and ρC is slightly larger (10–13°) than that of the tilt (7–9°), and together they illuminate the difficulty in describing these peptides by a single structural representation. The dynamic nature of interfacially bound peptides is an inherent behavior and worth determining since it may have functional meaning.28,33 Fourth, with consistency between the structures characterized by CD, dipolar waves, refinement of the NMR data, and MD, detailed comparisons of p1 and p3 in 3:1 PC/PG and 1:1 PE/PG bilayers can be confidently derived. Agreement also strengthens confidence in simulated atomistic characteristics of p1 and p3 such as their flexibility at G13 and depths of insertion. Moreover, since structures in MD trajectories cover a large conformational space while those refined from NMR represent an average, the MD simulations are useful in characterizing fluctuations on the orientation and depths of insertion of the peptide. Next, the orientations and bilayer positions of p1 and p3 are related to the amino acid content and bilayer composition.
Influence of Amino Acid Content on Peptide Orientation
In both PC/PG and PE/PG, the drive for optimized hydrophobic contacts between the bilayer hydrocarbon core and the peptides that have different amphipathicity at their N- and C-ends translates into tilted peptide orientations (τ ≠ 90°), a kink at the end of a G(X)4G motif, and sequence-dependent helical rotations on each side of this kink. Rotating and flexing are expected for these amphipathic peptides that are energetically driven to maximize their hydrophobic contacts with the membrane. The existence of a slight kink at G13 rather than an abrupt turn or bend is consistent with the finding by Lee et al.20 that the G13P mutation in p1 produces a peptide with lower helical content and impaired antimicrobial effects, as expected if the length of the C-terminal helix was critical to the peptide’s antimicrobial function. In general, p1 has a greater ρ than p3 indicating that piscidin’s orientation is dependent on the amino acid content. In this regard, aromatic residues known to be important for membrane partitioning and binding are very influential.64,73 Indeed, the average MD values of ρN (260°) and ρC (240°) for p1 (Table 2) allow H17 on the C-side of the kink to optimize its location in the hydrophilic region of the lipids, while the aromatic side chains of F1, F2 and F6 on the N-side of the kink are able to bury themselves in the hydrophobic core of the bilayer sitting 5–6 Å below the C2 atoms. A similar enhancement of interactions is displayed by p3 as a result of being kinked, but its ρN and ρC are smaller than those for p1, possibly because I2 in p3 does not need to be as centrally buried as F2 in p1 and F19 in p3 needs to be more buried than L19 in p1. The detailed characterization of p1 and p3 as done here in terms of not only secondary structure but also azimuthal rotation angles is important because it reveals that the glycine conserved at position 13 allows the two peptides to internally kink to the same extent (as measured by Δρ), while their different amino acid contents lead to differences in their absolute bilayer orientations (as measured by ρN and ρC). On one hand, the presence of a kink improves the amphipathicity of the peptides; on the other hand, it introduces an imperfection to their secondary structure.
Influence of Lipid Bilayer Composition and Peptide Amino Acid Content on Bilayer Positioning
To perform their membrane activity, AMPs need to find the right balance between hydrophobic and electrostatic interactions with the bilayer. These interactions define the bilayer position of each peptide and ultimately the degree of bilayer remodeling and disruption possible by the peptide. Here, the bilayer location of p1 and p3 is found to vary in two ways. First, the depth of insertion is influenced by the chemical nature of the amino acids on the hydrophobic and hydrophilic sides of the peptides. The highly conserved N-end of the peptide, which is more hydrophobic than the C-end, contains multiple membrane-anchoring phenylalanines as needed to satisfy strong hydrophobic interactions with the bilayer core. The more variable C-end contains two out of the three of the cationic residues and the only two nonhistidine polar residues. Moreover, it contains only three of the peptides’ 10 hydrophobic residues and therefore is likely responsible for membrane recognition and disruption involving the lipid headgroups. Separate use of the N- and C-ends for membrane anchoring and recognition has been observed in other AMPs.33,79,80 With the long arginine and lysine residues in the middle of the hydrophilic face and surrounded by ridges of shorter histidine residues, p1 and p3 may not be able to insert as deeply as if arginines and lysines rather than histidines were at the interface between hydrophobic and hydrophilic sectors. With regard to p1 in PE/PG, aromatic H17 compared to G17 in p3 and K14 compared to R14 in p3 may explain its shallower depth of insertion compared to p3. Second, bilayer composition affects the depth of insertion of piscidin, while its effects on Δρ and the hydrophobic moment are insignificant (Tables 2 and S3). Similar helical rotations for optimized hydrophobic interactions in PC/PG and PE/PG are understandable since these bilayers have similar hydrophobic cores that attract to them the nonpolar side of amphipathic peptides. It is intriguing that p1 and p3 are similarly active on S. aureus that lack PE but less active on E. coli that is abundant in PE. Arguably, this could be related to lesser insertion of the peptides in the PE-containing bacterial membranes.
Insights into the Mechanism of Antimicrobial Peptide Action
The mechanisms of action of AMPs, which are actively debated, include the barrel-stave or toroidal pores, the “grab and dip” carpet mechanism, and the disintegration of the bilayer via micellization; however these descriptions lack molecular details and rely on overly simplified peptide structures.1,3,12−16,81 Distinguishing between these mechanisms is not straightforward. For instance, the carpet model is described as an extension of the toroidal pore mechanism. Recently, Wimley,82 in the context of AMPs forming transient pores and preferring an equilibrium orientation parallel to the membrane, proposed interfacial activity as a semimolecular model that builds on the carpet and toroidal pore mechanisms and involves structures that are imperfectly amphipathic. Interfacial activity relies on the interfacial partitioning of an imperfectly amphipathic peptide to disrupt the vertical segregation between the lipid polar and nonpolar moieties and form transient pores comprised of peptides and lipids as well as solvent and solute molecules. In this view, permeabilization assays detect leakage from lipid vesicles occurring when solutes follow peptide molecules that are undergoing bilayer translocation until equilibrium concentrations are reached on each side of the membrane. In spite of the debate surrounding mechanistic studies of AMPs, there is consensus that depending on the bilayer composition of the targets, a given AMP could combine different mechanisms to achieve cell death more effectively. It is also understood that a given peptide may have to rely on different monomeric and possibly multimeric structures to support different steps of its mechanisms. This mechanistic adaptability of AMPs is consistent with the concept that the multiple structures and functions of AMPs are optimized in the specific pathogenic context faced by the host organism.83
It has been postulated that piscidin kills bacteria via a carpet mechanism leading to the formation of toroidal pores that incorporate lipids to line the hydrophilic side of the pore.19,28 More recently, p1 in 3:1 PC/PG bicellar preparations was shown to disrupt 6% of the phospholipids, which acquired mobility characteristic of micelles or small unilamellar vesicles, as expected in the carpet model.30 The results obtained here on both p1 and p3 in native-like membranes provide a unique opportunity to corroborate and add molecular details to these modes of action, at least in terms of monomeric form of the peptides at equilibrium. First, p1 and p3 are cationic and amphipathic, which allows them to adopt an in-plane orientation and accumulate on sticky anionic bilayer surfaces as needed to reach the threshold concentration beyond which they rupture the bilayers. Glukhov et al. described the in-plane orientation of AMPs as ideal to induce the greatest damage to bacterial cells because they can form “submarine-like” entities that can “unzip” bilayers.14 Second, p1 and p3 insert and rotate in the bilayer in a fashion that depends on the composition of the bilayer and amino acid content of the peptide. The strategic positions of aromatic residues allow the N-end of the peptides to insert more deeply in the bilayer than the C-end. Interestingly, members of the piscidin family invariably contain at least one phenylalanine at their amino end. In PE/PG, the stronger interactions with the lipid headgroups occur at a cost of reduced hydrophobic contacts with the hydrophobic core. Notably, the lesser insertion in 1:1 PE/PG than 3:1 PC/PG correlates with higher MICs in PE-containing bacteria. Thus, the nature of piscidin–lipid interactions involving the monomeric peptide and existing at equilibrium may prognosticate the degree of disruption under nonequilibrium conditions. Third, the kink at the end of the G(X)4G motif conserved in piscidin23,84−88 is preconditioned by the peptide’s amino acid sequences and reflects that the N- and C-ends have different orientational needs to interact optimally with and wedge themselves in the vertically segregated bilayer. While kinking in piscidin reduces the amphipathic imperfections of the helix, the imperfect amphipathicity reflected in the different orientations and locations of the N- and C-ends remains a source of bilayer distortion and thinning. Interestingly, a number of other AMPs studied in micelles also have kinks located at glycines from G(X)nG motifs.89−94 Most strikingly, magainin 2 has a G(X)4G motif between G13 and G18 and a central kink around G13,94 which occupies a helical wheel position similar to that of G13 in p1 and p3. Fourth, the equilibrium state of monomeric piscidin as described above is flexible at G13, which could play an important role in allowing the peptide to bend further, aggregate, and flip in the membrane to form transient pores. Fifth, structural and topological differences between p1 and p3 are helpful in identifying molecular determinants that may explain their different potencies. The combination of different amino acids at positions 14 and 17 of p1 as well as its larger volume (Figure S4) and lesser insertion compared to p3 may be related to its stronger biological activity in comparison to p3. The less inserted peptide in a given lipid system may be more disruptive because transient pores form more easily from peptides less embedded in the membrane. In addition, p1 is amphipathically less perfect than p3 in two ways. First, the particularly high concentration of aromatic residues at its N-end gives it an advantage in terms of further exacerbating the imbalance of amphipathicity between the N- and C-ends. Second, H17 in p1 increases its polar sector and clashes with the hydrophobic valine at position 10 (Figure 2 and green arrow in Figure S4). These “imperfections” may lead to enhanced ability to desegregate polar and nonpolar regions of the lipids and explain why p1 is a more efficacious AMP than p3. Further discussion of the bilayer response to the presence of p1 versus p3 and accompanying permeabilization assays in 3:1 PC/PG and 1:1 PE/PG will take place in a subsequent paper.
Conclusion
In this research, the high-resolution structures of p1 and p3 have been solved by OS ssNMR under biologically relevant conditions and with a level of detail that had not been previously achieved for purely amphipathic helices. MD simulations of the same peptides under the same conditions provide an independent determination of conformation and orientation. The approach presented here, which cross validates the NMR and MD results, provides a rigorous benchmark for the structural determination of interfacially bound α-helices and is very well suited for the study of other amphipathic peptides that fulfill important biological functions at the lipid bilayer. These atomic-level studies were necessary to reveal critical physicochemical features of the peptides that affect their amphipathicity and membrane-destabilizing potential, including a kink at the end of their G(X)4G motif, and different bilayer orientations and positions of their N- and C-ends. In biophysical terms, these studies provide an opportunity to revisit the model structures used to describe the mechanisms of action of AMPs. Specifically, results on p1 and p3 show that the peptides are preconditioned to flex at a central glycine so that their hydrophobic moment and amphipathicity are optimized in the bilayer and only the depth of insertion varies as function of lipid type. Imperfect amphipathicity results from differences in the physicochemical properties and bilayer positioning of the N- and C-segments. Flexing and tilting in the bilayer are advantageous to these peptides that need to adopt multiple structures and/or orientations as part of their mechanisms of action and adapt to changing environmental conditions. The sequences of p1 and p3 differ in several places, but the detailed structural studies presented here indicate that the higher potency of p1 may be achieved through strategic placement of two key residues: aromatic F2 in the N-terminus of the peptide enhances its anchoring capability and helical stability, while the polar H17 in the nonpolar sector creates the type of imperfection that enhances membrane distortion and thinning. Overall, this research reveals that p1 and p3 adopt disrupted α-helical structures that correct for the different amphipathicities of their N- and C-ends. Bound to different bacterial cell mimics, piscidin is tilted, kinked, and immersed in the bilayer, which enables its disruptive effects of diverse bilayers even in the monomeric state. For the first time, the atomic-level differences between p1 and p3 have become available to test new hypotheses about the modes of action of AMPs and facilitate the design of new drugs with enhanced antimicrobial activity and specificity for microbial membranes.
Acknowledgments
This research was supported in part by a grant from the National Science Foundation (CHE-0832571) and the Intramural Research Program of the NIH, National Heart, Lung and Blood Institute, and utilized the high-performance computational capabilities at the National Institutes of Health, Bethesda, MD (NHLBI LoBoS cluster). This work also utilized the Center for NMR Spectroscopy and Imaging of Proteins at the University of California, San Diego supported by the National Institutes of Health (P41EB002031). The authors are grateful for NMR time received at the National High Magnetic Field Laboratory (NHMFL) supported by the NSF Cooperative Agreement DMR-1157490, the State of Florida, and the U.S. Department of Energy.
Supporting Information Available
Figures and tables as well as details on sample preparation, NMR signal assignments, wave calculations, and statistical analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# Vanderbilt University Institute of Imaging Science (VUIIS), Department of Radiology, Nashville, Tennessee 37232
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fjell C. D.; Hiss J. A.; Hancock R. E. W.; Schneider G. Nat. Rev. Drug Discov. 2012, 11, 37–51. [DOI] [PubMed] [Google Scholar]
- Mookherjee N.; Hancock R. E. W. Cell. Mol. Life Sci. 2007, 64, 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haney E. F.; Hancock R. E. W. Biopolymers 2013, 6, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyansky A. A.; Chugunov A. O.; Vassilevski A. A.; Grishin E. V.; Efremov R. G. Curr. Protein Pept. Sci. 2012, 13, 644–657. [DOI] [PubMed] [Google Scholar]
- Last N. B.; Schlamadinger D. E.; Miranker A. D. Protein Sci. 2013, 22, 870–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. Pharmaceuticals 2013, 6, 728–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boman H. G. J. Intern. Med. 2003, 254, 197–215. [DOI] [PubMed] [Google Scholar]
- Zasloff M. Nature 2002, 415, 389–395. [DOI] [PubMed] [Google Scholar]
- Krauson A. J.; He J.; Wimley W. C. J. Am. Chem. Soc. 2012, 134, 12732–12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uematsu N.; Matsuzaki K. Biophys. J. 2000, 79, 2075–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig J. Biochim. Biophys. Acta 2004, 1666, 40–50. [DOI] [PubMed] [Google Scholar]
- Powers J. P.; Hancock R. E. Peptides 2003, 24, 1681–1691. [DOI] [PubMed] [Google Scholar]
- Yin L. M.; Edwards M. A.; Li J.; Yip C. M.; Deber C. M. J. Biol. Chem. 2012, 287, 7738–7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glukhov E.; Stark M.; Burrows L. L.; Deber C. M. J. Biol. Chem. 2005, 280, 33960–33967. [DOI] [PubMed] [Google Scholar]
- Dathe M.; Wieprecht T. Biochim. Biophys. Acta 1999, 1462, 71–87. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Guarnieri M. T.; Vasil A. I.; Vasil M. L.; Mant C. T.; Hodges R. S. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Li X.; Wang Z. Nucleic Acids Res. 2009, 37, D933–D937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todokoro Y.; Yumen I.; Fukushima K.; Kang S.-W.; Park J.-S.; Kohno T.; Wakamatsu K.; Akutsu H.; Fujiwara T. Biophys. J. 2006, 91, 1368–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagna S.; Saint N.; Molle G.; Aumelas A. Biochemistry 2007, 46, 1771–1778. [DOI] [PubMed] [Google Scholar]
- Lee S.-A.; Kim Y. K.; Lim S. S.; Zhu W. L.; Ko H.; Shin S. Y.; Hahm K.-S.; Kim Y. Biochemistry 2007, 46, 3653–3663. [DOI] [PubMed] [Google Scholar]
- Silphaduang U.; Noga E. Nature 2001, 414, 268–269. [DOI] [PubMed] [Google Scholar]
- Lauth X.; Shike H.; Burns J. C.; Westerman M. E.; Ostland V. E.; Carlberg J. M.; Olst J. C. V.; Nizet V.; Taylor S. W.; Shimizu C.; Bulet P. J. Biol. Chem. 2002, 277, 5030–5039. [DOI] [PubMed] [Google Scholar]
- Lauth X.; Shike H.; Burns J. C.; Westerman M. E.; Ostland V. E.; Carlberg J. M.; Van Olst J. C.; Nizet V.; Taylor S. W.; Shimizu C.; Bulet P. J. Biol. Chem. 2002, 277, 5030–5039. [DOI] [PubMed] [Google Scholar]
- Menousek J.; Mishra B.; Hanke M. L.; Heim C. E.; Kielian T.; Wang G. Int. J. Antimicrob. Agents 2012, 39, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Watson K. M.; Peterkofsky A.; Buckheit R. W. Jr. Antimicrob. Agents Chemother. 2010, 54, 1343–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W. S.; Lee J.; Lee D. G. Biol. Pharm. Bull. 2008, 31, 1906–1910. [DOI] [PubMed] [Google Scholar]
- Lin H.-J.; Huang T.-C.; Muthusamy S.; Lee J.-F.; Duann Y.-F.; Lin C.-H. Zool. Sci. 2012, 29, 327–332. [DOI] [PubMed] [Google Scholar]
- Chekmenev E. Y.; Vollmar B. S.; Forseth K. T.; Manion M. N.; Jones S. M.; Wagner T. J.; Endicott R. M.; Kyriss B. P.; Homem L. M.; Pate M.; He J.; Raines J.; Gor’kov P. L.; Brey W. W.; Mitchell D. J.; Auman A. J.; Ellard-Ivey M. J.; Blazyk J.; Cotten M. Biochim. Biophys. Acta 2006, 1758, 1359–1372. [DOI] [PubMed] [Google Scholar]
- Vogt T. C.; Bechinger B. J. Biol. Chem. 1999, 274, 29115–29221. [DOI] [PubMed] [Google Scholar]
- De Angelis A. A.; Grant C. V.; Baxter M. K.; McGavin J. A.; Opella S. J.; Cotten M. L. Biophys. J. 2011, 101, 1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekmenev E. Y.; Jones S. M.; Nikolayeva Y. N.; Vollmar B. S.; Wagner T. J.; Gor’kov P. L.; Brey W. W.; Manion M. N.; Daugherty K. C.; Cotten M. J. Am. Chem. Soc. 2006, 128, 5308–5309. [DOI] [PubMed] [Google Scholar]
- Fu R.; Gordon E. D.; Hibbard D. J.; Cotten M. J. Am. Chem. Soc. 2009, 131, 10830–10831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekmenev E. Y.; Vollmar B. S.; Cotten M. Biochim. Biophys. Acta 2010, 1798, 228–234. [DOI] [PubMed] [Google Scholar]
- Mihajlovic M.; Lazaridis T. Biochim. Biophys. Acta 2012, 1818, 1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmanpour A.; Ghahremanpour M. M.; Mehrnejad F.; Moghaddam M. E. J. Biomol. Struct. Dyn. 2012, 1–11. [DOI] [PubMed] [Google Scholar]
- Mehrnejad F.; Zarei M. J. Biomol. Struct. Dyn. 2010, 27, 551–559. [DOI] [PubMed] [Google Scholar]
- Fu R.; Cross T. A. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 235–268. [DOI] [PubMed] [Google Scholar]
- Opella S. J.; Marassi F. M. Chem. Rev. 2004, 104, 3587–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechinger B.; Salnikov E. S. Chem. Phys. Lipids 2012, 165, 282–301. [DOI] [PubMed] [Google Scholar]
- Hong M.; Su Y. Protein Sci. 2011, 20, 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesleh M. F.; Opella S. J. J. Magn. Reson. 2003, 163, 288–299. [DOI] [PubMed] [Google Scholar]
- Kovacs F. A.; Denny J. K.; Song Z.; Quine J. R.; Cross T. A. J. Mol. Biol. 2000, 295, 117–125. [DOI] [PubMed] [Google Scholar]
- Schwieters C. D.; Kuszewski J. J.; Tjandra N.; Marius Clore G. J. Magn. Reson. 2003, 160, 65–73. [DOI] [PubMed] [Google Scholar]
- Sharma M.; Yi M.; Dong H.; Qin H.; Peterson E.; Busath D. D.; Zhou H. X.; Cross T. A. Science 2010, 330, 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y.; Schwieters C. D.; Opella S. J.; Marassi F. M. J. Magn. Reson. 2012, 214, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. H.; Das B. B.; Casagrande F.; Tian Y.; Nothnagel H. J.; Chu M.; Kiefer H.; Maier K.; De Angelis A. A.; Marassi F. M.; Opella S. J. Nature 2012, 491, 779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban-MartÌn S.; Strandberg E.; Fuertes G.; Ulrich A. S.; Salgado J. S. Biophys. J. 2009, 96, 3233–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gor’kov P. L.; Chekmenev E. Y.; Li C.; Cotten M.; Buffy J. J.; Traaseth N. J.; Veglia G.; Brey W. W. J. Magn. Reson. 2007, 185, 77–93. [DOI] [PubMed] [Google Scholar]
- Blazyk J.; Wiegand R.; Klein J.; Hammer J.; Epand R. M.; Epand R. F.; Maloy W. L.; Kari U. P. J. Biol. Chem. 2001, 276, 27899–27906. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. III; MacKerell A. D. Jr.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D. Jr.; Bashford D.; M. Bellott R. L.; Dunbrack; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiorkiewicz-Kuczera J.; Yin D.; Karplus M. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable R. M.; Luo Y.; Gawrisch K.; Roux B.; Pastor R. W. J. Phys. Chem. B 2013, 117, 10183–10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. J. Comput. Chem. 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- Hoover W. G. Phys. Rev. A 1985, 31, 1695–1697. [DOI] [PubMed] [Google Scholar]
- Nose S. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar]
- Feller S. E.; Zhang Y. H.; Pastor R. W.; Brooks. B. R. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Perrin B. S. Jr.; Pastor R. W.; Cotten M. In Advances in Biological Solid-State NMR: Proteins and Membrane-Active Peptides; Separovic F., Naito A., Eds.; Royal Society of Chemistry: Cambridge, 2014, p 18–35. [Google Scholar]
- Wang J.; Denny J.; Tian C.; Kim S.; Mo Y.; Kovacs F.; Song Z.; Nishimura K.; Gan Z.; Fu R.; Quine J. R.; Cross T. A. J. Magn. Reson. 2000, 144, 162–167. [DOI] [PubMed] [Google Scholar]
- Kwan A. H.; Mobli M.; Gooley P. R.; King G. F.; Mackay J. P. FEBS J. 2011, 278, 687–703. [DOI] [PubMed] [Google Scholar]
- Comellas G.; Rienstra C. M. Annu. Rev. Biophys. 2013, 42, 515–536. [DOI] [PubMed] [Google Scholar]
- Wimley W. C.; White S. H. Nat. Struct. Mol. Biol. 1996, 3, 842–848. [DOI] [PubMed] [Google Scholar]
- Shaw J. E.; Epand R. F.; Hsu J. C. Y.; Mo G. C. H.; Epand R. M.; Yip C. M. J. Struct. Biol. 2008, 162, 121–138. [DOI] [PubMed] [Google Scholar]
- Hristova K.; Wimley W. C. J. Membr. Biol. 2011, 239, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beschiaschvili G.; Seelig J. Biochemistry 1992, 31, 10044–10053. [DOI] [PubMed] [Google Scholar]
- Sui S.-F.; Wu H.; Guo Y.; Chen K.-S. J. Biochem. 1994, 116, 482–487. [DOI] [PubMed] [Google Scholar]
- Heller W. T.; He K.; Ludtke S. J.; Harroun T. A.; Huang H. W. Biophys. J. 1997, 73, 239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romo T. D.; Bradney L. A.; Greathouse D. V.; Grossfield A. Biochim. Biophys. Acta 2011, 1808, 2019–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.-Y.; Lee M.-T.; Huang H. W. Biophys. J. 2002, 82, 908–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason A. J.; Chotimah I. N. H.; Bertani P.; Bechinger B. Mol. Membr. Biol. 2006, 23, 185–194. [DOI] [PubMed] [Google Scholar]
- Wieprecht T.; Beyermann M.; Seelig J. Biochemistry 1999, 38, 10377–10387. [DOI] [PubMed] [Google Scholar]
- Toke O. Pept. Sci. 2005, 80, 717–735. [DOI] [PubMed] [Google Scholar]
- Seto G. W. J.; Marwaha S.; Kobewka D. M.; Lewis R. N. A. H.; Separovic F.; McElhaney R. N. Biochim. Biophys. Acta 2007, 1768, 2787–2800. [DOI] [PubMed] [Google Scholar]
- Hallock K. J.; Lee D. K.; Ramamoorthy A. Biophys. J. 2003, 84, 3052–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell K. W.; Chekmenev E. Y.; Wittebort R. J. J. Am. Chem. Soc. 2004, 127, 9030–9035. [DOI] [PubMed] [Google Scholar]
- Park S.; Marassi F.; Black D.; Opella S. Biophys. J. 2010, 99, 1465–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S.; Sharma B.; Raghava G. BMC Bioinformatics 2007, 8, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kini R. M.; Evans H. J. Int. J. Pept. Protein Res. 1989, 34, 277–286. [DOI] [PubMed] [Google Scholar]
- Brogden K. A. Nat. Rev. Microbiol. 2005, 3, 239–250. [DOI] [PubMed] [Google Scholar]
- Wimley W. C. ACS Chem. Biol. 2010, 5, 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman M. R.; Yount N. Y. ASM News 2005, 71, 21–27. [Google Scholar]
- Sun B. J.; Xie H. X.; Song Y.; Nie P. J. Fish Dis. 2007, 30, 335–343. [DOI] [PubMed] [Google Scholar]
- Silphaduang U.; Noga E. J. Nature 2001, 414, 268–269. [DOI] [PubMed] [Google Scholar]
- Ruangsri J.; Fernandes J. M. O.; Rombout J.; Brinchmann M. F.; Kiron V. BMC Vet. Res. 2012, 8, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z.-X.; He W.; Chen W.-J.; Yan J.-H.; Yang J.-N.; Chan S.-M.; He J.-G. Aquaculture 2006, 253, 204–211. [Google Scholar]
- Niu S.-F.; Jin Y.; Xu X.; Qiao Y.; Wu Y.; Mao Y.; Su Y.-Q.; Wang J. Fish Shellfish Immunol. 2013, 35, 513–524. [DOI] [PubMed] [Google Scholar]
- Haney E. F.; Hunter H. N.; Matsuzaki K.; Vogel H. J. Biochim. Biophys. Acta 2009, 1788, 1639–1655. [DOI] [PubMed] [Google Scholar]
- Chi S.-W.; Kim J.-S.; Kim D.-H.; Lee S.-H.; Park Y.-H.; Han K.-H. Biochem. Biophys. Res. Commun. 2007, 352, 592–597. [DOI] [PubMed] [Google Scholar]
- Toke O.; Bánóczi Z.; Király P.; Heinzmann R.; Bürck J.; Ulrich A.; Hudecz F. Eur. Biophys. J. 2011, 40, 447–462. [DOI] [PubMed] [Google Scholar]
- Chia B. C. S.; Carver J. A.; Mulhern T. D.; Bowie J. H. Eur. J. Biochem. 2000, 267, 1894–1908. [DOI] [PubMed] [Google Scholar]
- Wang G. J. Biol. Chem. 2008, 283, 32637–32643. [DOI] [PubMed] [Google Scholar]
- Gesell J.; Zasloff M.; Opella S. J. Biomol. NMR 1997, 9, 127–135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.