

Abstract
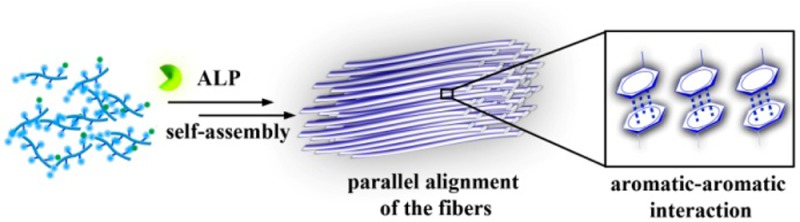
Anisotropy or alignment is a critical feature of functional soft materials in living organisms, but it remains a challenge for spontaneously generating anisotropic gel materials. Here we report a molecular design that increases intermolecular aromatic–aromatic interactions of hydrogelators during enzymatic hydrogelation for spontaneously forming an anisotropic hydrogel. This process, relying on both aromatic–aromatic interactions and enzyme catalysis, results in spontaneously aligned supramolecular nanofibers as the matrices of a monodomain hydrogel that exhibits significant birefringence. This work, as the first example of monodomain hydrogels formed via an enzymatic reaction, illustrates a new biomimetic approach for generating aligned anisotropic soft materials.
Anisotropy or alignment is a critical feature of functional materials in living organisms or man-made systems. For example, muscle cells rely on sophisticated anisotropic protein filaments to function, and liquid crystal display is based on the electrical static alignment of a simple anisotropic molecule (e.g., 4-cyano-4′-pentylbiphenyl1). Recognizing the importance of anisotropy, materials scientists are striving to create high quality anisotropic materials. While generating anisotropic materials from inorganic solids is relatively easy and successful,2a,2b the introduction of anisotropy to synthetic soft organic materials,3a−3d particularly hydrogels,4a−4c remains less developed and a considerable challenge despite the fact that biological anisotropic soft materials with substantial complexity prevail in living organisms.5 Although several approaches have shown effectiveness for inducing anisotropy in gels, including the use of an electrical field,6 the inclusion of liquid crystals,3c,3d and the utilization of mechanical stretching7a−7c or shear force from flow,4a each process still has its limitation for creating anisotropy in hydrogels because hydrogels contain a large amount of water that usually conducts electricity, excludes common liquid crystals, and favors relaxation to the isotropic state. Thus, better approaches are needed for spontaneously generating inherently anisotropic hydrogels.
Based on the anisotropic hydrogel in nature (e.g., corneal stroma formed mainly by regularly arranged collagen fibers5), one attractive approach is to use molecular self-assembly to generate anisotropic structures that immobilize water and form hydrogels. In fact, recent advances in the development of supramolecular gels8a−8d make this approach particular promising because the self-assembly of hydrogelators usually affords nanofibers,9a−9c which is inherently anisotropic. However, these nanofibers, being long and flexible, not only physically cross-link to form the matrices of gels but also adopt random entanglement, which results in isotropic hydrogels in most cases.8a Encouragingly, several exceptional examples of supramolecular hydrogels exhibit birefringence, thus offering useful hints for creating anisotropic hydrogels. For example, Stupp and co-workers demonstrated that a thermal pathway can convert isotropic solutions of peptide amphiphiles to a strongly birefringent hydrogel, in which molecules self-assemble to form large arrays of aligned nanofibers.4a Moreover, the addition of calcium ions into the solution of a phosphate containing peptide amphiphile results in a gel that exhibits birefringence, which coincides with the parallel alignment of the nanofibers shown in the transmission electron micrograph (TEM) of the gels.10 These results and other examples11a,11b suggest that the enhancement of interfiber interactions should be a reliable molecular approach to induce alignment of nanofibers and thus generate anisotropy.
Although ionic forces10,11b or overlaps between alkyl chains11a are able to increase the interfiber interactions, ionic interactions are intrinsically isotropic and interactions between alkyl chains are relatively weak and inefficient. Therefore, we decided to explore the use of aromatic–aromatic interactions to enhance interfiber interactions that are necessary for creating anisotropic supramolecular hydrogels because (i) aromatic–aromatic interaction is not only stronger than the van der Waal’s interaction (London dispersion force) between alkyl chains but also inherently directional due to the plane-to-plane or edge-to-plane orientation;12 (ii) nature has extensively used aromatic–aromatic interaction as a stabilizing force for generating ordered structures in proteins;12 and (iii) aromatic rings have relatively compact volumes; thus, the interactions between aromatic rings lead to a more predictable and efficient self-assembly of the molecules in aqueous phase for the formation of mechanically strong or stable supramolecular hydrogels.13
Besides the use of proper interfiber interactions, it is necessary to choose an appropriate pathway for molecular self-assembly. Instead of using a thermal pathway,4a we decided to use an enzymatic pathway because cells use enzymatic conversion to form anisotropic hydrogels5 and the enzymatic self-assembly process has emerged as a powerful method for generating ordered nanostructures14 and supramolecular hydrogels for a wide range of applications.15a,15b For example, enzymatic self-assembly has led to supramolecular hydrogels outside cells16 and inside cells,17a,17bin vitro and in vivo,18 and resulted in highly stable hydrogels that are unavailable via other pathways (e.g., thermal or pH).2a,4c,19 Recently, enzyme regulated self-assembly has provided a route for controlled drug release20a,20b and for the exploration of dynamic self-assembly.21 Despite these advancements, it remains unknown whether the enzymatic pathway can lead to aligned monodomain supramolecular hydrogels.
To address the above-mentioned question, we designed two pairs of precursor/hydrogelator (i.e., 1a/1b and 2a/2b) differing only in one phenylalanine (Phe) residue and examined their enzymatic hydrogelation by using polarized optical microscopy, transmission electron microscopy (TEM), and rheology. Our study shows that the addition of an enzyme (e.g., alkaline phosphatase (ALP)) into the solution of 1a affords an aligned monodomain supramolecular hydrogel of 1b, which consists of aligned nanofibers (Figure 1). Having one less phenylalanine residue, 2b forms a nonbirefringent hydrogel that consists of a network of randomly oriented nanofibers. In addition, the hydrogel of 1b (or 2b) formed by the change of pH exhibits little birefringence and consists of few aligned nanofibers. Thus, this work, as the first example of monodomain supramolecular hydrogels formed by an enzymatic reaction, illustrates a new, effective approach for generating anisotropic supramolecular soft materials, which ultimately may lead to the formation of aligned nanostructures directly in a cellular environment.
Figure 1.

(A) Illustration of aromatic–aromatic interactions, in a hydrogel (Gel1b) formed by treating the solution of a precursor (1a) with an enzyme (ALP), to enhance interfiber interactions that favors alignment of nanofibers and results in an inherently anisotropic hydrogel that causes optical retardance. (B) Structures of two hydrogelators, differing only in the number of phenylalanine residues.
Figure 1B shows the detailed molecular design of the two pairs of precursor/hydrogelator. The precursor 1a, as a pentapeptidic derivative, consists of a (naphthalene-2-ly)acetyl group (Nap), three phenylalanine residues (Phe-Phe-Phe), a lysine (Lys), and a tyrosine phosphate moiety (pTyr). The precursor 2a, as an analog of 1a, consists of Nap, Phe-Phe, Lys, and pTyr. Nap enhances intermolecular aromatic–aromatic interactions;22 the phenylalanine residues (Phe), besides providing aromatic–aromatic interactions, constitute the peptide backbone with Lys to provide the donors and acceptors for intermolecular hydrogen bonds. Since ALP catalytically dephosphorylates pTyr to Tyr to form the hydrogelators, 1b and 2b, the phosphate group on Tyr acts as a trigger for enzymatic hydrogelation.15a The hydrogelator 1b, bearing three phenylalanine residues, likely has stronger intermolecular aromatic–aromatic interactions than 2b (bearing two phenylalanine residues) does. After the molecular design, we used solid phase peptide synthesis (SPPS)23 to synthesize 1a, 2a, 1b, and 2b (Scheme S1).
As a soluble precursor, 1a (or 2a) (4 mg) dissolves in distilled water (0.5 mL) to a final concentration of 0.8 wt % and pH = 7.4. After the addition of ALP (2U/mL), the solution of 1a (or 2a) transforms to a transparent hydrogel (Figure S1, here denoted as Gel1b (or Gel2b)) within 2 h because the precursor 1a (or 2a) becomes the corresponding hydrogelator, 1b (or 2b). Gel1b, being placed between the cross polarizers, exhibits little birefringence when the vial is at 90° and 0° with polarizers (Figure 2A) but displays significant birefringence when the vial is at 45° with the polarizers (Figure 2B). This result indicates that the nanofibers of 1b in Gel1b have largely uniform alignment to result in a significant birefringence. In contrast, Gel2b hardly shows any birefringence in either of the orientations (90° and 0° in Figure 2C and 45° in Figure 2D). This result indicates that the nanofibers of 2b in Gel2b likely have random orientations. To further verify the alignment of nanofibers in Gel1b, we carried out the enzymatic gelation of Gel1b in two capillaries with a diameter of 0.3 mm. As shown in Figure 2E, F, Gel1b barely exhibits birefringence when the capillaries are at 90° and 0° with the polarizers (Figure 2E) but displays bright birefringence when the capillaries are at 45° with the polarizers (Figure 2F). In addition, light extinguishes at the crosspoint of two capillaries containing Gel1b (Figure 2G), confirming uniform alignments of the nanofibers of 1b in Gel1b. Although the change of the solution pH of 1b (or 2b) also results in a hydrogel (Figure S2A (or S3A), denoted as Gel1b′ (or Gel2b′)) at the concentration of 0.8 wt % and pH of 7.4, Gel1b′ shows sporadic, random, and orientation-independent birefringence (Figure S2B and S2C), and Gel2b′ hardly shows any birefringence (Figure S3B and S3C). These results indicate enzymatic hydrogelation as a feasible and effective pathway to generate an aligned, monodomain supramolecular hydrogel.
Figure 2.

Optical images of Gel1b and Gel2b, formed by treating the solutions of 1a and 2a with ALP (2 U/mL) at the concentration of 0.8 wt % and pH of 7.4 overnight. The images are taken with the vials or capillaries (d = 0.3 mm) placed between cross polarizers, illuminating by ambient light. (A, B) Gel1b in a vial and (C, D) Gel2b in a vial. (E, F, G) Gel1b formed in two capillaries that are placed at three different angles with the cross polarizers.
To quantify the birefringence in the hydrogels, we used polarized microscopy to image the hydrogels made of different samples (i.e., 1b and 2b) and by different pathways (i.e., enzyme or pH). Birefringence is a material property that derives from molecular alignment, which results in a fast and a slow axis in the material. The polarization of light changes due to a differential phase shift between two orthogonal polarized components, one of which, being parallel to the fast axis, travels faster than the other that is parallel to the slow axis. Retardance (Δn) signifies the differential phase shift between two wavefronts of the orthogonal components that have traveled through a birefringent material and is typically measured as a distance in the unit of nm (Figure 1A). Polarized microscopy with the OpenPolScope method24 measures the retardance of every resolved point of specimen and converts the results of computation into a color gradient of the retardance images. Thus, we used PolScope to obtain retardance images of Gel1b, Gel2b, Gel1b′, Gel2b′, and a solution of 1a or 2a. As shown in Figure 3A, the thin film of Gel1b contains largely uniform domains (with the sizes of hundreds of micrometers) that exhibit a retardance of approximately 35 nm or larger. Gel1b′, however, exhibits mostly multiple domains that have retardance between 0 and 25 nm (Figure 3C). As shown in Figure 3B and 3D, Gel2b and Gel2b′, under PolScope, give a retardance value of almost 0 nm, confirming that Gel2b and Gel2b′ are nonbirefringent. As expected, a solution of 1a or 2a at the concentration of 0.8 wt % and pH of 7.4 are nonbirefringent, as proven again by PolScope imaging (Figure S4). These results further support that the combination of strong aromatic–aromatic interactions and enzymatic hydrogelation facilitates the formation of anisotropic supramolecular hydrogels.
Figure 3.

Polarized optical microscopy retardance images (scale bar = 100 μm) of (A) Gel1b, (B) Gel2b, (C) Gel1b′, (D) Gel2b′. The images are taken with a sample thickness of 235 μm and concentration of 0.8 wt %.
Transmission electron microscopy (TEM) also confirms the orientation orders of the nanofibers in Gel1b. As shown in Figure 4A, the nanofibers in Gel1b have diameters (d) of 8 ± 2 nm and form exceptionally long, aligned bundles that stretch over many micrometers (Figure S5), suggesting strong interfiber interactions. In contrast, TEM shows that Gel2b contains a large amount of randomly entangled nanofibers (d = 8 ± 2 nm) that form a typical network (Figure 4B) as those found in conventional supramolecular hydrogels. A TEM image of Gel1b′ indicates a mixture of parallel aligned nanofibers and entangled nanofibers (d = 8 ± 2 nm) (Figure S6A and S6B). Gel2b′, however, contains entangled nanofibers (d = 8 ± 2 nm) to form a network, exhibiting the same morphology as that of Gel2b (Figure S6C and S6D). The agreement between the TEM images and PolScope images confirms that the alignment of the nanofibers formed during enzymatic hydrogelation is responsible for birefringence of Gel1b.
Figure 4.

TEM images of the nanofiber matrices of (A) Gel1b and (B) Gel2b. The scale bar is 250 nm.
To further verify the results in TEM images, we also compared the rheological properties of the hydrogels. Figure 5 shows the strain and frequency dependence of dynamic storage moduli (G′) and loss moduli (G′′) of Gel1b, Gel2b, Gel1b′, and Gel2b′ at a concentration of 0.8 wt %. The values of the storage moduli (G′) of all four gels are larger than those of their loss moduli (G′′), indicating that all the samples behave as viscoelastic gel materials. The values of G′ of the hydrogels change little during the frequency sweep (from 0.1 to 200 rad/s) (Figure S7), suggesting that the matrices of those gels have good tolerance to external shear force. In agreement with TEM data, Gel2b, which consists of entangled nanofibers (more cross-linking), shows stronger mechanical strength than Gel1b, which contains parallel aligned nanofibers (less cross-linking). On the other hand, when both of them have similar nanofiber networks (Figure S6), Gel1b′ exhibits a slightly higher storage moduli than Gel2b′, agreeing with that hydrogelator 1b has one more phenylalanine residue to provide stronger aromatic–aromatic interactions than 2b does.
Figure 5.
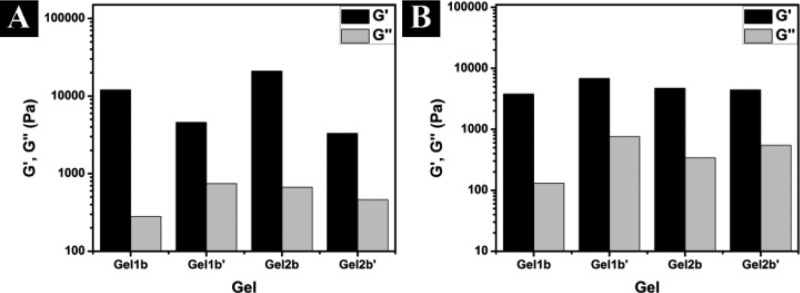
Rheological characterization of Gel1b, Gel2b, Gel1b′, and Gel2b′. (A) The strain dependence of the dynamic storage (G′) and loss storage (G″) is taken at a frequency equal to 6.28 rad/s, and (B) the frequency dependence is taken at a strain equal to 0.99%.
In conclusion, this communication describes a rational design that utilizes aromatic–aromatic interactions to promote interfiber contacts between nanofibers and to drive the alignment of nanofibers for producing inherently anisotropic supramolecular hydrogels via enzyme catalyzed molecular self-assembly. Although direct evidence of aromatic–aromatic interfiber interaction remains to be established, the crystal structure of the self-assembly motif (Nap-Phe-Phe)22 in 1 strongly supports the notion of aromatic–aromatic interfiber interactions. In addition, intermolecular aromatic–aromatic interaction from the overlapping of phenyl and/or naphthyl groups, which are responsible for forming single molecular width nanofibers,25 should favor interfiber interactions. The alignment of the nanofibers likely stems from the stereochemical cooperation between phosphatases and the precursors/hydrogelators because the use of the enantiomer of 1a for enzymatic hydrogelation hardly results in the alignment of nanofibers (Figure S7). Since the hydrogel directly prepared with 1b by a change in pH exhibits little birefringence and consists of few aligned nanofibers, enzymatic conversion is indispensable for the alignment of the nanofibers. The increase of the concentration of enzyme speeds up the gelation process, but shows little influence on birefringence or alignment. Our work, for the first time, illustrates PolScope imaging is a useful and effective method to study anisotropy of supramolecular hydrogels. By establishing enzyme catalysis as a new pathway to complement other processes for generating inherently anisotropic hydrogels, this work may lead to the production of aligned nanostructures in biological systems or living organisms.
Acknowledgments
This work was partially supported by NIH (R01CA142746) HFSP and a start-up fund from Brandeis University. We thank Brandeis EM and Optical Imaging facilities for TEM and Prof. Z. Dogic, Dr. Rudolf Oldenbourg, and Mr. Mark Zakhary for technical assistance on polarized microscopy.
Supporting Information Available
The details of the synthesis, NMR spectra and LC-MS data for all compounds, and rheological data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, 13 South Drive, Bethesda, Maryland 20892, United States.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gray G. W. Liq. Cryst. 1998, 24, 5. [Google Scholar]
- a Miyata H.; Fukushima Y.; Okamoto K.; Takahashi M.; Watanabe M.; Kubo W.; Komoto A.; Kitamura S.; Kanno Y.; Kuroda K. J. Am. Chem. Soc. 2011, 133, 13539. [DOI] [PubMed] [Google Scholar]; b Patzke G. R.; Krumeich F.; Nesper R. Angew. Chem., Int. Ed. 2002, 41, 2446. [DOI] [PubMed] [Google Scholar]
- a Cox J. R.; Simpson J. H.; Swager T. M. J. Am. Chem. Soc. 2013, 135, 640. [DOI] [PubMed] [Google Scholar]; b Hoogboom J.; Swager T. M. J. Am. Chem. Soc. 2006, 128, 15058. [DOI] [PubMed] [Google Scholar]; c Kato T.; Yasuda T.; Kamikawa Y.; Yoshio M. Chem. Commun. 2009, 729. [DOI] [PubMed] [Google Scholar]; d Kato T.; Mizoshita N.; Kanie K. Macromol. Rapid Commun. 2001, 22, 797. [Google Scholar]
- a Zhang S. M.; Greenfield M. A.; Mata A.; Palmer L. C.; Bitton R.; Mantei J. R.; Aparicio C.; de la Cruz M. O.; Stupp S. I. Nat. Mater. 2010, 9, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wu Z. L.; Kurokawa T.; Liang S.; Furukawa H.; Gong J. P. J. Am. Chem. Soc. 2010, 132, 10064. [DOI] [PubMed] [Google Scholar]; c Zhao F.; Gao Y. A.; Shi J. F.; Browdy H. M.; Xu B. Langmuir 2011, 27, 1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk D. E.; Fitch J. M.; Babiarz J. P.; Linsenmayer T. F. J. Cell Biol. 1988, 106, 999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshio M.; Shoji Y.; Tochigi Y.; Nishikawa Y.; Kato T. J. Am. Chem. Soc. 2009, 131, 6763. [DOI] [PubMed] [Google Scholar]
- a Wu Z. L.; Sawada D.; Kurokawa T.; Kakugo A.; Yang W.; Furukawa H.; Gong J. P. Macromolecules 2011, 44, 3542. [Google Scholar]; b Arifuzzaman M.; Wu Z. L.; Kurokawa T.; Kakugo A.; Gong J. P. Soft Matter 2012, 8, 8060. [Google Scholar]; c Lescanne M.; Colin A.; Mondain-Monval O.; Heuze K.; Fages F.; Pozzo J. L. Langmuir 2002, 18, 7151. [Google Scholar]
- a Estroff L. A.; Hamilton A. D. Chem. Rev. 2004, 104, 1201. [DOI] [PubMed] [Google Scholar]; b van Esch J. H.; Feringa B. L. Angew. Chem., Int. Ed. 2000, 39, 2263. [DOI] [PubMed] [Google Scholar]; c Johnson E. K.; Adams D. J.; Cameron P. J. J. Am. Chem. Soc. 2010, 132, 5130. [DOI] [PubMed] [Google Scholar]; d Terech P.; Weiss R. G. Chem. Rev. 1997, 97, 3133. [DOI] [PubMed] [Google Scholar]
- a Palmer L. C.; Stupp S. I. Acc. Chem. Res. 2008, 41, 1674. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Oda R.; Artzner F.; Laguerre M.; Huc I. J. Am. Chem. Soc. 2008, 130, 14705. [DOI] [PubMed] [Google Scholar]; c Brizard A.; Aime C.; Labrot T.; Huc I.; Berthier D.; Artzner F.; Desbat B.; Oda R. J. Am. Chem. Soc. 2007, 129, 3754. [DOI] [PubMed] [Google Scholar]
- Hartgerink J. D.; Beniash E.; Stupp S. I. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yamada N.; Ariga K.; Naito M.; Matsubara K.; Koyama E. J. Am. Chem. Soc. 1998, 120, 12192. [Google Scholar]; b Park J. S.; Jeong S.; Chang D. W.; Kim J. P.; Kim K.; Park E. K.; Song K. W. Chem. Commun. 2011, 47, 4736. [DOI] [PubMed] [Google Scholar]
- Burley S. K.; Petsko G. A. Science 1985, 229, 23. [DOI] [PubMed] [Google Scholar]
- Ma M. L.; Kuang Y.; Gao Y.; Zhang Y.; Gao P.; Xu B. J. Am. Chem. Soc. 2010, 132, 2719. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Gu H.; Fu D.; Gao P.; Lam K. J. K.; Xu B. Adv. Mater. 2006, 18, 545. [Google Scholar]
- a Yang Z.; Liang G.; Xu B. Acc. Chem. Res. 2008, 41, 315. [DOI] [PubMed] [Google Scholar]; b Williams R. J.; Mart R. J.; Ulijn R. V. Biopolymers 2010, 94, 107. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Liang G.; Ma M.; Gao Y.; Xu B. Small 2007, 3, 558. [DOI] [PubMed] [Google Scholar]
- a Yang Z.; Liang G.; Guo Z.; Guo Z.; Xu B. Angew. Chem., Int. Ed. 2007, 46, 8216. [DOI] [PubMed] [Google Scholar]; b Yang Z.; Xu K.; Guo Z.; Guo Z.; Xu B. Adv. Mater. 2007, 19, 3152. [Google Scholar]
- Yang Z.; Liang G.; Wang L.; Xu B. J. Am. Chem. Soc. 2006, 128, 3038. [DOI] [PubMed] [Google Scholar]
- Gao J.; Wang H.; Wang L.; Wang J.; Kong D.; Yang Z. J. Am. Chem. Soc. 2009, 131, 11286. [DOI] [PubMed] [Google Scholar]
- a Gao Y.; Kuang Y.; Guo Z.-F.; Guo Z.; Krauss I. J.; Xu B. J. Am. Chem. Soc. 2009, 131, 13576. [DOI] [PubMed] [Google Scholar]; b Minkenberg C. B.; Florusse L.; Eelkema R.; Koper G. J. M.; van Esch J. H. J. Am. Chem. Soc. 2009, 131, 11274. [DOI] [PubMed] [Google Scholar]
- Debnath S.; Roy S.; Ulijn Rein V. J. Am. Chem. Soc. 2013, 135, 16789. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Kuang Y.; Gao Y.; Xu B. Langmuir 2011, 27, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Chan W. C., White P. D., Eds.; Oxford University Press: New York, 2000. [Google Scholar]
- Shribak M.; Oldenbourg R. Appl. Opt. 2003, 42, 3009. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhang B.; Kuang Y.; Gao Y.; Shi J. F.; Zhang X. X.; Xu B. J. Am. Chem. Soc. 2013, 135, 5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.