

Abstract
Recent studies suggest that traumatic brain injury (TBI) and pesticide exposure increase the risk of Parkinson’s disease (PD), but the molecular mechanisms involved remain unclear. Using an in vitro model of TBI, we evaluated the role of mitochondrial membrane potential (ΔΨm) and mitochondrial reactive oxygen species (ROS) induced by stretch on dopaminergic cell death upon paraquat exposure. Human dopaminergic neuroblastoma SH-SY5Y cells grown on silicone membrane were stretched at mild (25%) and moderate (50%) strain prior to paraquat exposure. We observed that moderate stretch (50% strain) increased the vulnerability of cells to paraquat demonstrated by the loss of plasma membrane integrity (propidium iodide-uptake) and decreased mitochondrial activity (MTT assay). Mitochondrial depolarization occurred immediately after stretch, while mitochondrial ROS increased rapidly and remained elevated for up to 4 h after the stretch injury. Intracellular glutathione (GSH) stores were also transiently decreased immediately after moderate stretch. Cells treated with paraquat, or moderate stretch exhibited negligible mitochondrial depolarization at 48 h post treatment, whereas in cells stretched prior to paraquat exposure, a significant mitochondrial depolarization occurred compared to samples exposed to either paraquat or stretch. Moderate stretch also increased mitochondrial ROS formation, as well as exacerbated intracellular GSH loss induced by paraquat. Overexpression of manganese superoxide dismutase (MnSOD) markedly diminished the deleterious effects of stretch in paraquat neurotoxicity. Our findings demonstrate that oxidative stress induced by mitochondrial dysfunction plays a critical role in the synergistic toxic effects of stretch (TBI) and pesticide exposure. Mitigation of oxidative stress via mitochondria-targeted antioxidants appears an attractive route for treatment of neurodegeneration mediated by TBI.
Keywords: traumatic brain injury, paraquat, mitochondria, reactive oxygen species, MnSOD
1. Introduction
Parkinson’s disease (PD) affects more than 1% of the population over 65, and is characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta. It is widely acknowledged that PD etiology is multifactorial. The most important risk factors associated with sporadic forms of PD include age, genetic polymorphisms, pesticide exposure, and traumatic brain injury (TBI) (Freire and Koifman, 2012, Goldman et al., 2012, Kieburtz and Wunderle, 2013).
Various environmental agents, especially pesticides, and factors leading to increased exposure to these chemicals (e.g. farming, well-water drinking and rural living) have been proposed as potential risk factors for PD (Freire and Koifman, 2012). In the 1980s, it was discovered that exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a substance structurally similar to the herbicide paraquat, caused chronic and severe parkinsonism in humans resulting in the degeneration of dopaminergic neurons (Perez-Otano et al., 1994). Paraquat is frequently used as a neurotoxicant in experimental PD studies and an increasing body of evidence supports the strong correlation between herbicide exposure and PD (Abdulwahid Arif and Ahmad Khan, 2010, Allen and Levy, 2013, Tanner et al., 2011).
TBI has attracted tremendous attention as a PD risk factor (Szczygielski et al., 2005). TBI is the most common cause of death and impairment within the Western World population, affecting predominantly people in their productive age. Although the epidemiological data on the relationship between PD and TBI are inconsistent and the association remains a controversial subject, a number of studies have found substantial evidences supporting the hypothesis that there exists a positive TBI-PD correlation. A study on twins reported a 4-fold increased risk for developing PD among subjects self-reporting TBI experience, suggesting that mild closed-head injury is significantly related to PD in discordant twin pairs (Goldman et al., 2006). Bower and colleagues reported that patients suffering mild head trauma with a loss of consciousness or a severe head trauma had a significantly higher risk of developing PD late in life (Bower et al., 2003). Besides epidemiological research, extensive rapid accumulation of α-synuclein was reported in swollen axons in brain tissues from cases who died after TBI (Uryu et al., 2007). In experimental animals, TBI can induce the accumulation of α-synuclein (Uryu et al., 2003), neuroinflammatory response (Morales et al., 2005), progressive loss of nigrostriatal dopaminergic neurons (Hutson et al., 2011), and a reduction in tyrosine hydroxylase activity in the striatum (Onizuka et al., 2011).
An important question only partially addressed in recently published literature is whether there is synergistic effect between factors responsible for PD development. An animal study mimicking TBI by lateral fluid percussion (LFP) demonstrated that a single exposure to a mild TBI was sufficient to cause a progressive loss of nigrostriatal dopaminergic neurons (Hutson, Lazo, 2011). In this study, the number of dopaminergic neurons lost was much larger when TBI was accompanied by exposure to paraquat at a dose that by itself did not cause a significant loss of nigrostriatal dopaminergic neurons. In addition, greater induction of α-synuclein accumulation and inflammation were observed in animals in a combined model of paraquat exposure and head trauma (Hutson, Lazo, 2011). Another epidemiological research reported a 3-fold risk increase for subjects who reported a TBI insult and had also been exposed to paraquat at home and workplace, compared with persons who had never experienced TBI or been exposed to paraquat (Lee et al., 2012).
Recent research suggests that PD, TBI and neurotoxicant-induced neurodegeneration might share some common pathways (Chaturvedi and Flint Beal, 2013, Cheng et al., 2012). Loss of mitochondrial complex I catalytic activity in the electron transport chain (ETC) was reported in PD patients and animal models (Betarbet et al., 2000). Brain trauma and mechanical stretch injury of neurons derange mitochondrial function, initiate release of pro-apoptotic factors and thus result in cell death progression (Redell et al., 2013). Exposure to a mitochondrial/environmental toxin prior to a TBI insult produced a multifactorial injury with pathological changes greater than either insult alone. In a rat model, one week exposure to an environmental toxin, trichloroethylene, followed by TBI results in a synergistic dysfunction in striatal mitochondria (Sauerbeck et al., 2012). Although there have been previous reports linking TBI and pesticide exposure to PD, the molecular mechanisms involved remain unclear.
In the current study, we evaluated the synergistic effects of TBI (mechanical stretch injury) and pesticide exposure (paraquat) using an in vitro model. We report here that moderate stretch injury renders dopaminergic cells more vulnerable to subsequent paraquat toxicity. This exacerbation is associated with a high level of mitochondrial ROS formation triggered by mitochondrial dysfunction. Overexpression of manganese superoxide dismutase (MnSOD) diminishes the deleterious effect of stretch injury on paraquat exposure, supporting a selective role of superoxide anion in this process.
2. Materials and Methods
2.1 Materials
Paraquat stock solution was prepared in double distilled H2O (dd H2O, 0.5 M) and diluted to final concentration in DMEM media. All fluorescent probes stock solutions were prepared in dimethyl sulfoxide (DMSO) and diluted to their indicated final concentrations with DPBS or cell culture media with a final DMSO concentration of ≤ 0.1%.
2.2 Cell culture
We chose undifferentiated SH-SY5Y cells in current study. SH-SY5Y cells are frequently used to study neuron-like behavior in response to neurotoxins or mechanical injury. The SH-SY5Y cells can be used in both undifferentiated and differentiated state. However, it has been reported that differentiation by retinoic acid (RA) renders SH-SY5Y cells resistant to oxidative stress, alters mitochondrial function in SH-SY5Y cells, e.g. increases ΔΨm, levels of cytochorome c oxidase, MnSOD and bioenergic reserve capacity (Schneider et al., 2011). In addition, SH-SY5Y differentiation also renders cells resistant to PD toxins (Cheung et al., 2009). Thus, for this work, these cells were used in a non-differentiated stage differentiation. SH-SY5Y cells were maintained at 37°C under a humidified atmosphere containing 5% CO2 and cultured in DMEM media with 10% heat inactivated fetal bovine serum (FBS) and 100 μg/mL penicillin-streptomycin. Cells were seeded at a density of 10,000/cm2 on elastic transparent silicone membrane (polydimethylsiloxane, PDMS, Specialty Manufacturing Inc., Saginaw, MI) coated with poly-D-lysine overnight (0.05 mg/mL).
2.3 Stretch device
The stretch device consists of 3 main parts (Fig. 1): 1) a top cover with glass window, pressure probe and gas inlet; 2) a bottom plate and 3) a round stainless steel cell cultured plate with a circular opening (diameter: 20 mm). The culture plate is placed between the top cover and the bottom plate and the whole assembly is secured with 4 screws. At the bottom of the stainless steel cell culture plate the silicone (PDMS) membrane is affixed. The cells are attached in the internal part, and are stretched together with the membrane as it deforms outwards in response to the air pulse (inset in Fig. 1). The flow of the compressed air from the cylinder into the chamber is controlled by a pressure regulator (pressure magnitude controlling) and a solenoid valve (pulse duration controlling). The Matlab program (The MathWorks, Inc.) is used to control the solenoid valve and record the pressure profile inside of the stretch device.
Figure 1.

Main components of the cell stretching device: 1) a top plate with glass window, pressure probe and gas inlet: 2) a bottom plate and 3) a round stainless steel cell cultured plate with a circular opening (diameter: 20 mm). Inset: compressed air is introduced into an airtight chamber, which results in deformation of the PDMS membrane, and thus stretching the adherent cells. The PDMS with stochastic pattern was used only in strain calibration experiments (details are described elsewhere (Skotak, Wang, 2012)).
2. 4 Cell stretch injury model
The stretch tests were performed on the third day after cell seeding. A controlled air pulse was used to generate stretch to the cells cultured on PDMS membrane. According to the strain calibration results obtained in our previous work (Skotak et al., 2012), the discrete strain values in the 10-70% range were induced by air pulse with the following internal pressure (psi) – pulse duration (ms) characteristics: 1) 5-10 (10%); 2) 5-15 (25%); 3) 5-20% (35%); 4) 10-15 (50%); 5) 15-15 (70%). Before stretch all the media used for cell culture were discarded and 200 μL aliquot of fresh, warm (37 °C) DMEM were added to cover the cells, and prevent their dehydration. The stretch experiments were conducted at room temperature and finished within 1 min. Then, an aliquot of 3 mL warm media was added to the cell culture plate immediately after the stretch and the culture was returned to the incubator. When the membrane is stretched, a small central region is extended biaxially, therefore, all tests described here are subject to biaxial stretching. The strain gradient across the stretched membrane is a characteristic feature of this system, but the strain in the center region is relatively uniform.
2.5 Stretch injury model and paraquat exposure
It has been reported that mechanical stretch to primary cortical neurons resulted in a transient increase in plasma membrane permeability and that the plasma membrane is able to repair within several minutes after the initial stretch (Geddes et al., 2003). In our previous study (Skotak, Wang, 2012), we observed that the plasma membrane integrity completely recovered 2 h after 50% stretch. Thus, to completely prevent an increase in intracellular paraquat accumulation due to damaged plasma membrane, paraquat treatment was initiated 2 h after the stretch. Cells were treated with two different concentrations of paraquat (0.2 and 0.5 mM) after the stretch injury and were kept in the incubator for 48 h before assays.
2.6 Assessment of cell viability by mitochondrial MTT reduction assay
Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. MTT was dissolved in DPBS at 5 mg/mL and the solution was filtered with 0.22 μm syringe filters and stored at 4 °C in dark. The storage time was less than 1 week, otherwise new solution was made. 300 μL of working MTT solution was added to the cell culture plate with 3 mL media at a final concentration of 0.5 mg/mL and cells were incubated at 37 °C for 2 h. At the end of the incubation period, the media was aspirated and centrifuged and 1 mL dimethyl sulfoxide (DMSO) was added to each well to solubilize the formazan crystals on the membrane and in the centrifuge tube. 200 μL of DMSO formazan solution was loaded to 96-well plate. Each sample has 2 duplicate wells. Absorbance was read at 520 nm in a microplate reader. Results were expressed as a percentage of sample absorbance in relation to the control (untreated cells).
2.7 Analysis of cell membrane integrity by fluorescence activated cell sorting (FACS)
Propidium iodide (PI, Sigma) uptake was used as a marker for plasma membrane integrity loss. PI fluorescence was measured by flow cytometry. After treatment, both floating and attached cells were collected and washed in DPBS. Cells were centrifuged at 300 g for 5 min and cell pellets were re-suspended with DPBS containing PI (1 μg/mL). PI fluorescence was immediately recorded using a 561 nm excitation laser and 620/20 nm emission filter (Laser 1 FL3) in a BDFACSort (Cytek-DxP-10 upgrade). 10,000 cells were analyzed per sample and data analyzed using FlowJo 7.6.5 software.
2.8 Mitochondrial membrane potential (ΔΨm) measurement
ΔΨm was measured using the fluorescent dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide, Invitrogen). JC-1 is a metachromatic concentration-dependent fluorescent probe that exhibits potential-dependent accumulation in mitochondria as indicated by the red fluorescence emitted from healthy mitochondria with normal potential, whereas organelles with reduced potential emit green fluorescence. Cell cultures were pre-incubated at 37 °C with 2 μM JC-1 for 30 min. JC-1 fluorescence was recorded on Nikon Ti-E Eclipse microscope equipped with 130 W, high-pressure mercury lamp and filter cubes: 1) Semrock BrightLine FITC-3540C-NTE (ex/em: 460-500 nm/520-550 nm) and 2) Semrock BrightLine TxRed-4040C-NTE (ex/em: 530-580 nm/600-650 nm). The green and red channels were acquired separately using Nikon Plan Apo 10x (numerical aperture 0.45). Three random images with resolution of 1392 × 1040 pixels were acquired using (0.65 μm/pixel, corresponding to the imaging area of 0.905 × 0.676 mm). On average, three samples per predefined strain level and a total of a 600-800 of cells per sample were analyzed. The intensities of the images from both channels were measured using ImageJ software taking into account the background fluorescence, and the ratios of red and green fluorescence densities were calculated. In addition, flow cytometry was also used to evaluate changes in JC-1 fluorescence. Briefly, cells were harvested and incubated with 2 μM JC-1 15 min prior to FACS analysis and JC-1 green fluorescence was measured using 488 nm excitation and 530/30 nm emission filters (Laser 1 FL1).
2.9 Detection of mitochondrial reactive oxygen species (ROS) and intracellular glutathione (GSH)
For the measurement of mitochondrial ROS and intracellular GSH, the fluorescence probes MitoSOX Red (Molecular Probes, Invitrogen) and monochlorobimane (mBCl, Molecular Probes, Invitrogen) were used. The mBCl is a non-fluorescent substrate which can react with GSH in a reaction catalyzed by the enzyme, GSH-S-transferase to from a fluorescent conjugate. MitoSOX Red is a derivative of dihydroethidium with a cationic triphenylphosphonium substituent responsible for the electrophoretic uptake into actively respiring mitochondria. The cells were collected and incubated with reconstituted MitoSOX Red dye (5 μM) and mBCl (50 μM) for 15 min at 37°C prior to analysis. MitoSOX Red fluorescence was measured using 488 nm excitation and 620/20 nm emission filters (Laser 1 FL3) and the mBCl fluorescence was measured using 407 nm excitation and 450/50 nm emission filters (Laser 3 FL1). The final results were expressed as the percentage (or fold) of fluorescence compared with vehicle-treated controls.
2.10 Recombinant adenoviral vectors
Replication-deficient recombinant adenoviruses (Ad5CMV-MnSOD [Ad-MnSOD]) were used to overexpress MnSOD as described previously (Rodriguez-Rocha et al., 2013). Adenovirus containing only the CMV promoter (Ad-Empty) was utilized as control. Cells were infected with adenoviral vectors at a multiplicity of infection (MOI) of 0.15 and treated with experimental conditions at 24 h post-infection.
2.11 Statistical analysis
All experimental data points are independent and correspond to experiments performed on separated days. Data are presented as mean ± standard error of mean (SEM). All experiments were analyzed with a one-way analysis of variance (ANOVA), followed by post-hoc analysis with Fisher’s least significant difference (LSD) test. For all experiments statistical significance was considered at P<0.05. Flow cytometry plots presented are representative of at least four independent experiments.
3. Results
3.1 Stretch renders SH-SY5Y cells more vulnerable to a secondary insult with paraquat
In previous studies, we have established the dose-response strain curve with respect to injury in SH-SY5Y cells (Skotak, Wang, 2012). The dose-response curve for ε=0-100% presents three regions with different degrees of injury: (I) mild, with predominately live cells (ε=0-30%); (II) moderate, where the transition between healthy and injured cells takes place (ε=30-55%); and (III) severe, with mainly injured and dead cells (ε=55-100%). Temporal injury evaluation at four discrete strain values (ε=42%, 50%, 73% and 90%) shows that although cells suffered a significant injury after 42% and 50% stretch, they gradually recuperate and their recovery was almost complete 2 h post-injury. Cells injured at higher strains (ε=73% and 90%) partially recovered in initial 2-4 h, but at longer time (12-24 h) suffered from recurring plasma membrane integrity loss and relatively high numbers of dead cells, which is not the case of samples stretched at 42% and 50% strain. Hence, 25% and 50% strain were chosen as mild and moderate TBI stretch models in current study. First, we decided to evaluate the effect of stretch on cell death following exposure to paraquat by flow cytometry analyzing PI staining (Fig. 2A). 50% strain of stretch, 0.2 mM and 0.5 mM paraquat induced an increase in the mean fluorescence of PI (Fig. 2B). The stretch at 25% strain slightly increase paraquat (0.2 mM and 0.5 mM)-induced cell death, but it was not statistically significant. However, a 50% strain significantly increased 0.5 mM paraquat-induced cell death (Fig. 2B). Similar results were observed when cell viability was measured by MTT. The difference is that 50% strain of stretch significantly enhanced the toxicity of 0.2 mM paraquat, rather than 0.5 mM paraquat seen in PI staining. These results demonstrate that high strain stretch renders dopaminergic cells more vulnerable to a secondary insult with paraquat.
Figure 2. Effect of stretch on SH-SY5Y cell survival/death in the presence of paraquat (PQ).
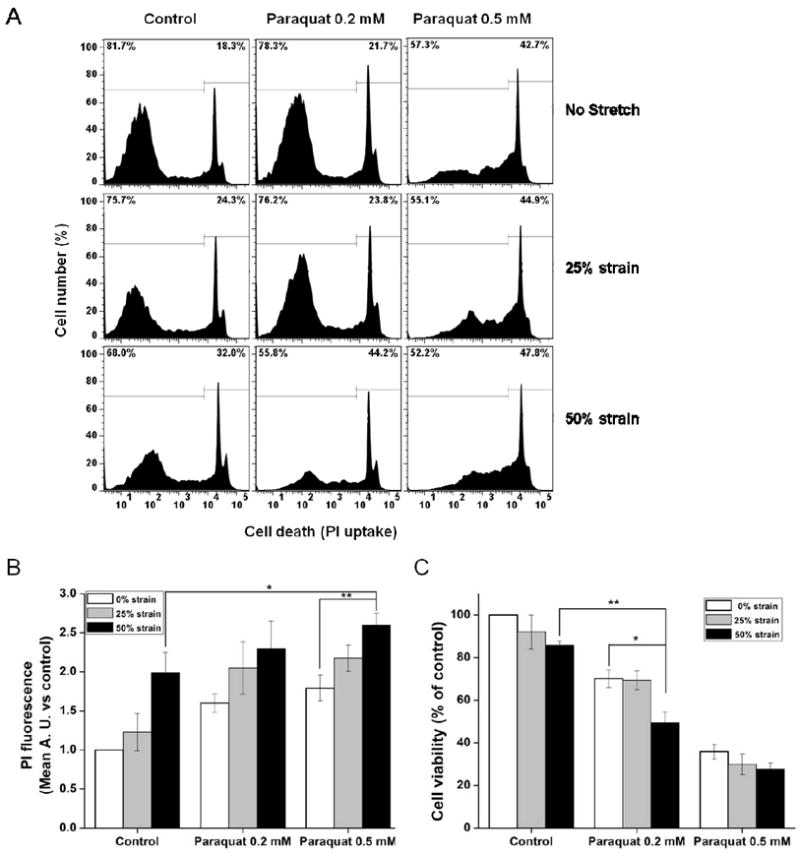
Cells were stretched at the indicated strain levels 2 h before exposure to paraquat. Cell death was evaluated 48 h after paraquat treatment by PI uptake and cell survival was analyzed by MTT. (A) Representative histograms of changes in PI fluorescence (loss of plasma membrane integrity is reflected as an increase in PI fluorescence). (B) PI uptake results are represented as the increase in mean PI fluorescence with respect to controls. (C) Cell viability of SH-SY5Y cells was determined by MTT. Data represent the mean ± SEM of five independent experiments (n=5). *P< 0.05 and **P< 0.01.
3.2 Stretch induces a reduction in ΔΨm
We then assessed the changes of ΔΨm induced by different levels of strain by fluorescence microscpy. Fig. 3A shows the fluorescent images of SH-SY5Y cells stained with JC-1. Immediately after a 50% or 70% stretch, cells presented a decrease in red fluorescence, which indicates ΔΨm depolarization. Fig. 3B shows the changes in JC-1 fluorescence induced by different strain levels (ratio of red to green) immediately after stretch. Mild stretch (10% and 25% strain) had no significant effect on mitochondrial membrane potential compared to controls. However, moderate (50% strain) and severe (70% strain) stretch significantly decreased the JC-1 red/green ratio (Fig. 3B). Time-dependent studies show that JC-1 red/green ratio was not altered after 25% strain stretch over the entire period of screening, i.e. 1, 2, 4, and 12 h (Fig. 3C). Immediately after a moderate strain, there is a significant loss in ΔΨm that subquently starts to recover with time progression after the injury. However, ΔΨm remained lower than control samples even after 12 h post stretch (Fig. 3D). This suggests that immediate mitochondrial dysfunction occurs with moderate stretch.
Figure 3. Effect of stretch on ΔΨm.
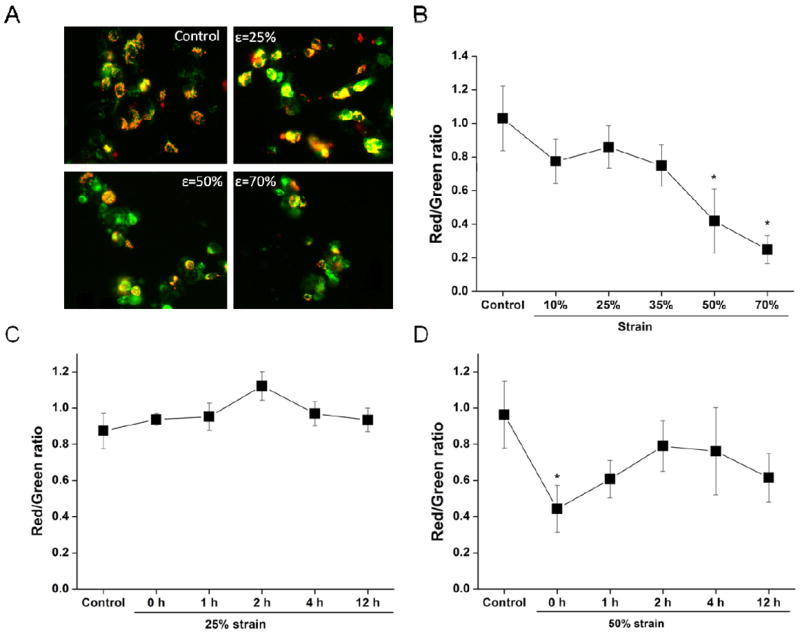
Cells were subject to the indicated strain levels. (A) Representative JC-1 fluorescence images for the indicated conditions. (B) Effects of strain on JC-1 staining (red/green ratio) at 0 h after insult measured by fluorescent microscopy. Temporal changes of ΔΨm after exposure to 25% (C) and 50% (D) strain of stretch (0, 1, 2, 4, 12 h). Data in graphs represent mean ± SEM of n = 4 experiments. *P< 0.05 compared with controls.
3.3 Stretch injury increases mitochondrial ROS and decreases intracellular GSH
Mitochondria are the main source of ROS and ROS overproduction is one of the early mitochondrial events following mitochondrial dysfunction. We used MitoSOX Red and mBCl to determine the alterations in mitochondrial ROS and intracellular GSH levels by flow cytometry after stretch injury. A 25% strain caused only a minor increase in mitochondrial ROS, but it was not statistically different from controls (Fig. 4A). This result is consistent with the ΔΨm data described in the previous section. The stretch at 50% strain significantly increased mitochondrial ROS production immediately after the injury (0 h), and high ROS levels persisted for up to 4 h (Fig. 4C). Along with the increase in ROS production, intracellular GSH also suffered a sharp decrease immediately after the stretch and the low GSH status lasted up till 4 h post-stretch (Fig. 4D). Interestingly, the decrease in GSH at 4 h after 25% strain stretch correlates with a minor increase in mitochondrial ROS formation. (Fig. 4B).
Figure 4. Time course of mitochondrial ROS generation and intracellular GSH levels in SH-SY5Y cells after stretch injury assessed by flow cytometry.
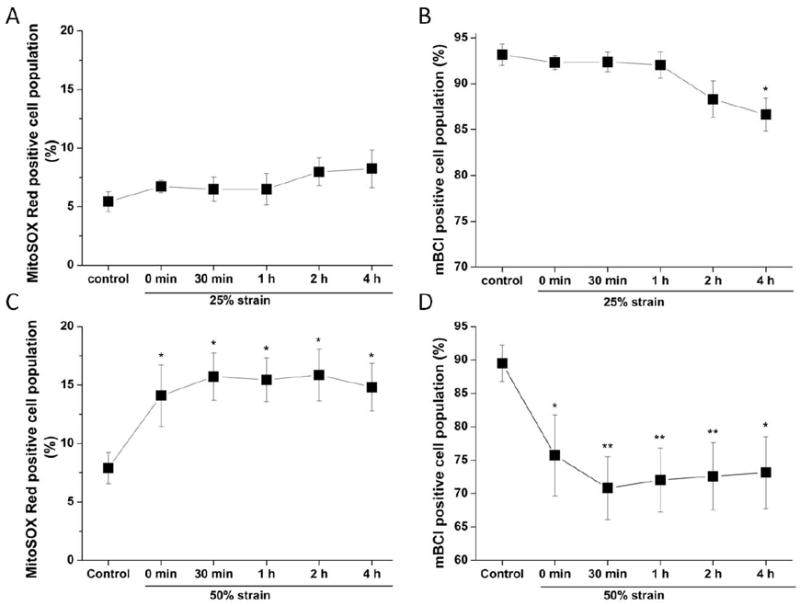
Cells were treated with 25% strain or 50% strain. Mitochondrial ROS and GSH were analyzed by FACS at the time indicated after stretch. In A (25% strain) and C (50% strain), data are expressed as mitochondrial ROS production (MitoSOX Red positive cell population, %). In B (25% strain) and D (50% strain), data are presented as intracellular GSH (mBCl positive cell population, %) normalized against control values. Data in graphs represent mean ± SEM of n = 5 experiments. *P< 0.05 and **P< 0.01 compared with control.
3.4 Moderate stretch increases mitochondrial depolarization induced by paraquat
Mitochondrial dysfunction has been previously reported to occur after paraquat exposure (Rodriguez-Rocha, Garcia-Garcia, 2013). Accordingly, we examined the effect of combining two different modes of injury on mitochondrial dysfunction (stretch with subsequent paraquat exposure) by ΔΨm measurements (Fig. 5A). In the absence of any insult, SH-SY5Y cells maintain their ΔΨm throughout the 48 h observation period. Cultures exposed to 50% strain stretch or 0.2 mM paraquat for 48 h exhibited slight mitochondrial depolarization, reflected as an increase in JC-1 green fluorescence, but this decrease is not statistically significant compared to untreated cultures (Fig. 5B). However, treatment with 0.5 mM paraquat alone caused a significant depolarization of the mitochondria. Cells injured with stretch at 50% strain were subsequently challenged with 0.2 mM paraquat, and we observed a statistically significant decrease in ΔΨm, with respect to stretch-only or paraquat-only insults. 50% strain stretch did not further exacerbate the loss of ΔΨm induced by 0.5 mM paraquat alone.
Figure 5. Effect of stretch on ΔΨm of SH-SY5Y cells in the presence or absence of paraquat assessed by flow cytometry.
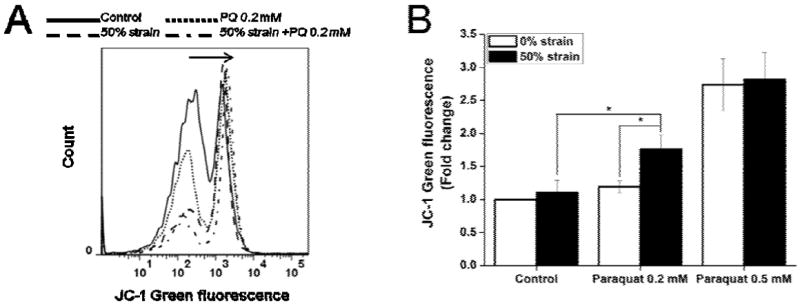
Cells were stretched at 50% strain 2 h before exposure to paraquat (PQ). JC-1 green fluorescence was evaluated 48 h after paraquat treatment by FACS. A. Representative histograms depict the changes in the JC-1 mean green fluorescence. B. Data are expressed as JC-1 green fluorescence normalized against control values. Data in the graph represent mean ± SEM of n = 5 experiments. *P< 0.05.
3.5 Moderate stretch injury increases mitochondrial ROS production and GSH depletion induced by paraquat
Mitochondria are significant sources of ROS in paraquat induced injury (Rodriguez-Rocha, Garcia-Garcia, 2013) and ROS mediate cell injury at the early stages following the traumatic event (Abdul-Muneer et al., 2013). Because cell death in stretch- plus paraquat-treated cells was associated with loss of mitochondrial function, we questioned whether this could be related to ROS. Fig. 6A and 6B shows that treatment of cells with 0.5 mM paraquat for 48 h caused an expected increase in mitochondrial ROS, while exposure to 0.2 mM of paraquat alone, or 50% strain stretch only slightly increased the mitochondrial ROS production. However, a combined effect of 50% strain stretch injury followed by treatment with 0.2 mM paraquat resulted in the production of higher amounts of mitochondrial ROS. This finding correlates with the MTT analysis of mitochondrial function (Fig. 2C) and mitochondrial depolarization (Fig. 5B). However, 50% strain stretch followed by treatment with 0.5 mM paraquat did not evoke any substantial mitochondrial ROS formation compared to cultures treated only with 0.5 mM paraquat (Fig. 6B). In contrast, 50% strain stretch exacerbated GSH depletion induced by both 0.2 mM and 0.5 mM parauqat (Fig. 6C).
Figure 6. Effect of stretch on mitochondrial ROS and intracellular GSH levels in SH-SY5Y cells treated with paraquat.
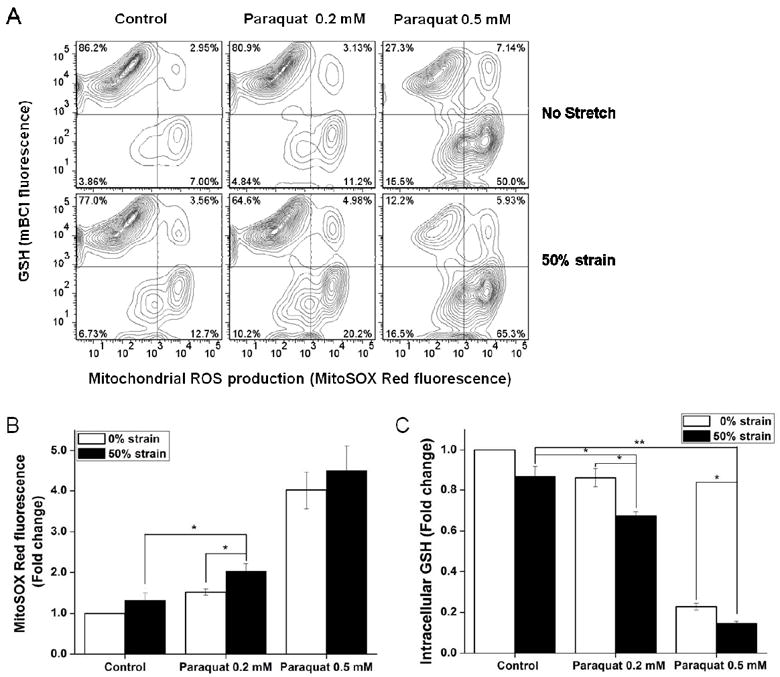
Cells were stretched at 50% strain 2 h before exposure to paraquat. MitoSOX and mBCl fluorescence was evaluated 48 h after paraquat treatment by FACS. A. Representative histograms depict the changes in MitoSOX and mBCl fluorescence in response to the indicated treatment. In B and C, data are expressed as mitochondrial ROS production (MitoSOX Red fluorescence) normalized against control values. Data in graphs represent mean ± SEM of n = 5 experiments. *P< 0.05 and **P< 0.01.
3.6 Overexpression of MnSOD decreases the stimulatory effects of stretch in paraquat toxicity
Paraquat toxicity is mediated primarily by mitochondrial superoxide anion formation (Rodriguez-Rocha, Garcia-Garcia, 2013). Superoxide dismutases (SODs) convert superoxide anion to the relatively more stable and less toxic hydrogen peroxide. MnSOD is a mitochondrial antioxidant enzyme located in the mitochondrial matrix. We overexpressed MnSOD using adenoviral vectors and determined its effect on cell viability in stretched cells treated with paraquat. MnSOD overexpression (Ad-MnSOD) significantly increased cell survival in cells treated with stretch at 50% strain stretch and paraquat compared with the Ad-Empty cells (Fig. 7). This result demonstrates that stretch-induced exacerbation of paraquat-induced toxicity are directly dependent on the generation of mitochondrial superoxide.
Figure 7. Effect of MnSOD overexpression on paraquat induced cytotoxicity following stretch injury.
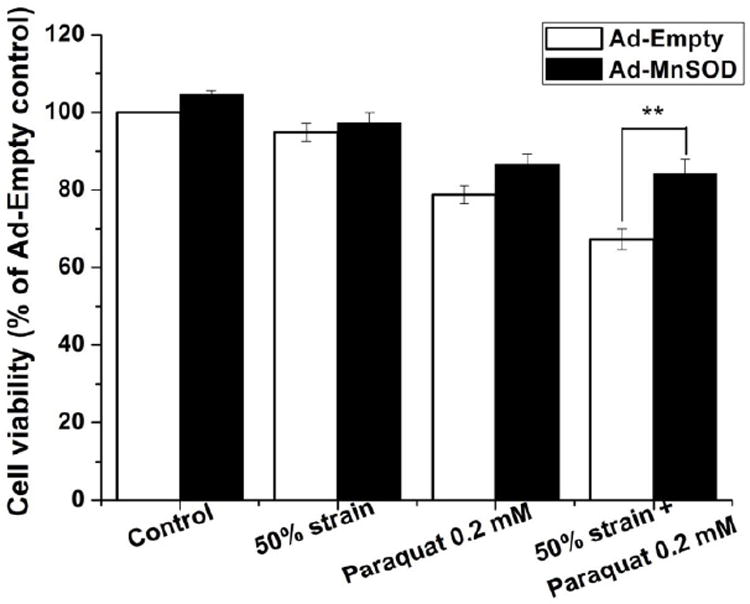
Cells were infected with Empty adenovirus or adenovirus encoding MnSOD as indicated in materials and methods, and treated with the experimental conditions at 24 h post-infection. Data in the graph represent mean ± SEM of n = 5 experiments. **P< 0.01.
4. Discussion
Epidemiological studies have correlated the potential relationship between TBI and PD (Lee, Bordelon, 2012). However, a clear causative link between TBI and PD is difficult to establish and as a result it has been proposed that a multifactorial mechanism may be involved (Sauerbeck, Hunter, 2012). According to a recent study, TBI results in impairments of the activity of tyrosine hydroxylase (TH) in the striatum which could facilitate additional damage to the dopaminergic system, the core system affected in PD (Shin et al., 2011). Furthermore, a recent dual injury model in rats based on TBI and paraquat exposure has shown that TBI potentiated deficits that occur following a subsequent toxin exposure and led to changes characteristic for PD (Hutson, Lazo, 2011). In the current study, we used an in vitro dual injury model to test the synergistic effect of occupational risk (TBI) and environmental exposure (paraquat) on dopaminergic cell death. We demonstrated that stretch induced mitochondrial dysfunction and mitochondrial superoxide anion formation potentiates paraquat-induced toxicity in dopaminergic cells.
Mechanical stretch induced injury has been used to study the effects of trauma on neurons (Arundine, 2004) and astrocytes (Ahmed et al., 2000) in vitro. This injury model replicates many of the post-traumatic responses observed in vivo, including increases in cell permeability (Geddes, Cargill, 2003), calcium concentration (Geddes and Cargill, 2001) and mitochondrial dysfunction (Ahmed, Rzigalinski, 2000). Stretch model has been used to explore whether multifacotrial injury (TBI and activation of NMDA receptors) can produce a greater neuronal damage (Arundine, 2004). Recently, we established an in vitro stretch injury model and current work relies on the injury thresholds measured in that study (Skotak, Wang, 2012). In order to understand if exposure to paraquat is capable of leading to greater cell death following stretch injury, SH-SY5Y cells were stretched 2 h before exposing to paraquat. Our results revealed that mild stretch (25% strain) does not contribute to paraquat toxicity, whereas moderate stretch (50% strain) does (Fig. 2B, 2C). Similarly, in neuronal cultures, cells subjected to a sub-lethal mechanical stretch are more sensitive to NMDA insult (Arundine, 2004). We observed that moderate stretch significantly increases cell death in cultures treated with 0.5 mM paraquat, but not in cultures exposed to 0.2 mM parauquat. In contrast, moderate stretch only significantly decreased mitochondrial function of in cultures treated with 0.2 mM PQ, but not in cultures treated with 0.5 mM PQ. These results suggest that mitochondrial function precedes cell injury in response to stretch and PQ.
Mitochondrial dysfunction is a major pathological mechanism involved in TBI and PD, and is an early event in experimental PD models such as paraquat and 1-methyl-4-phenylpyridinium ion (MPP+) (Cheng, Kong, 2012, Stack et al., 2008). Previous studies have shown that mechanical stretch induces mitochondrial dysfunction and ATP release in neurons using a Flexercell stretch model (Ahmed, Rzigalinski, 2000). To explore the mechanism involved in the effects of stretch in paraquat toxicity, we first focused on alterations in ΔΨm induced by stretch. Mild stretch (up to 25% strain) does not affect ΔΨm, whereas moderate (50% strain) and severe stretch (70% strain) cause significant depolarization of the mitochondria (Fig. 3B) immediately after stretch. Mild stretch (25% strain) neither produced immediate or delayed changes in ΔΨm, whereas moderate stretch (50% strain) caused an acutely significant decrease in ΔΨm (Fig. 3C, 3D) followed by slow 30 min post stretch recovery. These data suggest that stretch depolarizes ΔΨm in a strain- and time- dependent manner. In Ahmed’s research, a similar injury pattern (strain-dependent) was also reported in Flexercell stretch model (Ahmed, Rzigalinski, 2000). However, in their model, moderate stretch did not cause an acute ΔΨm depolarization, but caused a significant decrease in ΔΨm at 48 h post-stretch. In addition, in their model, severe stretch did significantly depolarize ΔΨm in neurons in the acute recovery phase (15 min post stretch). The discrepancy between our results and theirs can be explained by a number of factors: 1) the different response of cell types used in both studies, 2) arbitrarily chosen injury thresholds, and 3) the lack of standardized criteria to describe injury severity on a cellular level. These limitationis of contemporary in vitro stretch injury (TBI) models make comparison of the results between different laboratories extremely challenging.
The net production of ROS is an important mechanism by which mitochondria are thought to contribute to TBI pathology (Abdul-Muneer, Schuetz, 2013). In our in vitro model, mild stretch did not increase mitochondrial ROS level, which correlates well with the lack of effect of mild stretch on mitochondrial membrane potential (Fig. 4A, 4B). However, in cells subjected to moderate stretch, induction of immediate mitochondrial ROS burst lasting for up to 4 h after stretch was evident (Fig. 4D, 4E). These results suggest that acute mitochondrial dysfunction can induce a long-term mitochondrial ROS production. In a Flexercell stretch model, the ROS levels of neurons were gauged over 60 min using dihydrorhodamine (DHR) and results showed that lethal stretch caused a rapid increase in ROS production (Arundine, 2004), which is similar to our results.
GSH plays a key role in the maintenance of intracellular oxidative balance (Franco and Cidlowski, 2012). This tripeptide can function as an antioxidant in the presence of glutathione peroxidase by reducing hydroperoxides to less toxic substances. Thus, GSH constitutes the first line of defense in the protection of neurons from oxidant-mediated damage (Schulz et al., 2000). In a clinical study, it has been reported that GSH in cerebrospinal fluid (CSF) increased on day 1 and then progressively decreased from day 2 to day 7 after TBI (Bayir et al., 2002). Cyclic stretch in A549 cells caused a transient depletion of GSH at 1 h followed by a significant increase in intracellular GSH 4 h after the injury (Jafari et al., 2004). Moderate stretch caused a rapid decrease in GSH, indicating alterations in the redox balance by stretch (Fig. 4D, 4F). However, GSH levels recovered slowly after stretch. Interestingly, in spite mild stretch doesn’t affect plasma membrane integrity or alters the mitochondrial ROS levels (only a minor but not significant increase was detected compared to control), it causes depletion of intracellular GSH at later time points (4 h) (Fig. 4A, 4C). These results suggest that lower levels of stretch may trigger ROS production in the cytosol, as well to deplete intracellular GSH. It is also possible that stretch might be affecting GSH synthesis and metabolism or that GSH depletion might also be mediated by stretch activated efflux pathways (Franco and Cidlowski, 2012, Sabirov et al., 2013).
A number of recently published papers have been demonstrated that exposure to paraquat causes a significant impairment in mitochondrial function (Abdulwahid Arif and Ahmad Khan, 2010, Rodriguez-Rocha, Garcia-Garcia, 2013). We aimed to understand if exposure to paraquat and stretch injury exerts a synergistic effect at the mitochondrial level. We have shown earlier that ΔΨm decreases significantly right after after the stretch injury, while recovery begins as early as 30 minutes post insult. Indeed, moderate stretch combined with 0.2 mM paraquat caused a significant degree of mitochondrial depolarization. No significant difference was noticed between cells treated with 0.5 mM paraquat combined with moderate stretch, compared to cells treated only with paraquat. It appears that at high paraquat concentrations ΔΨm is severely altered by action of paraquat alone, and stretch-injury has no effect for the gross outcome of the two combined modes of injury. These findings are also consistent with the MTT results and suggest that exposure to a mitochondrial toxin after TBI injury produces a greater alteration in mitochondrial function than either insult alone. A similar result was reported in an in vivo model where 1 week exposure to the environmental/mitochondrial toxin, trichloroethylene, in combination with TBI, exerted a significant reduction of mitochondria bioenergetic function in the striatum 6 hours after TBI injury (Sauerbeck, Hunter, 2012).
In stretched cells treated with paraquat, a significant increase in mitochondrial ROS was observed, which is consistent with the result that TBI and mitochondria-targeted toxins can interact at the mitochondrial level. Another possibility is that after stretch, cells are in a high oxidative stress status, which makes them more vulnerable to secondary injuries. Moderate stretch enhances the loss of GSH induced by 0.5 mM paraquat, which does not correlate with mitochondrial ROS production. These results suggest that glutathione system may be altered by stretch.
MnSOD, a specific antioxidant enzyme for superoxide, is a primary cellular defense enzyme to protect cells from oxidative stress. MnSOD overexpression detoxifies paraquat-induced superoxide radicals (Hussain et al., 2004, Rodriguez-Rocha, Garcia-Garcia, 2013). We overexpressed MnSOD before exposing cells to stretch and paraquat injury (Fig. 7). Cells overexpressing MnSOD are resistant to the synergistic toxicity induced by stretch at 50% strain and prolonged exposure to 0.2 mM paraquat. These results demonstrate that the action of mitochondrial superoxide is a major player in enhancing paraquat toxicity upon treatment for an extended period of time.
In summary, we report that mitochondrial ROS generation (superoxide anion primarily) induced by mitochondrial dysfunction in an in vitro TBI model enhances the toxicity of paraquat in dopaminergic cells. These results also provide further evidence supporting the notion that mitochondrial targeted antioxidant therapy might be a promising approach against TBI-induced neurodegeneration.
Highlights.
We studied the mechanisms by which cell stretch (TBI) regulates paraquat toxicity
Paraquat-induced mitochondrial depolarization and ROS was enhanced by stretch
MnSOD diminished the deleterious effects of stretch in paraquat neurotoxicity
ROS/mitochondrial dysfunction mediate the synergistic toxicity of stretch/paraquat
Acknowledgments
This work was supported by the US Army Research Office project “Army-UNL Center of Trauma Mechanics” (Contract No.W911NF08-10483) and the National Institutes of Health Grant P20RR17675 Centers of Biomedical Research Excellence (COBRE). We are grateful to Dr. Charles A. Kuszynski, and Zhi Hong Gill at the Nebraska Center for Virology for their help with flow cytometry analyses.
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S, et al. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic Biol Med. 2013;60:282–91. doi: 10.1016/j.freeradbiomed.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulwahid Arif I, Ahmad Khan H. Environmental toxins and Parkinson’s disease: Putative roles of impaired electron transport chain and oxidative stress. Toxicology and Industrial Health. 2010;26:121–8. doi: 10.1177/0748233710362382. [DOI] [PubMed] [Google Scholar]
- Ahmed SM, Rzigalinski BA, Willoughby KA, Sitterding HA, Ellis EF. Stretch-induced injury alters mitochondrial membrane potential and cellular ATP in cultured astrocytes and neurons. J Neurochem. 2000;74:1951–60. [PubMed] [Google Scholar]
- Allen MT, Levy LS. Parkinson’s disease and pesticide exposure - a new assessment. Crit Rev Toxicol. 2013;43:515–34. doi: 10.3109/10408444.2013.798719. [DOI] [PubMed] [Google Scholar]
- Arundine M. Vulnerability of Central Neurons to Secondary Insults after In Vitro Mechanical Stretch. Journal of Neuroscience. 2004;24:8106–23. doi: 10.1523/JNEUROSCI.1362-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Tyurina YY, Tyurin V, Ruppel RA, Adelson PD, et al. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr Res. 2002;51:571–8. doi: 10.1203/00006450-200205000-00005. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–6. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Bower JH, Maraganore DM, Peterson BJ, McDonnell SK, Ahlskog JE, Rocca WA. Head trauma preceding PD: a case-control study. Neurology. 2003;60:1610–5. doi: 10.1212/01.wnl.0000068008.78394.2c. [DOI] [PubMed] [Google Scholar]
- Chaturvedi RK, Flint Beal M. Mitochondrial diseases of the brain. Free Radic Biol Med. 2013;63:1–29. doi: 10.1016/j.freeradbiomed.2013.03.018. [DOI] [PubMed] [Google Scholar]
- Cheng G, Kong RH, Zhang LM, Zhang JN. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br J Pharmacol. 2012;167:699–719. doi: 10.1111/j.1476-5381.2012.02025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung YT, Lau WK, Yu MS, Lai CS, Yeung SC, So KF, et al. Effects of all-trans-retinoic acid on human SH-SY5Y neuroblastoma as in vitro model in neurotoxicity research. NeuroToxicology. 2009;30:127–35. doi: 10.1016/j.neuro.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Franco R, Cidlowski JA. Glutathione efflux and cell death. Antioxid Redox Signal. 2012;17:1694–713. doi: 10.1089/ars.2012.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire C, Koifman S. Pesticide exposure and Parkinson’s disease: Epidemiological evidence of association. NeuroToxicology. 2012;33:947–71. doi: 10.1016/j.neuro.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Geddes DM, Cargill RS., 2nd An in vitro model of neural trauma: device characterization and calcium response to mechanical stretch. J Biomech Eng. 2001;123:247–55. doi: 10.1115/1.1374201. [DOI] [PubMed] [Google Scholar]
- Geddes DM, Cargill RS, 2nd, LaPlaca MC. Mechanical stretch to neurons results in a strain rate and magnitude-dependent increase in plasma membrane permeability. J Neurotrauma. 2003;20:1039–49. doi: 10.1089/089771503770195885. [DOI] [PubMed] [Google Scholar]
- Goldman SM, Kamel F, Ross GW, Jewell SA, Bhudhikanok GS, Umbach D, et al. Head injury, alpha-synuclein Rep1, and Parkinson’s disease. Annals of Neurology. 2012;71:40–8. doi: 10.1002/ana.22499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SM, Tanner CM, Oakes D, Bhudhikanok GS, Gupta A, Langston JW. Head injury and Parkinson’s disease risk in twins. Annals of Neurology. 2006;60:65–72. doi: 10.1002/ana.20882. [DOI] [PubMed] [Google Scholar]
- Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I, et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004;64:2350–6. doi: 10.1158/0008-5472.can-2287-2. [DOI] [PubMed] [Google Scholar]
- Hutson CB, Lazo CR, Mortazavi F, Giza CC, Hovda D, Chesselet M-F. Traumatic Brain Injury in Adult Rats Causes Progressive Nigrostriatal Dopaminergic Cell Loss and Enhanced Vulnerability to the Pesticide Paraquat. Journal of Neurotrauma. 2011;28:1783–801. doi: 10.1089/neu.2010.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari B, Ouyang B, Li LF, Hales CA, Quinn DA. Intracellular glutathione in stretch-induced cytokine release from alveolar type-2 like cells. Respirology. 2004;9:43–53. doi: 10.1111/j.1440-1843.2003.00527.x. [DOI] [PubMed] [Google Scholar]
- Kieburtz K, Wunderle KB. Parkinson’s disease: Evidence for environmental risk factors. Movement Disorders. 2013;28:8–13. doi: 10.1002/mds.25150. [DOI] [PubMed] [Google Scholar]
- Lee PC, Bordelon Y, Bronstein J, Ritz B. Traumatic brain injury, paraquat exposure, and their relationship to Parkinson disease. Neurology. 2012;79:2061–6. doi: 10.1212/WNL.0b013e3182749f28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales DM, Marklund N, Lebold D, Thompson HJ, Pitkanen A, Maxwell WL, et al. Experimental models of traumatic brain injury: Do we really need to build a better mousetrap? Neuroscience. 2005;136:971–89. doi: 10.1016/j.neuroscience.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Onizuka S, Yonaha T, Tamura R, Kasiwada M, Shirasaka T, Tsuneyoshi I. Lidocaine depolarizes the mitochondrial membrane potential by intracellular alkalization in rat dorsal root ganglion neurons. Journal of Anesthesia. 2011;25:229–39. doi: 10.1007/s00540-010-1079-y. [DOI] [PubMed] [Google Scholar]
- Perez-Otano I, Oset C, Luquin MR, Herrero MT, Obeso JA, Del Rio J. MPTP-induced parkinsonism in primates: pattern of striatal dopamine loss following acute and chronic administration. Neurosci Lett. 1994;175:121–5. doi: 10.1016/0304-3940(94)91094-4. [DOI] [PubMed] [Google Scholar]
- Redell JB, Moore AN, Grill RJ, Johnson D, Zhao J, Liu Y, et al. Analysis of Functional Pathways Altered after Mild Traumatic Brain Injury. Journal of Neurotrauma. 2013;30:752–64. doi: 10.1089/neu.2012.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rocha H, Garcia-Garcia A, Pickett C, Li S, Jones J, Chen H, et al. Compartmentalized oxidative stress in dopaminergic cell death induced by pesticides and complex I inhibitors: Distinct roles of superoxide anion and superoxide dismutases. Free Radical Biology and Medicine. 2013;61:370–83. doi: 10.1016/j.freeradbiomed.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabirov RZ, Kurbannazarova RS, Melanova NR, Okada Y. Volume-sensitive anion channels mediate osmosensitive glutathione release from rat thymocytes. PLoS ONE. 2013;8:e55646. doi: 10.1371/journal.pone.0055646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauerbeck A, Hunter R, Bing G, Sullivan PG. Traumatic brain injury and trichloroethylene exposure interact and produce functional, histological, and mitochondrial deficits. Exp Neurol. 2012;234:85–94. doi: 10.1016/j.expneurol.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Giordano S, Zelickson BR, Johnson MS, Benavides GA, Ouyang X, et al. Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radical Biology and Medicine. 2011;51:2007–17. doi: 10.1016/j.freeradbiomed.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267:4904–11. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- Shin SS, Bray ER, Zhang CQ, Dixon CE. Traumatic brain injury reduces striatal tyrosine hydroxylase activity and potassium-evoked dopamine release in rats. Brain Res. 2011;1369:208–15. doi: 10.1016/j.brainres.2010.10.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotak M, Wang F, Chandra N. An in vitro injury model for SH-SY5Y neuroblastoma cells: Effect of strain and strain rate. Journal of Neuroscience Methods. 2012;205:159–68. doi: 10.1016/j.jneumeth.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Stack EC, Ferro JL, Kim J, Del Signore SJ, Goodrich S, Matson S, et al. Therapeutic attenuation of mitochondrial dysfunction and oxidative stress in neurotoxin models of Parkinson’s disease. Biochim Biophys Acta. 2008;1782:151–62. doi: 10.1016/j.bbadis.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Szczygielski J, Mautes A, Steudel WI, Falkai P, Bayer TA, Wirths O. Traumatic brain injury: cause or risk of Alzheimer’s disease? A review of experimental studies. Journal of Neural Transmission. 2005;112:1547–64. doi: 10.1007/s00702-005-0326-0. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, et al. Rotenone, Paraquat, and Parkinson’s Disease. Environmental Health Perspectives. 2011;119:866–72. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Chen X-H, Martinez D, Browne KD, Johnson VE, Graham DI, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Experimental Neurology. 2007;208:185–92. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Giasson BI, Longhi L, Martinez D, Murray I, Conte V, et al. Age-dependent synuclein pathology following traumatic brain injury in mice. Exp Neurol. 2003;184:214–24. doi: 10.1016/s0014-4886(03)00245-0. [DOI] [PubMed] [Google Scholar]