

Abstract
Internalization into cancer cells is a significant challenge in the delivery of many anticancer therapeutics. Drug carriers can address this challenge by facilitating cellular uptake of cytotoxic cargo in the tumor, while preventing cellular uptake in healthy tissues. Here we describe an extrinsically controlled drug carrier, a nanopeptifier, that amplifies cellular uptake by modulating the activity of cell-penetrating peptides with thermally toggled self-assembly of a genetically encoded polypeptide nanoparticle. When appended with a proapoptotic peptide, the nanopeptifier creates a cytotoxic switch, inducing apoptosis only in its self-assembled state. The nanopeptifier provides a new approach to tune the cellular uptake and activity of anticancer therapeutics by an extrinsic thermal trigger.
Keywords: Cell-penetrating peptide, elastin-like polypeptide, self-assembly, cancer, targeted drug delivery
Anticancer therapeutics face major transport barriers between their site of administration and their intended site of action in the tumor. Most systemically delivered therapies must overcome barriers to prevent clearance of drug from the circulation, enhance the penetration and accumulation of drug in the tumor tissue, and achieve internalization of drug in cancer cells, a final barrier that is critical to overcome for drugs with intracellular therapeutic targets. Cellular uptake can be a significant challenge for therapeutics, such as peptide drugs, as the cell membrane may act as an impermeable barrier to their entry and thereby prevent internalization.
The challenge of enhancing cellular uptake of drug in the tumor can be addressed with drug carriers that selectively maximize their uptake by cancer cells, while minimizing their uptake by cells in healthy tissues. Affinity targeting is one approach to specifically enhance uptake in the tumor by functionalizing drug carriers with targeting ligands that preferentially interact with receptors overexpressed on cancer cells and induce internalization of the bound complex. However, this approach is limited in its application to specific types of cancer1 and subsets of patients2−5 that reliably express the targeted receptor at levels that are significantly above healthy tissues. An alternative approach that nonspecifically enhances cellular uptake relies on cell-penetrating peptides (CPPs) that can ferry diverse cargo such as peptides, proteins, liposomes, and micelles across cell membranes by a receptor-independent mechanism.6−9 This nonspecific means of cellular uptake, however, results in a lack of selectivity, as the promiscuity of CPPs will lead to uptake into most cells that they encounter as they traverse the body.
Approaches to harness the cellular uptake of CPPs for tumor-targeted internalization typically seek to activate CPP function in response to a tumor-specific trigger, such as overexpressed tumor enzymes or low tumor pH.10 These approaches include activation of CPP function by the removal of stealth coatings to reveal CPPs,11−15 the activation of molecular actuators to expose CPPs beyond protective coronas,16 or the release of CPPs from ionically associated inhibitors.17−21 These approaches successfully control CPP activity but rely on intrinsic features of the tumor and thus are subject to limitations of tumor heterogeneity, similar to affinity targeting.
Herein, we demonstrate a method to extrinsically tune CPP activity with thermally toggled self-assembly of a genetically encoded polypeptide nanoparticle and thereby control the intracellular delivery of a cytotoxic cargo. We call this platform the nanopeptifier, by analogy to an electronic amplifier (Figure 1). The nanopeptifier consists of three modular, genetically encoded peptide elements. The first element is an externally triggered digital switch, which consists of a temperature-responsive elastin-like polypeptide diblock copolymer (ELPBC) that self-assembles into a spherical micelle above a critical temperature.22 This digital switch of morphology from unimer to micelle controls the activity of the second element, a CPP that is appended to the hydrophilic terminus of the ELPBC. Working in concert, the ELPBC and CPP act as an amplifier of cellular uptake by modulating the density and thus the activity of the CPP on the nanoparticle surface and enhancing the cellular uptake of the micelle (high CPP density), as compared to the unimer (low CPP density). The third element of the nanopeptifier is a therapeutic payload. A proapoptotic peptide drug was chosen as a stringent test of the nanopeptifier due to the inherently poor intracellular delivery of many peptide drugs.
Figure 1.
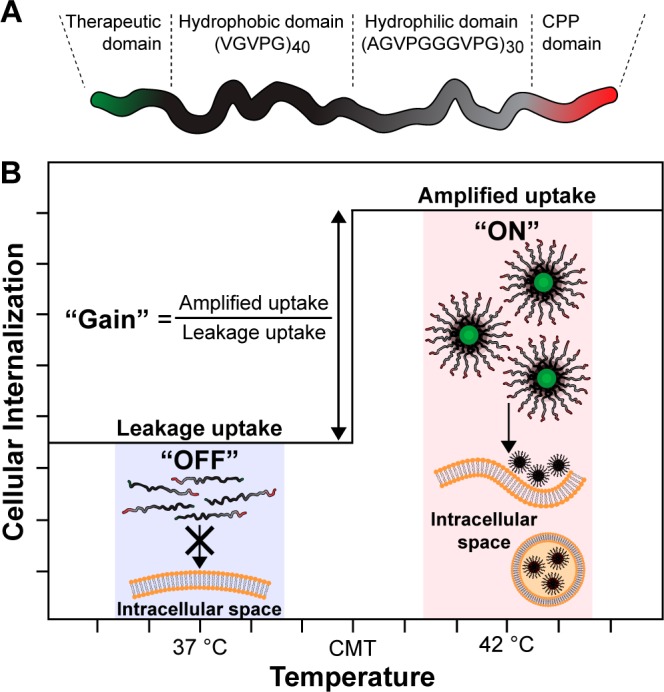
Design and function of an ELPBC nanopeptifier. (A) Each nanopeptifier is a ternary fusion of a therapeutic payload, an ELPBC comprised of a hydrophobic and hydrophilic ELP domain, and a CPP. (B) At physiologic temperature (“off” state, 37 °C), nanopeptifiers are soluble unimers, displaying a single CPP on their hydrophilic terminus. At conditions of mild clinical hyperthermia (“on” state, 42 °C), nanopeptifiers self-assemble into spherical micelles, displaying a high density of CPPs on the micelle corona. The increased CPP density amplifies cellular uptake in the micelle state, as compared to the unimer state. Important nanopeptifier parameters include: leakage uptake, “off” state uptake at 37 °C; amplified uptake, “on” state uptake at 42 °C; gain, ratio of “on” to “off” state uptake.
The nanopeptifier provides controlled and tunable intracellular delivery of a therapeutic payload by the modulation of cellular uptake triggered by an external thermal input. The choice of the CPP element (ranging from weak to strong in its cell-penetrating ability) and the modulation of its interfacial density by nanoparticle self-assembly creates a system in which three important parameters can be controlled: (1) the “off” state-leakage-uptake that controls systemic off-target toxicity, (2) the “on” state-amplified-uptake that controls targeted anticancer cell toxicity, and (3) the “gain” in cellular uptake, defined as the fold-increase between the amplified uptake and leakage uptake that controls the selectivity of intracellular delivery between target and off-target cells (Figure 1). The nanopeptifier platform thus provides a library of stimulus-responsive carriers that can probe physiological questions critical to the success of actively targeted drug carriers by examining the importance of limiting cellular uptake in the carrier’s “off” state, increasing uptake in the “on” state and providing sufficient amplification between these two states to tune the controlled delivery of a cytotoxic payload.
The nanopeptifier significantly advances our previous efforts toward building a digital switch of cellular uptake.23 In this previous study, ELPBCs functionalized with a penta-arginine (Arg5) CPP were designed to self-assemble in response to conditions of mild hyperthermia for which 42 °C is a typical temperature achieved in the clinical setting.24,25 We chose mild hyperthermia as the extrinsic trigger of self-assembly because it can be applied to tumors in a variety of anatomical locations by the use of focused microwaves, radiofrequency, or ultrasound.26−28 In cell culture, raising the temperature from 37 to 42 °C triggered the self-assembly of the Arg5-ELPBC into spherical micelles, thereby resulting in a high interfacial density of Arg residues, which enhanced intracellular accumulation of the micelles at 42 °C, as compared to the Arg5-ELPBC unimers at 37 °C. These preliminary results showed that intracellular uptake could be controlled by the manipulation of CPP density with hyperthermia-triggered nanoparticle assembly.23 However, with only a single CPP-ELPBC used in this previous study, none of the three parameters of interest, the leakage uptake, amplified uptake, or gain, could be tuned and the lack of a therapeutic payload precluded the functional evaluation of these parameters in the context of targeted drug delivery and anticancer efficacy.
The nanopeptifier library investigated herein is composed of an ELPBC that is genetically appended with different CPPs at the C-terminus of the hydrophilic ELPBC domain, such that the CPP is displayed on the nanoparticle corona upon micelle assembly under conditions of mild hyperthermia (Table 1). In order to modulate the performance of the nanopeptifier, we chose a diverse set of CPPs that include peptides that are nonfunctional when presented as a monovalent entity, such as Arg5,29−31 peptides that are potent even in a monovalent state, such as Arg8 and TAT, and a peptide with intermediate potency, such as a modified TAT sequence (RTAT), which conserves the Arg residues in the TAT CPP while replacing all other residues with glycine.
Table 1. Library of CPP-ELPBC Nanopeptifiers.
| nanopeptifier | CPP functionalization | MW (kDa) | RH at 37 °C (nm)a | RH at 42 °C (nm)a |
|---|---|---|---|---|
| Arg5-ELPBC | RRRRR | 40.50 | 5.7 ± 0.3 | 20.8 ± 1.0 |
| Arg8-ELPBC | RRRRRRRR | 40.97 | 5.4 ± 0.4 | 21.2 ± 0.3 |
| TAT-ELPBCb | YGRKKRRQRRR | 41.26 | 5.7 ± 0.3 | 21.4 ± 0.3 |
| RTAT-ELPBCc | YGRGGRRGRRR | 41.05 | 6.5 ± 0.2 | 21.8 ± 0.3 |
| ELPBC | none | 39.72 | 6.2 ± 0.1 | 20.3 ± 0.3 |
Hydrodynamic radius determined by dynamic light scattering of ELPBC at 10 μM in PBS. Data represents mean of three measurements ± SEM.
TAT sequence corresponds to residues 47–57 from the transactivator of transcription of HIV.
RTAT was modified to explore the effects of arginine architecture on CPP function, where all nonarginine residues of TAT were replaced with neutral glycine residues.
Genes encoding the nanopeptifier library of CPP-ELPBCs (Supporting Information Figure S1) were synthesized by recursive directional ligation by plasmid reconstruction,32 expressed in E. coli from a plasmid-borne gene and purified by inverse transition cycling.33 The size and purity of the CPP-ELPBCs was confirmed by SDS-PAGE (Supporting Information Figure S2A) and the thermal properties of the CPP-ELPBCs were measured by temperature-regulated turbidimetry and dynamic light scattering (DLS). Turbidimetry measurements of CPP-ELPBC solutions at 15 μM, as a function of solution temperature, revealed a modest increase in optical density (OD at 350 nm of <0.1), which is indicative of the transition from unimer to micelle between 38 and 41 °C, defined as the critical micellization temperature (CMT). Increasing the temperature beyond ∼54–66 °C resulted in a significantly greater optical density (OD at 350 nm of >1.5) that signals the aggregation of CPP-ELPBC micelles into micrometer size coacervates (Figure 2). The incorporation of a CPP at the hydrophilic terminus of the ELPBC did not perturb temperature-triggered self-assembly of the CPP-ELPBC, as confirmed by DLS, since all CPP-ELPBCs and a nonfunctionalized ELPBC control existed as unimers at 37 °C with a hydrodynamic radius (RH) < 10 nm, while a RH of ∼20 nm was measured at 42 °C, indicating self-assembly into micelles (Table 1). Although CPP-functionalization did not perturb the CMT of the ELPBC, it did influence the temperature at which micelle aggregation occurred. Interestingly, the Arg5 functionalization depressed the temperature at which micelle aggregation was observed, as compared to nonfunctionalized ELPBC (Figure 2). This effect likely stems from the relative hydrophobicity of the arginine amino acid, despite its polar cationic character, due to the delocalized charge of its guanidinium headgroup.34−36 However, the effect of arginine functionalization is likely a complex combination of hydrophobic and electrostatic factors, as increasing the net charge from Arg5 to Arg8 functionalization did not further depress the micelle aggregation temperature but rather increased slightly the temperature at which micelle aggregation occurred.
Figure 2.

Characterization of CPP-ELPBC nanopeptifiers by temperature-regulated turbidimetry. A moderate increase in optical density (OD < 0.1) of solutions at 15 μM is suggestive of self-assembly of ELPBC unimers into spherical micelles between 38 and 41 °C. A drastic increase in optical density indicates coacervation of CPP-ELPBCs into micrometer size aggregates between approximately 54 and 66 °C. Arg5- and Arg8-ELPBCs are shown here as representative examples of the thermal behavior of the CPP-ELPBC library, demonstrating that CPP-functionalization does not perturb the CMT, as compared to the nonfunctionalized ELPBC control, but does influence the temperature at which aggregation of the micelles occurs.
Controlled cellular uptake as a function of temperature-triggered CPP-ELPBC micelle assembly was visualized by live cell confocal fluorescence microscopy. HeLa cervical cancer cells were incubated for 1 h with 15 μM of Alexa Fluor 488-labeled CPP-ELPBCs at 37 or 42 °C. Nonfunctionalized ELPBC showed no visible uptake at either thermal condition (Figure 3A,B), demonstrating that in the absence of a CPP moiety self-assembly of the ELPBCs alone results in negligible amplification of internalization. The ELPBC can thus serve as a useful scaffold for self-assembly wherein the performance parameters of the nanopeptifier can be encoded primarily by the CPP moiety. Arg5-ELPBC demonstrated little uptake at 37 °C in its unimer state but exhibited significantly greater cellular uptake at 42 °C (Figure 3C,D). These results are consistent with our previous study in which we showed that although Arg5 is a nonfunctional CPP as a single copy presented by an ELP unimer, it shows evidence of cell-penetrative ability when presented at a high interfacial density on the micelle corona.23
Figure 3.
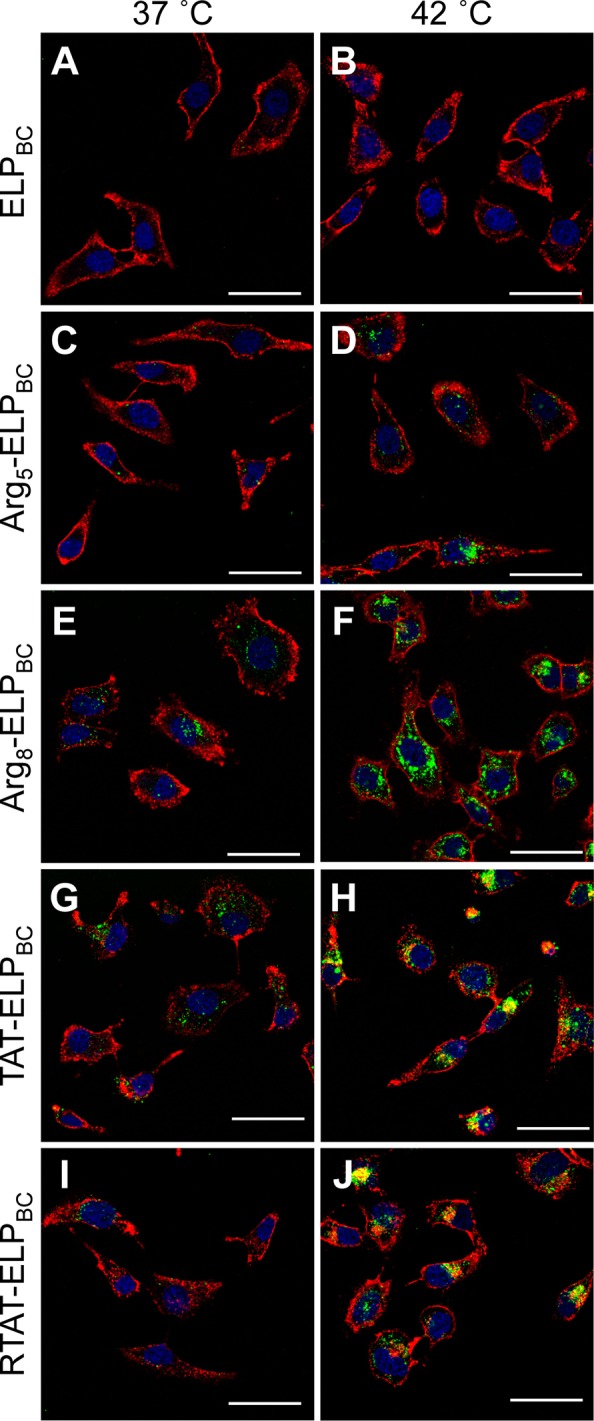
Visualization of controlled cellular uptake of CPP-ELPBCs by confocal fluorescence microscopy. HeLa cells were incubated for 1 h at 37 or 42 °C with 15 μM ELPBC (A,B), Arg5-ELPBC (C,D), Arg8-ELPBC (E,F), TAT-ELPBC (G,H), or RTAT-ELPBC (I,J). Cells were briefly incubated with Alexa 594 wheat agglutinin and Hoechst 33342 to stain the cell membrane and cell nuclei, respectively. At 42 °C, the CPP-ELPBCs self-assembled into micelles displaying enhanced CPP density on the micelle corona, which resulted in amplified cellular uptake, though to differing extents, for the CPP-ELPBCs, as compared to 37 °C, at which all CPP-ELPBCs existed as soluble unimers presenting only a single CPP per polypeptide. In contrast, the nonfunctionalized ELPBC control showed no difference in cellular uptake between 37 and 42 °C. Red, cell membrane; blue, cell nuclei; green, ELPBC; scale bars, 50 μm.
In contrast, when the ELPBC was appended with a functional CPP, such as Arg8 (Figure 3E), TAT (Figure 3G), or RTAT (Figure 3I), there was visible uptake even at the “off” state of 37 °C, which is consistent with the known ability of a single copy of these CPPs to cause cellular uptake. Cellular internalization was dramatically amplified when a high density of these CPPs, Arg8, TAT, and RTAT, were presented by the CPP-ELPBC micelles, as seen by the strong intracellular fluorescence at 42 °C (Figure 3F,H,J). Nanopeptifiers that display functional CPPs thus provide greatly enhanced uptake at their “on” state but at the cost of increased uptake in their “off” state, which is particularly pronounced for Arg8- and TAT-ELPBCs. The characteristic punctate appearance of internalized CPP-ELPBCs indicates that uptake of all nanopeptifiers occurs by endocytic mechanisms at both 37 and 42 °C.
Quantification of cellular uptake by flow cytometry confirmed the tunable intracellular delivery by thermally triggered CPP-ELPBC micelle assembly. All CPP-ELPBCs demonstrated an amplification of uptake at 42 °C, as compared to 37 °C, while the nonfunctionalized ELPBC control showed no significant difference in uptake between the “off” (37 °C) and “on” (42 °C) state (Figure 4A). This supported our hypothesis that the interfacial CPP density that is controlled by micelle assembly controls the amplification in cellular uptake, and that mild hyperthermia alone does not result in the observed effect. While all CPP-ELPBCs demonstrated an amplification of cellular uptake with temperature-triggered micelle assembly, the amplitude of uptake at the “off” and “on” temperatures varied greatly depending on the potency of the nanopeptifier’s CPP domain. The gain, calculated as the ratio of cellular fluorescence at 42 °C as compared 37 °C, demonstrated the controlled internalization afforded by temperature-triggered micelle assembly of the nanopeptifier, while also revealing the variation in nanopeptifier performance depending on the CPP functionalization (Figure 4B).
Figure 4.

Quantification of controlled cellular uptake of CPP-ELPBCs by flow cytometry. HeLa cells were incubated with 10 μM CPP-ELPBCs for 1 h at 37 or 42 °C. (A) Internalization was quantified by cellular fluorescence in live cells. All CPP-ELPBCs showed significant increase in cellular fluorescence at 42 °C, as compared to 37 °C, whereas nonfunctionalized ELPBC showed no difference in uptake between the two temperatures. *Indicates p < 0.01 between thermal conditions (two-way ANOVA, Bonferroni posthoc test for multiple comparisons). (B) Gain in cellular uptake was defined as the ratio of cellular fluorescence at 42 and 37 °C. *Indicates p < 0.0125 versus ELPBC control (ANOVA, Bonferroni posthoc test for multiple comparisons). Data represents mean of three experiments ± SEM.
Flow cytometry quantified the cellular uptake and provided quantitative figures-of-merit (FOM) for the nanopeptifier that are likely to control the partitioning of CPP-ELPBC between healthy and tumor tissue by focused mild hyperthermia (Table 2). The intracellular uptake at 37 and 42 °C for each CPP-ELPBC, as expressed in arbitrary units of cellular fluorescence, was defined as the leakage and amplified uptake, respectively. The ratio of amplified to leakage uptake was defined as the gain achieved by each CPP-ELPBC. Definition of these parameters provided a self-consistent approach to comparing nanopeptifier performance between CPP-ELPBCs. The range of values for each FOM suggested that these three critical parameters could be tuned across one to two orders of magnitude within this nanopeptifier library. To our knowledge, no other system for selective cell uptake shows such tunability.
Table 2. Nanopeptifier Figures-of-Merit Quantified by Flow Cytometry.
| nanopeptifier | CPP functionalization | leakage uptakea | amplified uptakea | gainb |
|---|---|---|---|---|
| Arg5-ELPBC | RRRRR | 2.5 | 12.5 | 5.0 |
| Arg8-ELPBC | RRRRRRRR | 11.0 | 129.4 | 11.8 |
| TAT-ELPBC | YGRKKRRQRRR | 10.0 | 101.9 | 10.2 |
| RTAT-ELPBC | YGRGGRRGRRR | 2.9 | 23.0 | 7.9 |
| ELPBC | none | 1.0 | 0.9 | 0.9 |
Measured in arbitrary units of cellular fluorescence, normalized to leakage uptake of nonfunctionalized ELPBC.
Defined as the ratio of amplified to leakage uptake.
Next, we explored the tunable amplification of cellular uptake for controlled therapeutic delivery by appending a cytotoxic payload to the nanopeptifier. Arg5- and Arg8-ELPBCs were selected as the carrier platforms as they encompassed the extremes in nanopeptifier FOM that are likely to control the performance of CPP-ELPBCs as drug carriers. These CPP-ELPBCs were functionalized with a biologic drug, the BH3 peptide (MGQVGRQLAIIGDDINRRY), corresponding to residues 71–89 of the proapoptotic Bak protein.37,38 The BH3 peptide sensitizes cells to apoptosis by inhibiting pro-survival proteins and has been shown to induce cytotoxic effects in HeLa cells when administered by CPP-assisted delivery.38−40 This drug was chosen as a stringent test for CPP-ELPBC carriers, as the intracellular delivery of peptide drugs remains one of the grand challenges of drug delivery, since peptide drugs typically suffer from limited efficacy due to fast plasma clearance, susceptibility to degradation, and difficulty in crossing the hydrophobic cell membrane to reach their intracellular therapeutic targets.41
The BH3 peptide was genetically appended to the N-terminus of the ELPBC such that it would be sequestered in the micelle core upon self-assembly. To enable intracellular release of free BH3 peptide, an RVRR peptide linker, cleavable by furin and cathepsin B proteases,42−44 was included between the BH3 peptide and the CPP-ELPBC. Furin is primarily localized in the trans-Golgi network from which it gains access to endosomal and lysosomal compartments42,45,46 whereas cathepsin B is present in early and late endosomes as well as lysosomes.47 These enzymes thus serve as good candidates for the intracellular release of peptide drug following the endocytic uptake of CPP-ELPBCs.
Arg5-ELPBC, Arg8-ELPBC, and a nonfunctionalized ELPBC control were genetically appended with BH3 and an intervening furin- and cathepsin B-cleavable RVRR peptide linker (denoted cBH3). A noncleavable control, lacking the RVRR linker (denoted BH3), was synthesized to evaluate the importance of intracellular release of the BH3 peptide from the nanopeptifier. Additionally, a control appended with the RVRR linker but lacking the BH3 peptide was synthesized to confirm that the peptide linker had no bioactivity. The size and purity of CPP-ELPBC drug conjugates and controls were confirmed by SDS-PAGE (Supporting Information Figure S2B). Incorporation of the BH3 peptide and cleavable linker into the CPP-ELPBC design did not perturb temperature-triggered micelle assembly, as demonstrated by temperature-regulated turbidimetry measurements (Supporting Information Figure S3) and DLS (Table 3). The release of free BH3 peptide was confirmed by incubation of CPP-ELPBC drug conjugates with furin and visualization of the cleavage products by SDS-PAGE. Free BH3 peptide was released from all CPP-ELPBC-cBH3 carriers, whereas CPP-ELPBC-BH3 lacking the cleavable RVRR linker did not release the BH3 peptide in the presence of furin (Supporting Information Figure S4).
Table 3. Library of CPP-ELPBC Nanopeptifier Drug Conjugates.
| nanopeptifier | CPP functionalization | BH3 drug | RVRR linker | RH at 37 °C (nm)a | RH at 42 °C (nm)a |
|---|---|---|---|---|---|
| ELPBC-cBH3 | none | + | + | 9.7 ± 0.1 | 23.6 ± 0.8 |
| Arg5-ELPBC-cBH3 | RRRRR | + | + | 7.8 ± 0.2 | 24.1 ± 1.7 |
| Arg8-ELPBC-cBH3 | RRRRRRRR | + | + | 8.2 ± 0.1 | 27.5 ± 0.3 |
| Arg8-ELPBC-BH3 | RRRRRRRR | + | – | 7.4 ± 0.3 | 22.1 ± 0.4 |
Hydrodynamic radius determined by DLS of CPP-ELPBC drug conjugates at 15 μM in PBS. Data represents mean of three measurements ± SEM.
The cytotoxicity of CPP-ELPBC drug conjugates was assessed by a modified survival assay in which HeLa cells were incubated with drug carrier or controls at 37 or 42 °C for 1 h, after which the cells were washed and returned to complete media. After 24 h, cell survival was quantified by MTS assay. This assay is an extremely stringent test of the ability of the CPP-ELPBC drug conjugates to induce a cytotoxic effect, as it relies on the intracellular delivery of BH3 peptide drug cargo over the course of only 1 h, mimicking the typical in vivo duration of mild clinical hyperthermia.24
Arg8-ELPBC-cBH3 induced significant cell death at 42 °C, as compared to cells at 37 °C, while no significant changes in cell survival were observed with Arg5-ELPBC-cBH3 or with nonfunctionalized ELPBC-cBH3 control (Figure 5A). The difference in cell survival between 37 and 42 °C was much less pronounced for Arg8-ELPBC-BH3, which lacked a cleavable peptide linker, and thus could not release free BH3 following internalization. Free BH3 peptide alone showed no cytotoxicity at either thermal condition, likely due to poor intracellular delivery caused by the impermeability of the cell membrane to this peptide. Furthermore, treatment with mild hyperthermia alone, CPP-ELPBC carrier without drug or Arg8-ELPBC-RVRR resulted in no significant difference in cell survival between 37 and 42 °C (Supporting Information Figures S5 and S6). These results clearly indicate that the intracellular delivery afforded by Arg8-ELPBC-cBH3 and subsequent intracellular release of free peptide drug by cleavage of the RVRR linker are necessary features that control the functional performance of a nanopeptifier armed with proapoptotic BH3 peptide cargo.
Figure 5.

Quantification of cytotoxicity of CPP-ELPBC drug carriers. (A) Cytotoxicity of CPP-ELPBC carriers appended with BH3 peptide drug was evaluated by a modified cell survival assay. HeLa cells were incubated with Arg5-ELPBC-cBH3, Arg8-ELPBC-cBH3, or ELPBC-cBH3 with cleavable peptide drug cargo for 1 h at 37 or 42 °C. HeLa cells were also incubated with controls including Arg8-ELPBC-BH3 with noncleavable peptide drug cargo and free BH3 peptide alone. Cells were washed and returned to complete media for 24 h before cell survival was quantified by MTS assay. *Indicates p < 0.005 between thermal conditions (two-way ANOVA, Bonferroni posthoc test for multiple comparisons). (B) Induction of apoptosis was evaluated by quantification of caspase-3 activity. Caspase-3 activity was quantified in HeLa cell lysates after treatment with ELPBC-cBH3, Arg5-ELPBC-cBH3, Arg8-ELPBC-cBH3, or free BH3 peptide for 1 h at 37 or 42 °C. *Indicates p < 0.01 between thermal conditions (two-way ANOVA, Bonferroni posthoc test for multiple comparisons). Data represents mean of three experiments ± SEM.
Comparing the performance of Arg5- and Arg8-ELPBC carriers of BH3 peptide drug allowed the definition of nanopeptifier FOM required for thermally controlled cytotoxicity. Specifically, amplified uptake between 12 and 130 arbitrary units was necessary to induce a therapeutic effect with BH3 peptide drug cargo (as defined in Table 2), because Arg5-ELPBC-cBH3 with an amplified uptake of ∼12 showed no efficacy while Arg8-ELPBC-cBH3 with an amplified uptake of ∼130 showed significant cell death at 42 °C. Leakage uptake of less than 11 arbitrary units was sufficient to avoid cell death, as cells were safely spared by treatment at 37 °C with Arg8-ELPBC-cBH3, which has a leakage uptake of 11. These parameters thus defined the FOM that induced significant cell death at 42 °C, while safely sparing cells at 37 °C for cells treated with nanopeptifiers armed with BH3 peptide drug. Other therapeutic payloads, however, will likely require different FOM for optimized therapeutic effect, further supporting the need for a tunable amplifier of cellular uptake.
Next, the role of apoptosis in the mechanism of cytotoxicity induced by Arg8-ELPBC-cBH3 was investigated by the activation of the effector enzyme caspase-3. Enzymatic activity of caspase-3 was evaluated in cells treated with CPP-ELPBC drug conjugates using an assay that quantifies fluorescent products from the cleavage of caspase-specific targets. Arg8-ELPBC-cBH3 showed a significant increase in caspase-3 activity at 42 °C, as compared to 37 °C (Figure 5B), corroborating that the controlled cytotoxicity demonstrated in Figure 5A was due to the induction of apoptosis by the BH3 peptide cargo. Caspase-3 activity was elevated to a lesser extent in Arg8-ELPBC-cBH3 treated cells at 37 °C, however, this activation did not correlate with enhanced cytotoxicity, suggesting that a threshold of caspase-3 activity was not met to induce significant cell death at this temperature. No significant changes in caspase-3 activity were seen between cells treated with Arg5-ELPBC-cBH3, nonfunctionalized ELPBC-cBH3 control, or free BH3 peptide at 37 or 42 °C. Additionally, hyperthermia alone was not responsible for changes in caspase-3 activity (Supporting Information Figure S7), supporting the conclusion that the amplified drug delivery afforded by the Arg8-ELPBC carrier was responsible for controlled cell death by apoptosis.
While our results clearly indicate that enhanced intracellular delivery and release of BH3 peptide drug with Arg8-ELPBC-cBH3 is the primary factor that leads to temperature-controlled cytotoxicity of cancer cells, several ancillary factors may also contribute to the controlled cytotoxicity observed for the Arg8-ELPBC nanopeptifier. First, the cationic charge contributed to the BH3 peptide by the residual RVRR peptide following cleavage at the C-terminus of the linker could potentially enhance its cytotoxicity by electrostatically localizing the peptide to the mitochondrial membrane.48 This could lead either to increased interaction of BH3 with prosurvival proteins anchored in the mitochondrial membrane or directly amplify the apoptosis cascade by destabilizing the mitochondrial membrane.49 Second, mild hyperthermia could directly contribute to the cytotoxic effect by influencing components in the apoptotic pathway. Although high lethal temperatures can directly activate proapoptotic proteins such as Bax and Bak,50 lower temperatures typical of mild hyperthermia may contribute to the apoptotic cascade by down-regulation of apoptosis-related proteins, such as antiapoptotic Bcl-2,51 or by destabilization of the mitochondrial membrane,52 resulting in cytochrome c release that can initiate activation of effector enzymes in the apoptotic cascade. We did not observe that mild hyperthermia alone led to apoptosis, suggesting that the effects of heat synergized with the delivery of BH3 peptide, which further disturbed the balance of apoptosis-related proteins and led to significant cell death. Together these factors, enhanced proapoptotic BH3 peptide delivery, charge conferred by the RVRR peptide linker, and mild hyperthermia, may act synergistically to provide controlled cytotoxicity at conditions of mild hyperthermia while sparing cells at physiologic temperature.
The nanopeptifier platform described here provides two novel advances to the field of cancer-targeted cellular uptake. First, the nanopeptifier provides controlled cellular uptake via an extrinsic thermal trigger that is independent of the heterogeneity of intrinsic tumor characteristics exploited by other tumor-targeted delivery systems, such as upregulated receptors, overexpressed enzymes, and depressed tumor pH. Second, the modular design of the nanopeptifier affords tunable amplification of cellular uptake that can provide a selective therapeutic effect specific to a drug cargo of interest. This genetically encoded drug delivery system thus has the potential to control the intracellular delivery and cytotoxicity of a variety of anticancer therapeutics across a range of solid tumors.
Acknowledgments
This work was supported by funding from the NIH (R01EB007205) to A.C. and by the NSF’s Research Triangle MRSEC (DMR-1121107).
Glossary
Abbreviations
- CPP
cell-penetrating peptide
- ELPBC
elastin-like polypeptide diblock copolymer
- Arg
arginine
- TAT
transactivator of transcription
- MW
molecular weight
- DLS
dynamic light scattering
- OD
optical density
- CMT
critical micellization temperature
- FOM
figures-of-merit
Supporting Information Available
Materials, recombinant synthesis, expression, and purification of ELPs, fluorescent labeling of ELPs and their thermal characterization, cell culture methods, confocal microscopy, flow cytometry, cytotoxicity assay, caspase activity assay, and statistical analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
S.R.M planned and performed experiments, analyzed results, and wrote the manuscript. A.C. planned experiments and edited the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Parker N.; Turk M. J.; Westrick E.; Lewis J. D.; Low P. S.; Leamon C. P. Anal. Biochem. 2005, 338, 284–293. [DOI] [PubMed] [Google Scholar]
- Muss H. B.; Thor A. D.; Berry D. A.; Kute T.; Liu E. T.; Koerner F.; Cirrincione C. T.; Budman D. R.; Wood W. C.; Barcos M.; et al. N. Engl. J. Med. 1994, 330, 1260–1266. [DOI] [PubMed] [Google Scholar]
- Fink-Retter A.; Gschwantler-Kaulich D.; Hudelist G.; Mueller R.; Kubista E.; Czerwenka K.; Singer C. F. Oncol. Rep. 2007, 18, 299–304. [PubMed] [Google Scholar]
- Taniguchi K.; Okami J.; Kodama K.; Higashiyama M.; Kato K. Cancer Sci. 2008, 99, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter D. L.; Leith J. T. J. Clin. Oncol. 1986, 4, 244–257. [DOI] [PubMed] [Google Scholar]
- Bidwell G. L. 3rd; Perkins E.; Hughes J.; Khan M.; James J. R.; Raucher D. PLoS One 2013, 8, e55104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia J. S.; Dowdy S. F. Adv. Drug Delivery Rev. 2005, 57, 579–596. [DOI] [PubMed] [Google Scholar]
- Qin Y.; Chen H.; Zhang Q.; Wang X.; Yuan W.; Kuai R.; Tang J.; Zhang L.; Zhang Z.; Liu J.; He Q. Int. J. Pharm. 2011, 420, 304–312. [DOI] [PubMed] [Google Scholar]
- Sawant R. R.; Torchilin V. P. Int. J. Pharm. 2009, 374, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEwan S. R.; Chilkoti A. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2013, 5, 31–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren E.; Apte A.; Jani A.; Torchilin V. P. J. Controlled Release 2012, 160, 264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok H.; Bae K. H.; Ahn C. H.; Park T. G. Langmuir 2009, 25, 1645–1650. [DOI] [PubMed] [Google Scholar]
- Harris T. J.; von Maltzahn G.; Lord M. E.; Park J. H.; Agrawal A.; Min D. H.; Sailor M. J.; Bhatia S. N. Small 2008, 4, 1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; Kate P.; Torchilin V. P. ACS Nano 2012, 6, 3491–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethuraman V. A.; Lee M. C.; Bae Y. H. Pharm. Res. 2008, 25, 657–666. [DOI] [PubMed] [Google Scholar]
- Lee E. S.; Gao Z.; Kim D.; Park K.; Kwon I. C.; Bae Y. H. J. Controlled Release 2008, 129, 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goun E. A.; Shinde R.; Dehnert K. W.; Adams-Bond A.; Wender P. A.; Contag C. H.; Franc B. L. Bioconjugate Chem. 2006, 17, 787–796. [DOI] [PubMed] [Google Scholar]
- Watkins G. A.; Jones E. F.; Scott Shell M.; VanBrocklin H. F.; Pan M. H.; Hanrahan S. M.; Feng J. J.; He J.; Sounni N. E.; Dill K. A.; Contag C. H.; Coussens L. M.; Franc B. L. Bioorg. Med. Chem. 2009, 17, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson E. S.; Jiang T.; Aguilera T. A.; Nguyen Q. T.; Ellies L. G.; Scadeng M.; Tsien R. Y. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 4311–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Q. T.; Olson E. S.; Aguilera T. A.; Jiang T.; Scadeng M.; Ellies L. G.; Tsien R. Y. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 4317–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. T.; Kwon Y. M.; Spangrude G. J.; Liang J. F.; Chung H. S.; Park Y. J.; Yang V. C. J. Biomed. Mater. Res., Part A 2009, 91, 209–220. [DOI] [PubMed] [Google Scholar]
- Dreher M. R.; Simnick A. J.; Fischer K.; Smith R. J.; Patel A.; Schmidt M.; Chilkoti A. J. Am. Chem. Soc. 2008, 130, 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEwan S. R.; Chilkoti A. Nano Lett. 2012, 12, 3322–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer R. B. Annu. Rev. Biomed. Eng. 1999, 1, 347–376. [DOI] [PubMed] [Google Scholar]
- Falk M. H.; Issels R. D. Int. J. Hyperthermia 2001, 17, 1–18. [DOI] [PubMed] [Google Scholar]
- Jones E.; Thrall D.; Dewhirst M. W.; Vujaskovic Z. Int. J. Hyperthermia 2006, 22, 247–253. [DOI] [PubMed] [Google Scholar]
- James J. R.; Gao Y.; Soon V. C.; Topper S. M.; Babsky A.; Bansal N. Int. J. Hyperthermia 2010, 26, 79–90. [DOI] [PubMed] [Google Scholar]
- O’Neill B. E.; Li K. C. Int. J. Hyperthermia 2008, 24, 506–520. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Mitchell D. J.; Pattabiraman K.; Pelkey E. T.; Steinman L.; Rothbard J. B. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbard J. B.; Jessop T. C.; Lewis R. S.; Murray B. A.; Wender P. A. J. Am. Chem. Soc. 2004, 126, 9506–9507. [DOI] [PubMed] [Google Scholar]
- Mitchell D. J.; Kim D. T.; Steinman L.; Fathman C. G.; Rothbard J. B. J. Pept. Res. 2000, 56, 318–325. [DOI] [PubMed] [Google Scholar]
- McDaniel J. R.; Mackay J. A.; Quiroz F. G.; Chilkoti A. Biomacromolecules 2010, 11, 944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D. E.; Chilkoti A. Nat. Biotechnol. 1999, 17, 1112–1115. [DOI] [PubMed] [Google Scholar]
- Dougherty D. A. J. Nutr. 2007, 137, 1504S–1508S. [DOI] [PubMed] [Google Scholar]
- Moon C. P.; Fleming K. G. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 10174–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud F.; Karlin S. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 12494–12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M.; Liang H.; Nettesheim D.; Meadows R. P.; Harlan J. E.; Eberstadt M.; Yoon H. S.; Shuker S. B.; Chang B. S.; Minn A. J.; Thompson C. B.; Fesik S. W. Science 1997, 275, 983–986. [DOI] [PubMed] [Google Scholar]
- Holinger E. P.; Chittenden T.; Lutz R. J. J. Biol. Chem. 1999, 274, 13298–13304. [DOI] [PubMed] [Google Scholar]
- Duvall C. L.; Convertine A. J.; Benoit D. S.; Hoffman A. S.; Stayton P. S. Mol. Pharm. 2010, 7, 468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albarran B.; Hoffman A. S.; Stayton P. S. React. Funct. Polym. 2011, 71, 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craik D. J.; Fairlie D. P.; Liras S.; Price D. Chem. Biol. Drug Des. 2013, 81, 136–147. [DOI] [PubMed] [Google Scholar]
- Thomas G. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustapa M. F.; Grosse S. M.; Kudsiova L.; Elbs M.; Raiber E. A.; Wong J. B.; Brain A. P.; Armer H. E.; Warley A.; Keppler M.; Ng T.; Lawrence M. J.; Hart S. L.; Hailes H. C.; Tabor A. B. Bioconjugate Chem. 2009, 20, 518–532. [DOI] [PubMed] [Google Scholar]
- Gordon V. M.; Klimpel K. R.; Arora N.; Henderson M. A.; Leppla S. H. Infect. Immun. 1995, 63, 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari J.; Helenius A. EMBO J. 2011, 30, 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy S. S.; Thomas L.; VanSlyke J. K.; Stenberg P. E.; Thomas G. EMBO J. 1994, 13, 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha S.; Padh H. Indian J. Biochem. Biophys. 2008, 45, 75–90. [PubMed] [Google Scholar]
- Emanuelsson O.; von Heijne G.; Schneider G. Methods Cell Biol. 2001, 65, 175–187. [DOI] [PubMed] [Google Scholar]
- Frohlich E. Int. J. Nanomed. 2012, 7, 5577–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliari L. J.; Kuwana T.; Bonzon C.; Newmeyer D. D.; Tu S.; Beere H. M.; Green D. R. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 17975–17980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed K.; Zhao Q. L.; Matsuya Y.; Yu D. Y.; Salunga T. L.; Nemoto H.; Kondo T. Int. J. Hyperthermia 2007, 23, 353–361. [DOI] [PubMed] [Google Scholar]
- Zukiene R.; Nauciene Z.; Ciapaite J.; Mildaziene V. Int. J. Hyperthermia 2010, 26, 56–66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.