

Abstract

Hydrogels with predictable degradation are highly desired for biomedical applications where timely disintegration of the hydrogel (e.g., drug delivery, guided tissue regeneration) is required. However, precisely controlling hydrogel degradation over a broad range in a predictable manner is challenging due to limited intrinsic variability in the degradation rate of liable bonds and difficulties in modeling degradation kinetics for complex polymer networks. More often than not, empirical tuning of the degradation profile results in undesired changes in other properties. Here we report a simple but versatile hydrogel platform that allows us to formulate hydrogels with predictable disintegration time from 2 to >250 days yet comparable macroscopic physical properties. This platform is based on a well-defined network formed by two pairs of four-armed polyethylene glycol macromers terminated with azide and dibenzocyclooctyl groups, respectively, via labile or stable linkages. The high-fidelity bioorthogonal reaction between the symmetric hydrophilic macromers enables robust cross-linking in water, phosphate-buffered saline, and cell culture medium to afford tough hydrogels capable of withstanding >90% compressive strain. Strategic placement of labile ester linkages near the cross-linking site within this superhydrophilic network, accomplished by adjustments of the ratio of the macromers used, enables broad tuning of the disintegration rates precisely matching with the theoretical predictions based on first-order linkage cleavage kinetics. This platform can be exploited for applications where a precise degradation rate is targeted.
Hydrogels, referring to cross-linked water-swollen polymer networks, have been exploited for a wide range of applictions.1 For advanced biomedical applications, such as guided tissue regeneration5 and drug delivery,6 biocompatible hydrogels with controlled degradation rates and robust physical properties are highly desired. Numerous degradable hydrogels have been reported,5,6 where the degradability is conferred by linkages liable to hydrolysis,7 photoirradiation,10 redox reaction,14 or enzymes.16 Hydrogel degradation is a complex process, dictated by not only the chemical composition but also the structure of the polymer network. Limited control over degradation rate has been realized by either incorporating liable linkages with varying cleavage rates or altering the polymer network structures containing the same labile linkages (which often causes undesired changes in other macroscopic properties), or both. The concept of tailoring the polarity/charge/structure of neighboring groups to affect the hydrolysis rate of labile linkages18 has seen some success in degradable hydrogel designs. Achieving broadly tunable degradation rates for a given polymer network, however, remains difficult due to the complexity and ill-defined relationship between most polymer network structures and their chemical compositions. This is the case even for chemically simple, widely utilized hydrogel systems such as photopolymerized (meth)acrylated polyethylene glycol (PEG) hydrogels,21 where the poorly defined networks resulting from uncontrolled radical polymerization led to inconsistent degradation, mechanical, and biological properties.
Here we report a simple and robust strategy for achieving widely tunable and predictable degradation rates within hydrogels with consistent macroscopic properties by strategic placement of liable ester linkages within a well-defined network. We hypothesize that, in a homogenously cross-linked network where all polymer chains are fully tethered with evenly spaced netpoints, the degradation behavior becomes much easier to predict when a single liable linkage is precisely positioned between neiboring netpoints (Scheme 1). Cleavage of the labile linkages within such a network in a highly swollen state can be treated as a pseudo-first-order reaction, where the remaining intact linkage fraction (P) over time can be described by a very simple model:
| 1 |
where kd is the rate constant of the labile linkage cleavage, t is time, and [linkage]0 and [linkage]t are the intact linkage concentration prior to degradation and at time t, respectively. When P reaches a critical value (Pc) where the infinite network no longer exists, the hydrogel disintegrates. This critical value is the same as the critical gelling point during the cross-linking, defined by the macromer structure and the cross-linking chemistry. Therefore, the disintegration time (tc) for such a degradable network is determined by Pc and kd:
| 2 |
Scheme 1. Degradation of an Ideally Cross-Linked and Highly Swollen Homogeneous Network Containing a Single Labile Linkage between Neighboring Netpoints.
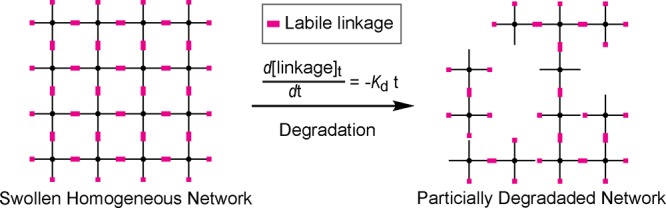
Cleavage of the labile linkages is assumed to occur independently in a first-order kinetics.
Similarly, if two liable linkages with varying cleavage rates are incorporated within such a network, the remaining intact linkage fraction over time can be described as
| 3 |
where r is the percentage of the faster-degrading labile linkage among the total labile network linkages, while kdf and kd are the cleavage rate constants of the faster- and slower-degrading labile linkages, respectively. The disintegration time will thus be determined by three intrinsic parameters, Pc, kdf, and kd, and one formulation parameter, r. By changing r, the disintegration time could be tuned between −(ln Pc)/kdf and −(ln Pc)/kd. This concept can be extended to incorporate multiple liable linkages with varying susceptibility to provide even more flexible tuning of tc.
To test this hypothesis, we chose 4-armed PEG with Mn = 20 000 g/mol (4-armPEG) as the base macromer structure due to its well-defined symmetric structure, high hydrophilicity, and commercial availability and strain-promoted azide/alkyne cycloaddition (SPAAC) as the cross-linking chemistry due to its high reactivity and established bioorthogonality (tolerance to biological species) under physiological conditions23 (Scheme 2). We first synthesized two groups of macromers, with azide (N3) and dibenzocyclooctyl (DBCO) end groups attached to the 4-armPEG via a labile ester or stable (e.g., amide) linkage, respectively. Nearly complete end-group functionization was accomplished: 100% conversion based on 1H NMR integration, further validated by the disappearance of characteristic 13C NMR signals upon conversion using highly concentrated samples and extremely long 13C NMR acquisitions (Figures S1–S12).
Scheme 2. Structures and Naming of Macromers and the Orthogonally Cross-Linked ClickGel Networks.
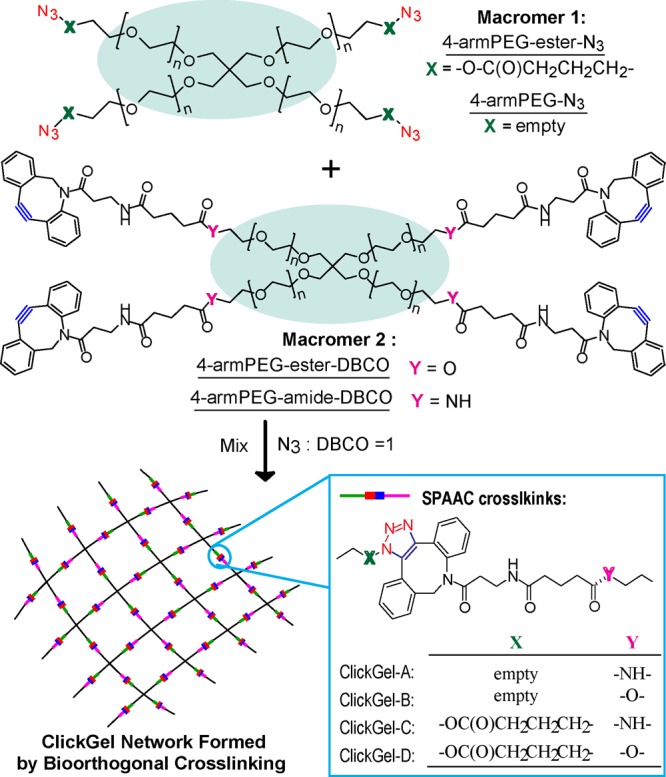
Four hydrogels (ClickGel-A, -B, -C, and -D) were prepared by combinatorial mixing of one N3- and one DBCO-terminated macromer in equal molar ratio. All formulations gelled in as rapidly as 5 min, and the degree of cross-linking was nearly 100% after 20 h, as evidenced by the complete conversion of N3 and DBCO end groups into SPAAC cross-links, as confirmed by FTIR and UV/vis, respectively (Figure S13).
All four hydrogels exhibited comparable equilibrated swelling ratios of ∼1.50 (Figure 1A), with ClickGel-A and -C prepared from 4-armPEG-N3 swelling slightly more than those prepared from 4-armPEG-ester-N3. Unconfined compressive testing (Figure 1B) showed that all four hydrogels withstood up to 90% compressive strain without breaking, exhibiting nearly identical stress/strain curves with the moduli sharply increasing with increasing stains, typical of ideal elastic networks. ClickGel-B and -D formed from 4-armPEG-ester-N3 (Figure 1B) showed slightly higher moduli at larger deformations than the hydrogels formed from 4-armPEG-N3, likely due to some degrees of hydrobobic interactions between the esters.
Figure 1.
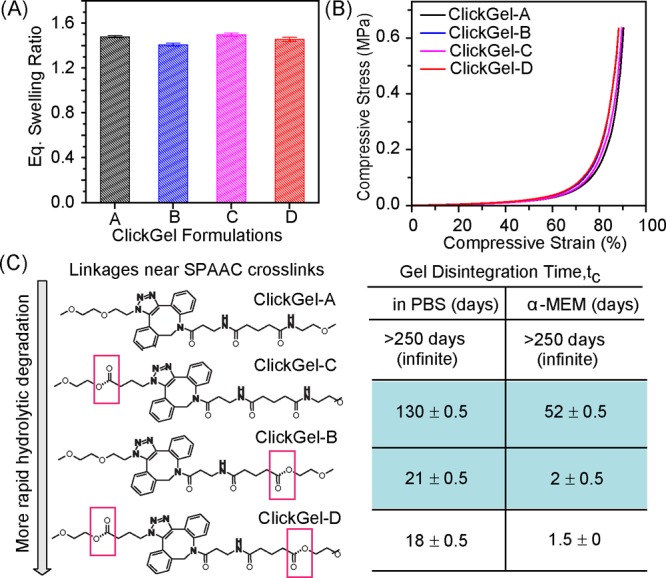
Four hydrogels cross-linked from different combinations of N3- and DBCO-terminated macromers, showing similar macroscopic properties but distinct network disintegration rates. (A) Equilibrium gel swelling ratio (by weight) in PBS (pH 7.4) at 37 °C. (B) Stress/strain curves from unconfined compressive testing. (C) Distinct disintegration time (at least three specimens for each hydrogel) in PBS and α-MEM.
Despite comparable swelling and mechanical properties, the four hydrogels exhibited distinctly different disintegration rates. In PBS, ClickGel-A was stable for a very long time (>250 days), while ClickGel-B, -C, and -D disintegraded in 21, 130, and 18 days, respectively. Since the comparable macroscopic properties of these hydrogels support similar network structures, the drastic differences in the degradation rates of these hydrogels can be ascribed to the presence and specific positioning of the liable ester linkages within the otherwise identical SPAAC cross-linked network. ClickGel-A does not contain any labile linkages, thus was stable over a long period in both PBS and cell culture media continining a rich source of nucleophiles (α-MEM). Only one type of liable linkage, the ester linkage from 4-armPEG-ester-DBCO or 4-armPEG-ester-N3, existed in ClickGel-B and -C, making eq 1 suitable for describing the degradation kinetics of these two hydrogels.
According to the Flory–Rehner gelation theory on networks formed by step-polymerization,25 the critical gelling point for an equal molar mixture of mutually reactive 4-arm macromers is Pc = 1/3 (see Supporting Information). With the experimentally determined critical gel disintegration time for ClickGel-B and -C (e.g., tc = 21 and 131 days in PBS, respectively, Figure 1C), the apparent cleavage rate constants for the two liable ester linkages could thus be calculated by eq 2 as kdN3 = 52.3 × 10–3 day–1 and kd = 8.5 × 10–3 day–1 in PBS (pH 7.4). In α-MEM, ClickGel-B and -C both degraded much more rapidly, but with the same relative rates as observed in PBS, with respective degradation constants of 0.549 and 0.021 day–1. For the non-degradable network chain, kd = 0 in both aqueous media.
According to our hypothesis, it is possible to alter the ratio of the non-labile amide-DBCO vs labile ester-DBCO linkages, to prepare hydrogels ranging from having a disintegration time of 21 days (−ln(1/3)/0.0523) to being non-degradable (infinite degradation time, −ln(1/3)/0) in PBS, or from 2 days (−ln(1/3)/0.549) to non-degradable in α-MEM, respectively. To test this hypothesis, we prepared a series of hydrogels by varying the ratio of 4-armPEG-ester-DBCO and 4-armPEG-amide-DBCO (formulation parameter r) mixed with 4-armPEG-N3 while keeping [DBCO]/[N3] = 1. These hydrogels exhibited similar macroscopic mechanical properties, as expected, and their experimentally determined disintegration time in PBS precisely matched with those theoretically predicted over a wide range of formulation parameters (r = 0–1, Figure 2A,C). The excellent match between experimental and predicted values was also observed in α-MEM (Figure 2B,D) despite the relatively larger standard deviations of the experimental data, likely due to the more complex nucleophiles present in the media (e.g., primary amine, thiol, hydroxyl, and phenol groups from amino acids, vitamins, ribonucleosides, deoxyribonucleosides, and phenol red in α-MEM). These observations support our hypothesis that hydrogel degradation could be controlled through strategic placement of liable linkages within an ideally structured homogeneous network and precisely predicted by a simple model. Although the mechanism of linkage cleavage may vary in different media, the modular hydrogel platform and this validated prediction model could still guide the formulation of hydrogels to achieve specific disintegration rates, as long as the labile linkage cleavage rate constant can be experimentally derived for the specific medium of interest using a ClickGel containing only the labile linkage of interest (e.g., GlickGel-B or -C in this case).
Figure 2.
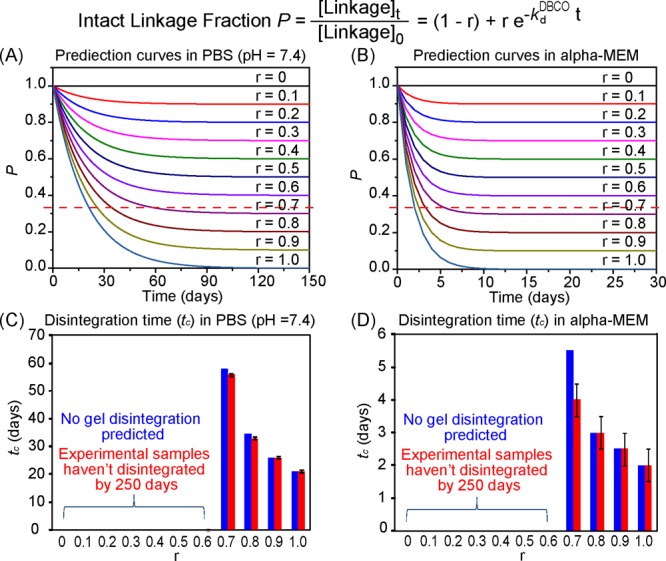
Disintegration time (tc) hydrogels prepared from 4-armPEG-N3 with varying ratios of 4-armPEG-ester-DBCO and 4-armPEG-amide-DBCO (r) predicted by the theoretical model and validated by experimental data. (A,B) Prediction curves of the intact linkage fraction P vs time in PBS (pH 7.4) and α-MEM, respectively. The red dotted line represents the critical intact linkage fraction of the network (Pc = 1/3), and its crosspoint with each curve indicates the predicted tc for the specific formulation. (C,D) Predicted (blue) and experimentally observed (red) tc in PBS (pH 7.4) and α-MEM, respectively.
The subtle difference in the location of the hydrolytically labile ester linkage in ClickGel-B vs -C (on either side of the SPAAC cross-link) resulted in a significant difference in their gel disintegration times (Figure 1C). Why the ester linkage on the DBCO side of the SPAAC cross-link is more labile than the one on the N3 side is a topic of ongoing investigations. Using the same strategy, hydrogels with disintegration time ranging from 130 days to infinitely long were prepared by altering the ratio of 4-armPEG-ester-N3 and 4-armPEG-N3 (formulation parameter r) mixed with 4-armPEG-amide-DBCO while keeping [DBCO]/[N3] = 1. Similarly, the experimentally determined disintegration time of these hydrogels agreed well with the predicted values over a wide range of r = 0–1 in both PBS (Figure 3A,C) and α-MEM (Figure 3B,D).
Figure 3.

Disintegration times (tc) of hydrogels prepared from 4-armPEG-amide-DBCO with varying ratios of 4-armPEG-ester-N3 and 4-armPEG-N3 (r) predicted by the theoretical model and validated by experimental data. (A,B) Prediction curves of the intact linkage fraction P vs time in PBS (pH 7.4) and α-MEM, respectively. The red dotted line represents the critical intact linkage fraction of the network (Pc = 1/3), and its crosspoint with each curve indicates the predicted tc for the specific formulation. (C,D) Predicted (blue) and experimentally observed (red) tc in PBS (pH 7.4) and α-MEM, respectively.
In the two systems described above, the labile ester linkage was strategically positioned near the SPAAC cross-links to ensure that the degradation process can be viewed as the playback of the cross-linking process in a slow motion. This is an indispensable design element, without which the mathematical adoption of the critical gelling point (Pc) for the prediction of the critical hydrogel disintegration time would not have been valid.
It should also be noted that the two systems described above offer not only different ranges of possible gel disintegration time (e.g., 21 days and above in PBS for the system described in Figure 2 vs 130 days and above for the one described in Figure 3) but also a wide range of degradation rates prior to reaching the network disintegration (slope of the prediction curves). For instance, although it is feasible to formulate a hydrogel with disintegration time >130 days using either system, one could enable more gradual degradation than the other (Figure S14). This may be particularly useful for applications where a gradual loss in mass or mechanical integrity of the network is required. For instance, scaffold-guided tissue regeneration in older or metabolically challenged patients may take longer than in younger/normal patients, thus requiring more extended structural/mechanical support of a resorbable tissue scaffold.
Unlike ClickGel-B or -C, ClickGel-D possesses labile ester linkages on both sides of the SPAAC cross-links. Assuming that the cleavage of these linkages proceeds independently from each other, the labile linkage cleavage kinetics in ClickGel-D could be described as
| 5 |
Applying the kdN3 and kd experimentally determined from ClickGel-B and -C, respectively, the disintegration time for ClickGel-D is thus predicted as 18.1 days in PBS or 1.9 days in α-MEM, which precisely matched with the theroretical prediction (Figure 1C), validating our proposed model.
All scenarios described thus far involve the use of no more than three of the four designer macromers. When necessary, the use of all four macromers could provide an even more versatile platform to formulate hydrogels with far more refined degradation profiles, as described by
 |
6 |
where rN3 and rDBCO are the ratios of ester-containing macromers in the total azido- and DBCO-terminated macromers, respectively, and kdN3 and kd are the cleavage rate constants of the ester linkage positioned on the N3 and DBCO side of the SPAAC cross-links, respectively. According to eq 6, it should be possible to prepare hydrogels with disintegration time >18 days in PBS or >2 days in α-MEM using this platform by simply changing the formulation parameters rN3 and rDBCO (selected prediction curves shown in Figure S15).
In summary, the modular hydrogel platform based on two pairs of well-defined 4-armPEG macromers, the robust and cytocompatible SPAAC cross-linking chemistry, and the strategic positioning of labile ester linkages enable unprecedented predictive design of hydrogels with consistent macroscopic physical properties yet highly tunable degradation profiles over a broad range. This work underscores the importance of network structure in controlling degradation rates. It accomplishes predictive tuning of degradation rates without the need for introducing complex degradable components via tedious multistep syntheses, which may also result in hard-to-define degradation products. We envision that this design concept can be extended to other degradable polymer systems and significantly benefit the development of advanced resorbable materials for personalized medicine.
Acknowledgments
This work was supported by the National Institutes of Health (R01AR055615, R01GM088678).
Supporting Information Available
Detailed macromer synthesis, hydrogel preparation and characterization methods, and Figures S1–S15. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hoffman A. S. Adv. Drug Delivery Rev. 2012, 64, 18–23. [Google Scholar]
- Kharkar P. M.; Kiick K. L.; Kloxin A. M. Chem. Soc. Rev. 2013, 42, 7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppas N. A.; Bures P.; Leobandung W.; Ichikawa H. Eur. J. Pharm. Biopharm. 2000, 50, 27. [DOI] [PubMed] [Google Scholar]
- a van Dijk-Wolthuis W. N. E.; Hoogeboom J. A. M.; van Steenbergen M. J.; Tsang S. K. Y.; Hennink W. E. Macromolecules 1997, 30, 4639. [Google Scholar]; b Zustiak S. P.; Leach J. B. Biomacromolecules 2010, 11, 1348. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Li X.; Tsutsui Y.; Matsunaga T.; Shibayama M.; Chung U.-i.; Sakai T. Macromolecules 2011, 44, 3567. [Google Scholar]
- a Griffin D. R.; Kasko A. M. J. Am. Chem. Soc. 2012, 134, 13103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fairbanks B. D.; Singh S. P.; Bowman C. N.; Anseth K. S. Macromolecules 2011, 44, 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]; c DeForest C. A.; Anseth K. S. Nat. Chem. 2011, 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kloxin A. M.; Benton J. A.; Anseth K. S. Biomaterials 2010, 31, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dunn S. S.; Tian S. M.; Blake S.; Wang J.; Galloway A. L.; Murphy A.; Pohlhaus P. D.; Rolland J. P.; Napier M. E.; DeSimone J. M. J. Am. Chem. Soc. 2012, 134, 7423. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yang F.; Wang J.; Cao L.; Chen R.; Tang L.; Liu C. J. Mater. Chem. B 2014, 2, 295. [DOI] [PubMed] [Google Scholar]
- a Lutolf M. P.; Lauer-Fields J. L.; Schmoekel H. G.; Metters A. T.; Weber F. E.; Fields G. B.; Hubbell J. A. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 5413. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ehrbar M.; Rizzi S. C.; Schoenmakers R. G.; San Miguel B.; Hubbell J. A.; Weber F. E.; Lutolf M. P. Biomacromolecules 2007, 8, 3000. [DOI] [PubMed] [Google Scholar]
- a Rydholm A. E.; Anseth K. S.; Bowman C. N. Acta Biomater. 2007, 3, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jo Y. S.; Gantz J.; Hubbell J. A.; Lutolf M. P. Soft Matter 2009, 5, 440. [Google Scholar]; c Ashley G. W.; Henise J.; Reid R.; Santi D. V. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lin C. C.; Anseth K. S. Pharm. Res. 2009, 26, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nguyen Q. T.; Hwang Y.; Chen A. C.; Varghese S.; Sah R. L. Biomaterials 2012, 33, 6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Agard N. J.; Prescher J. A.; Bertozzi C. R. J. Am. Chem. Soc. 2004, 126, 15046. [DOI] [PubMed] [Google Scholar]; b Xu J. W.; Filion T. M.; Prifti F.; Song J. Chem. Asian J. 2011, 6, 2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Flory P. J. Chem. Rev. 1946, 39, 137. [DOI] [PubMed] [Google Scholar]; b Flory P. J.; Rehner J. J. Chem. Phys. 1943, 11, 521. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.